Covalent Polymer-Drug Conjugates

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Introduction
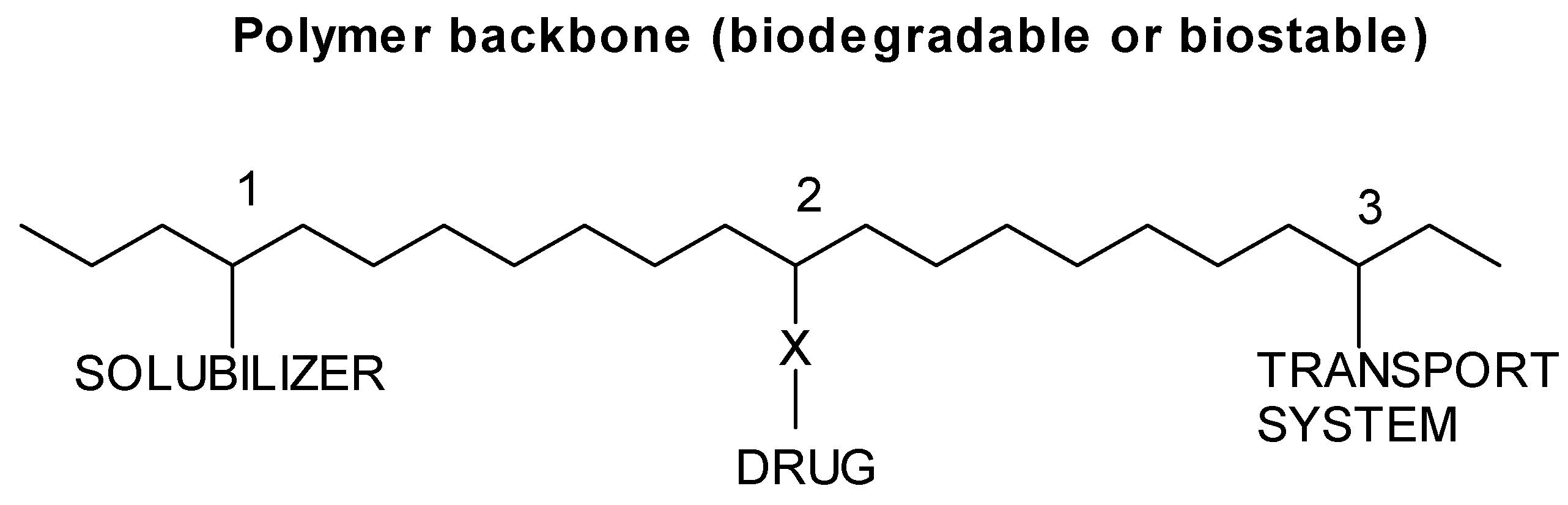
The Ringsdorf model, synthetic polymer-drug conjugates
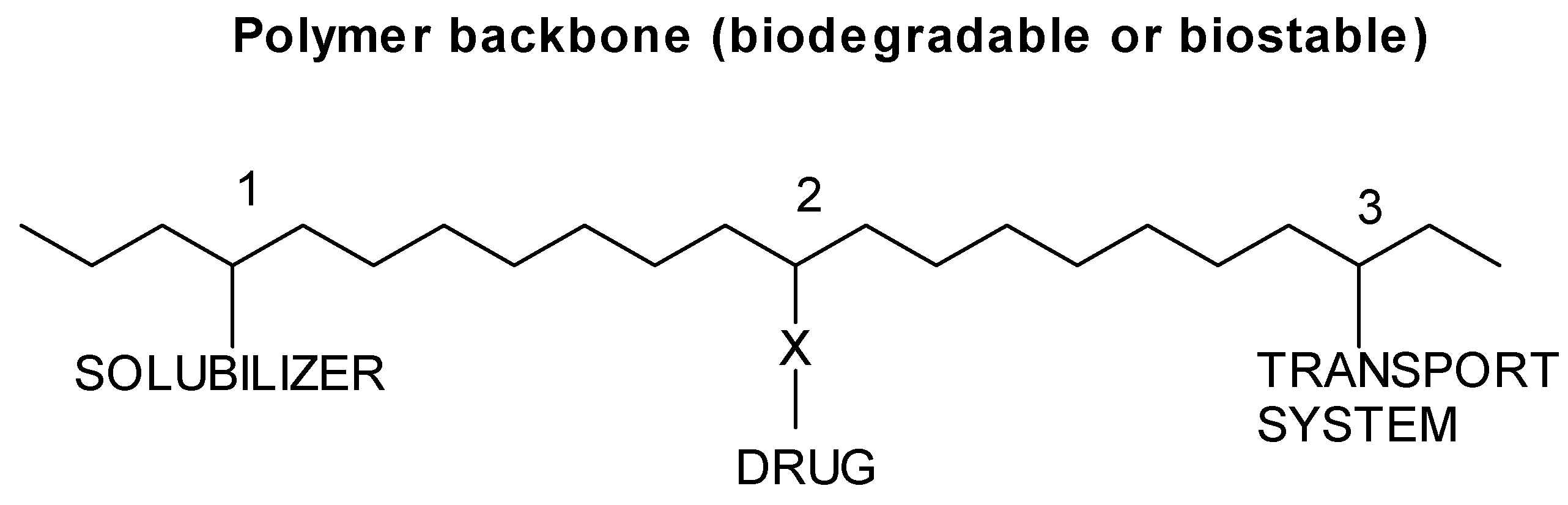
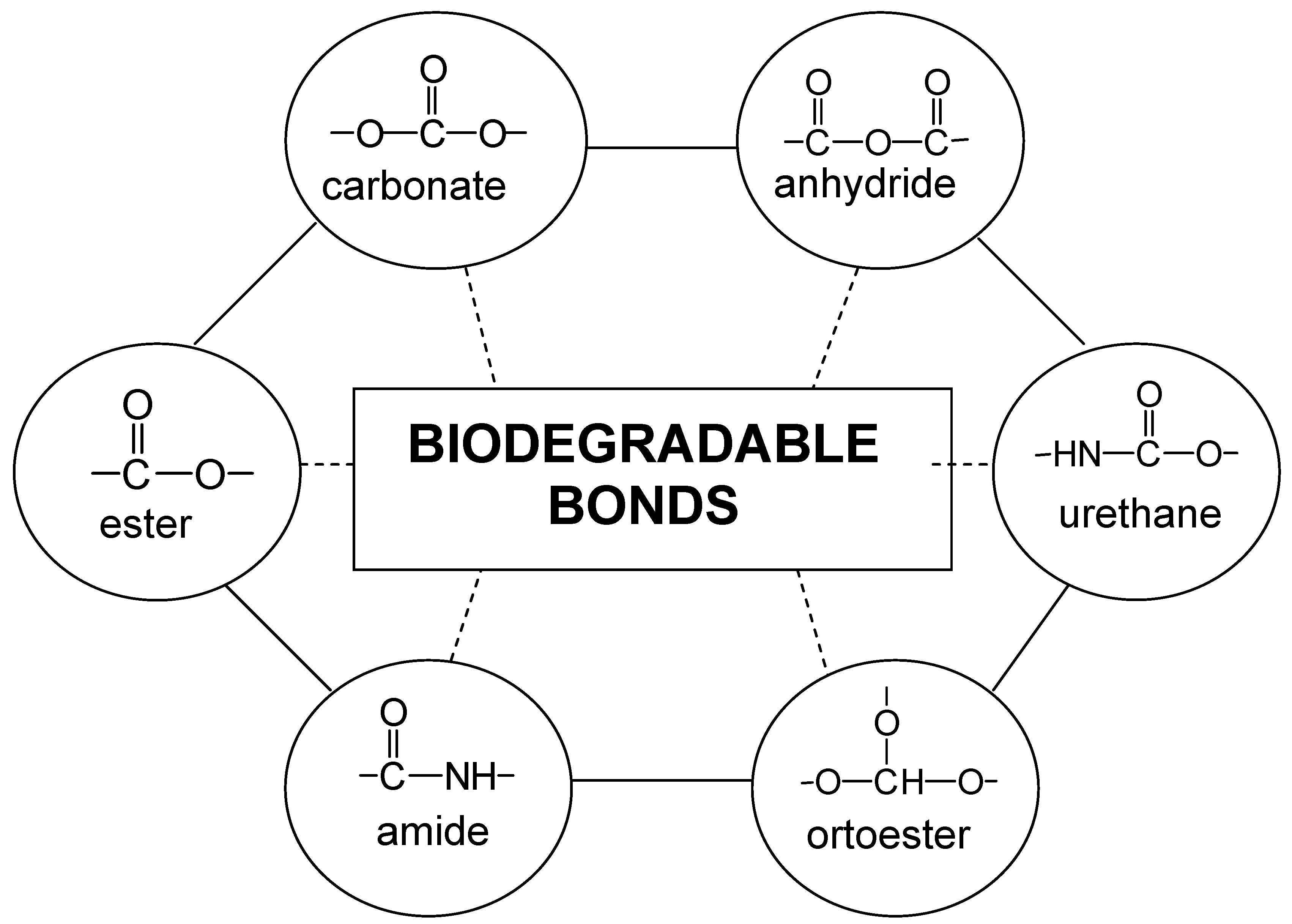
Types of polymer-drug conjugates
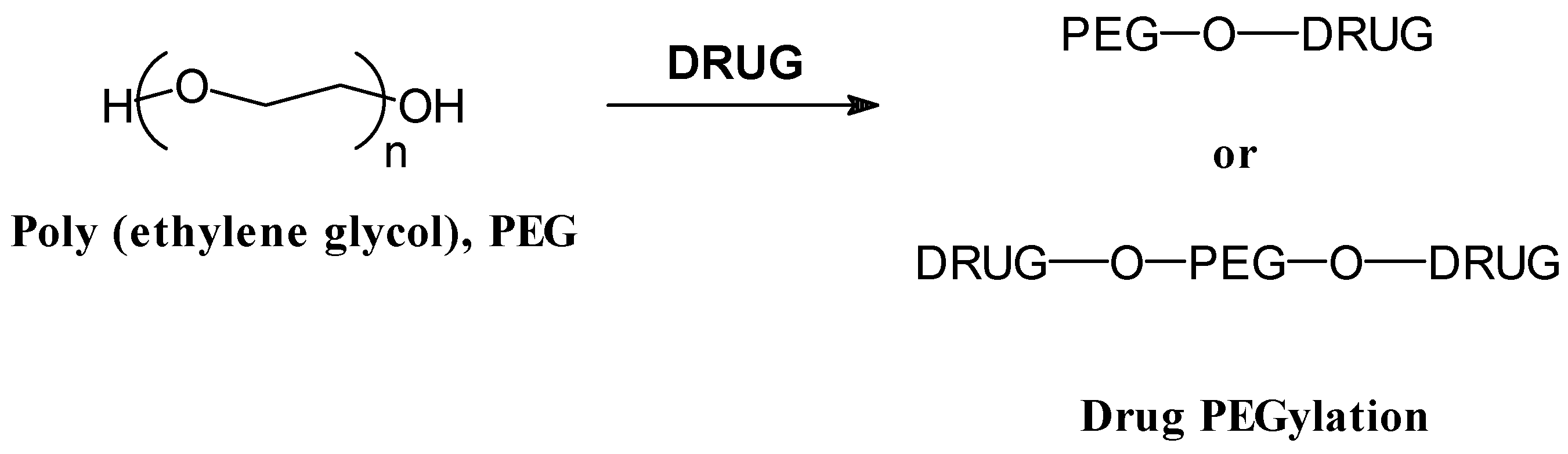
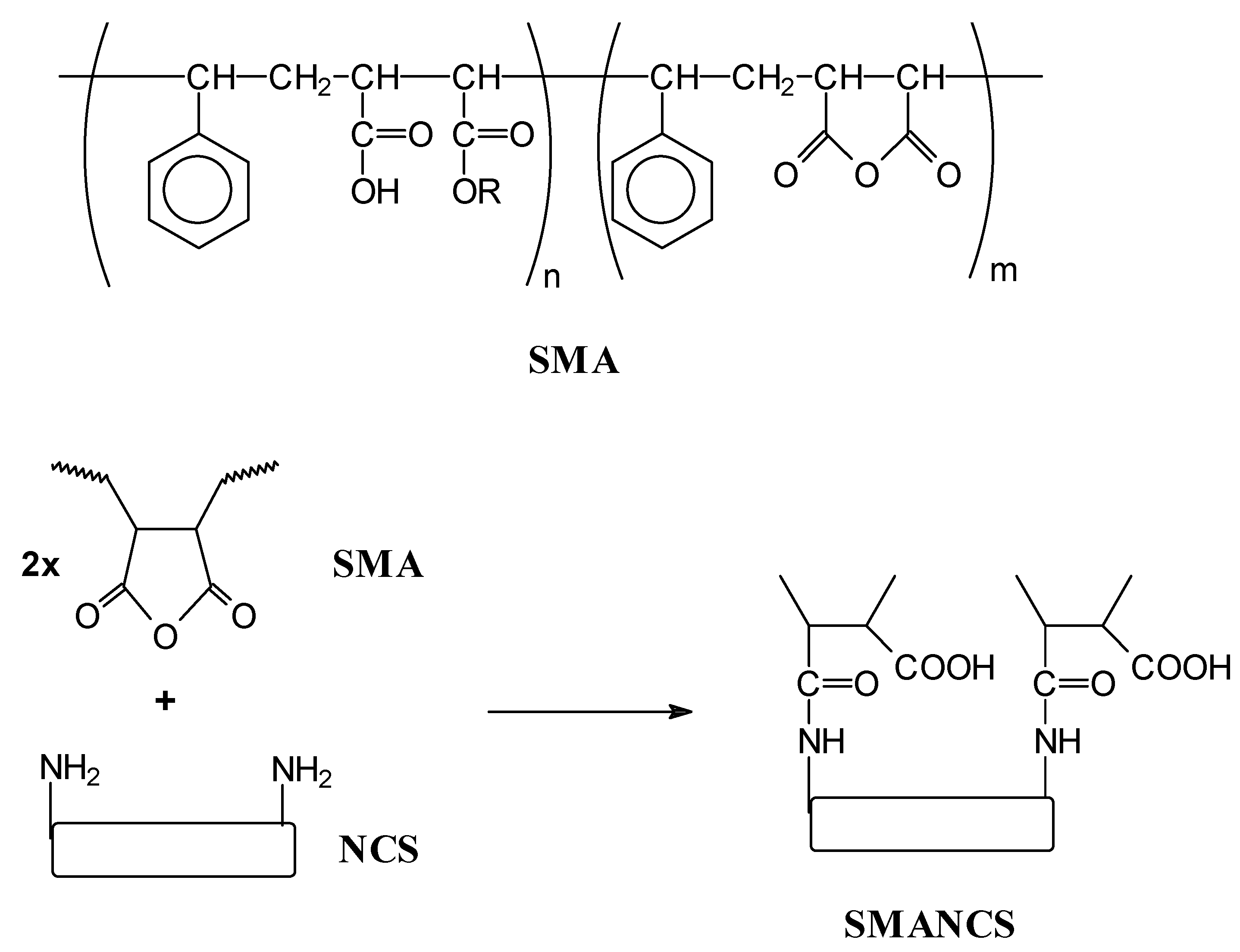
End group systems
Pendant group systems
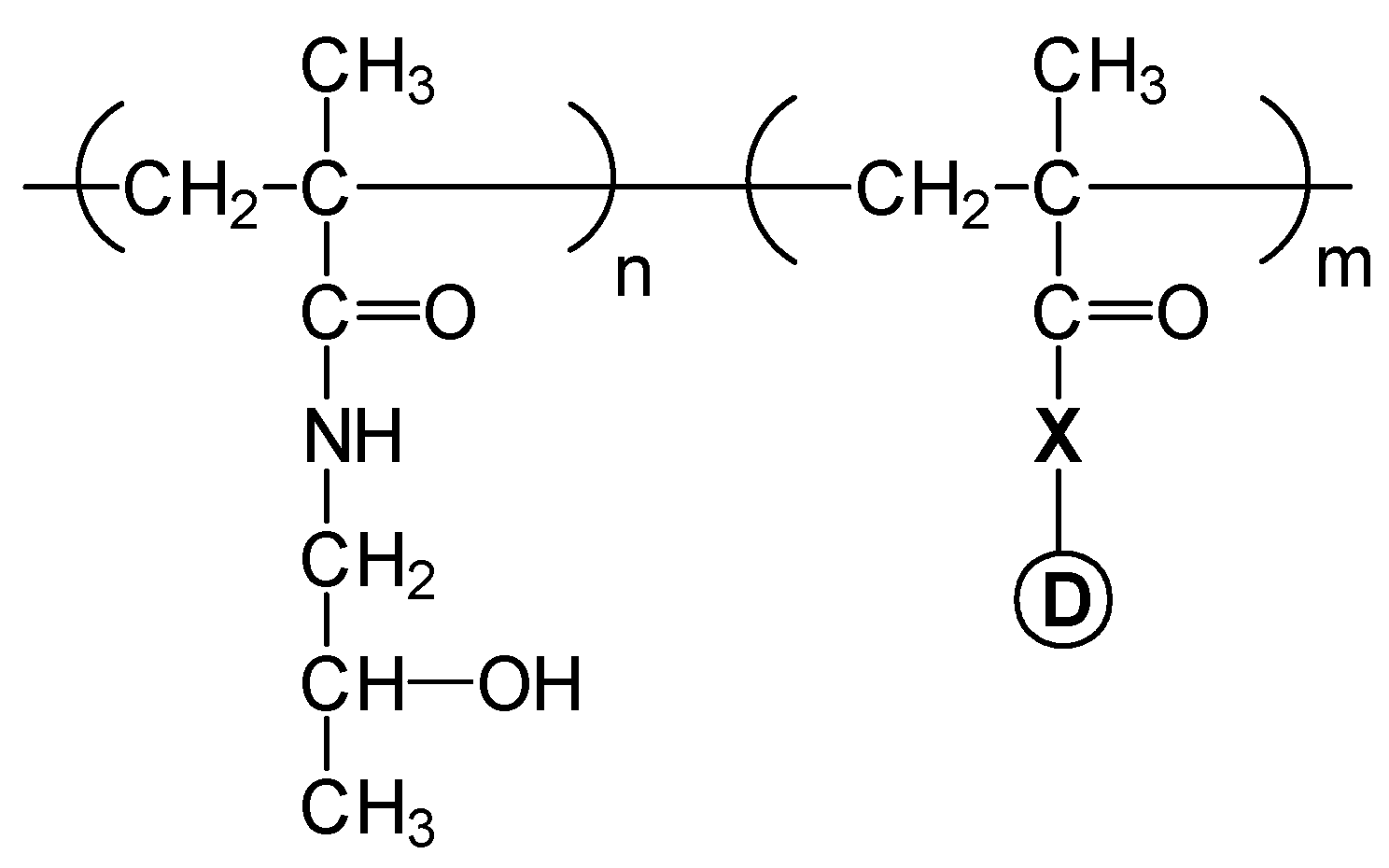
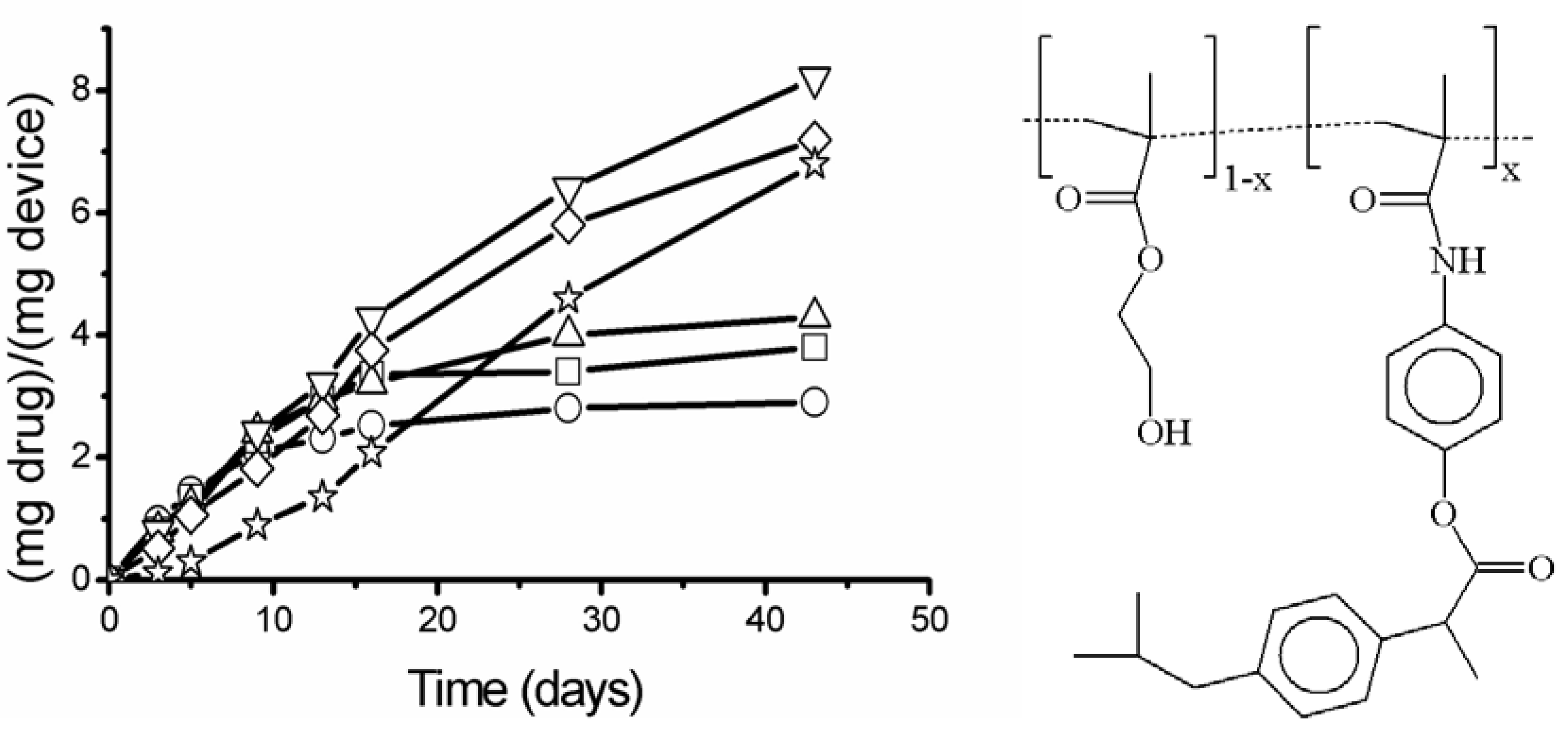
 ), 20 (
), 20 (  ) and 30 % (
) and 30 % (  ).
), 20 ( ) and 30 % ( ).
).
), 20 ( ) and 30 % ( ).
Conclusions
Acknowledgements
References
- Mathiowitz, E. Encyclopedia of Controlled Drug Delivery; Wiley-Interscience: New York, 1999. [Google Scholar]
- Kydonieus, A. Treatise on Controlled Drug Delivery; Marcel Dekker: New York, 1992. [Google Scholar]
- Baker, R. Controlled Release of Biologically Active Agents; Academic Press: New York, 1987. [Google Scholar]
- Shiino, D.; Murata, Y.; Kataoka, K.; Koyama, Y.; Yokoyama, M.; Okano , T.; Sakurai, Y. Preparation and characterization of a glucose-responsive insulin-releasing polymer device. Biomaterials 1994, 15, 121–128. [Google Scholar]
- Bromberg, L.E.; Ron, E.S. Temperature-responsive gels and thermogelling polymer matrices for protein and peptide delivery. Adv. Drug Deliv. Rev. 1998, 31, 197–221. [Google Scholar]
- Qiu, Y.; Park, K. Environment-sensitive hydrogels for drug delivery. Adv. Drug Deliv. Rev. 2001, 53, 321–339. [Google Scholar]
- Ringsdorf, H. Synthetic Polymeric Drugs. In Polymeric Delivery Systems; Kostelnik, R.J., Ed.; Gordon and Breach Science Publishers, Inc.: New York, 1978; pp. 197–225. [Google Scholar]
- Ringsdorf, H. Structure and properties of pharmacologically active polymers. J. Polym. Sci., Symp. 1975, 51, 135–153. [Google Scholar]
- Christie, R.J.; Grainger, D.W. Design strategies to improve soluble macromolecular delivery constructs. Adv. Drug Deliv. Rev. 2003, 55, 421–437. [Google Scholar]
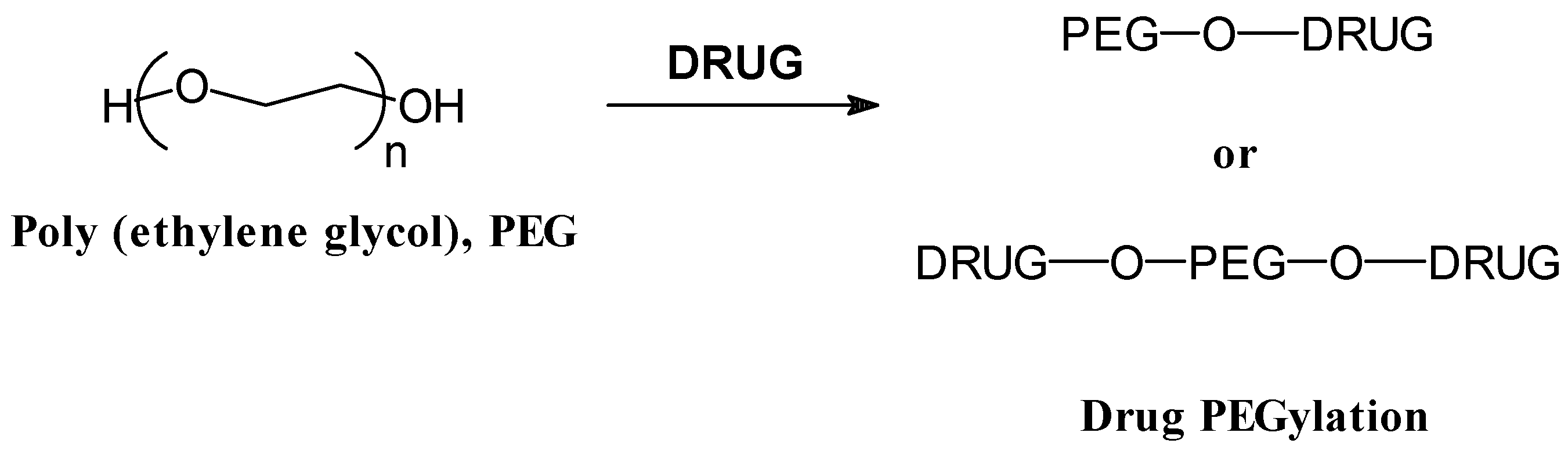
- Abuchowski, A.; Van Es, T.; Palzuk, N.C.; Davis, F.F. Effect of covalent attachment of polyethylene glycol on immunogenicity and circulating life of bovine liver catalase. J. Biol. Chem. 1977, 2, 3578–3581. [Google Scholar]
- Hooftman, G.; Herman, S.; Schacht, E. Review: poly (ethylene glycol)s with reactive endgroups. II. Practical consideration for the preparation of protein-PEG conjugates. J. Bioact. Compat. Polymers 1996, 11, 135–159. [Google Scholar]
- Zalipsky, S. Chemistry of polyethylene glycol conjugates with biologically active molecules. Adv. Drug Delivery Rev. 1995, 16, 157–182. [Google Scholar]
- Fuke, I.; Toshio, H.; Tabata, Y.; Ikada, Y. Synthesis of PEG derivatives with different branching and their use for protein modification. J. Control. Rel. 1994, 30, 27–34. [Google Scholar]
- Greenwald, R.B.; Choe, Y.H.; McGuire, J.; Conover, C.D. Effective drug delivery by PEGylated drug conjugates. Adv. Drug Deliv. Rev. 2003, 55, 217–250. [Google Scholar]
- Greenwald, R.B. PEG drugs: an overview. J. Control. Rel. 2001, 74, 159–171. [Google Scholar]
- Hershfield, M.S. Adenosine deaminase deficiency: clinical expression, molecular basis and therapy. Semen. Hematol. 1998, 35, 291–298. [Google Scholar]
- Ho, P.P.K; Milkin, E.B.; Bobbitt, J.L.; Grinnan, E.L.; Burck, P.J.; Frank, B.H.; Boeck, L.V.D.; Squires, R.W. Crystalline L-asparaginase from Escherichia coli B. J. Biol. Chem. 1970, 245, 3706–3715. [Google Scholar]
- Grace, M.; Youngster, S.; Gitlin, G.; Sydor, W.; Xie, L.; Westreich, L.; Jacobs, S.; Brassard, D.; Bausch, J.; Bordens, R. Structural and biological characterization of PEGylated recombinant IFN-α-2b. J. IFN Cytikine Res. 2001, 21, 1103–1115. [Google Scholar]
- Esslinger, H.U.; Haas, S.; Maurer, R.; Lassman, A.; Dübbers, K.; Müller-Peltzer, H. Pharmacodynamic and safety results of PEG-Hirudin in healthy volunteers. Thromb. Haemost. 1997, 77, 911–919. [Google Scholar]
- Yoo, S.D.; Jun, H.; Shin, B.S.; Lee, H.S.; Park, M.O.; Peluca, P.D.; Lee, K.C. Pharmacokinetic disposition of polyethylene glycol-modified salmon calcitonins in rats. Chem. Pharm. Bull. 2000, 48, 1921–1924. [Google Scholar]
- Greenwald, R.B.; Pendri, A.; Bolikal, D. Highly water soluble taxol derivatives. 7-Polyethylene glycol carbamates and carbonates. J. Org. Chem. 1995, 60, 331–336. [Google Scholar]
- Greenwald, R.B.; Choe, Y.H.; McGuire, J.; Conover, C.D. Effective drug delivery by PEGylated drug conjugates. Adv. Drug Deliv. Rev. 2003, 55, 217–250. [Google Scholar]
- Rapozzi, V.; Cogoi, S.; Soessotto, P.; Risso, A.; Bonora, G.M.; Quadrifoglio, F.; Xodo, L.E. Antigene effect in K562 cells of a PEG-conjugated triples-forming oligonucleotide targeted to bcrlabl oncogene. Biochemistry 2002, 41, 502–510. [Google Scholar]
- Hurwitz, E.; Klapper, L.N.; Wilchek, M.; Yarden, Y.; Sela, M. Inhibition of tumour growth by poly(ethylene glycol) derivatives of anti-ErbB2 antibodies. Cancer Immunol. Immunother. 2000, 49, 226–234. [Google Scholar]
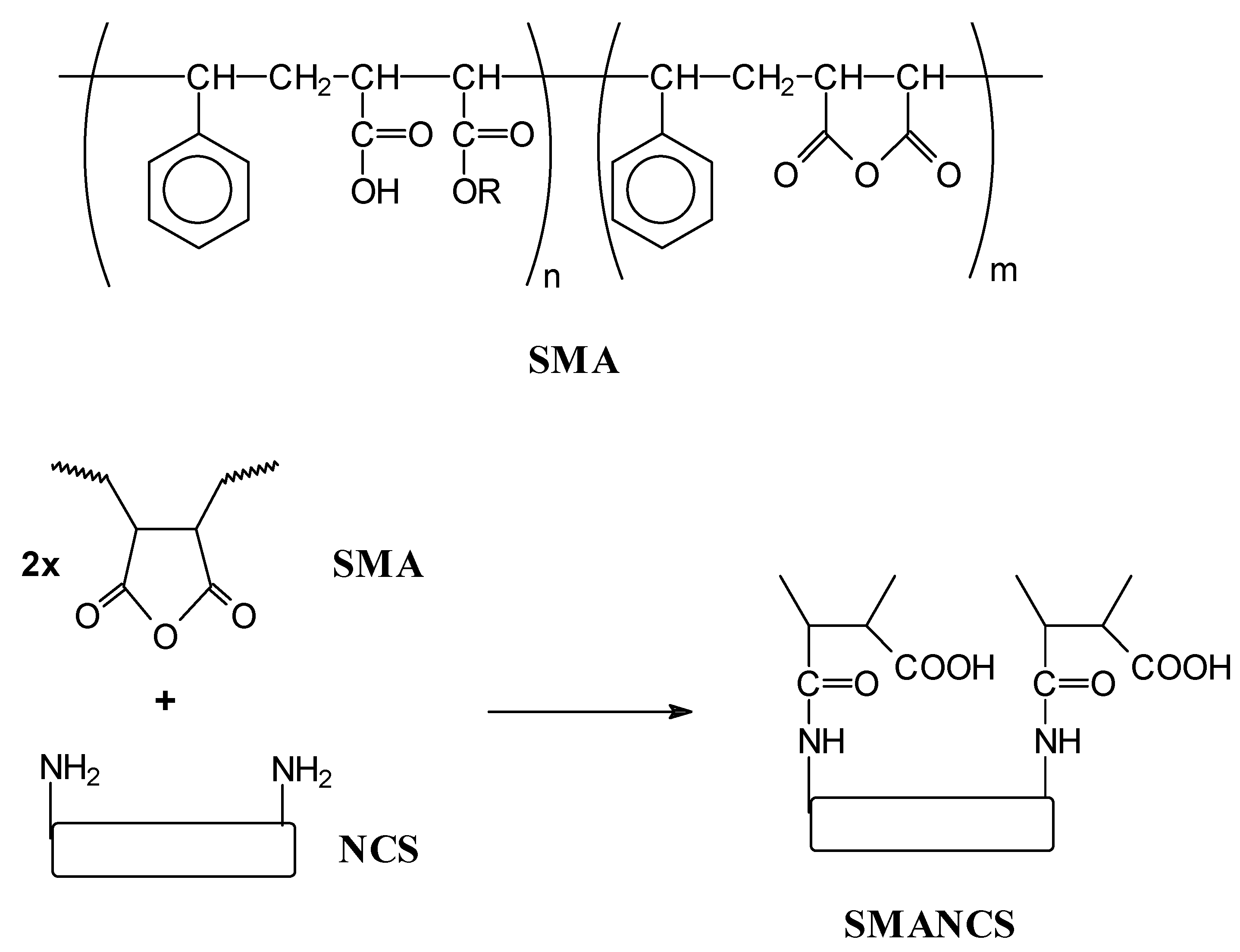
- Maeda, H. SMANCS and polymer-conjugated macromolecular drugs: advantages in cancer chemotheraphy. Adv. Drug Deliv. Rev. 1991, 6, 181–202. [Google Scholar]
- Maeda, H. SMANCS and polymer-conjugated macromolecular drugs: advantages in cancer chemotheraphy. Adv. Drug Deliv. Rev. 2001, 46, 169–185. [Google Scholar]
- Iwai, K.; Maeda, H.; Konno, T. Use of oily contrast medium for selective drug targeting to tumour: enhanced therapeutic effect and X-ray image. Cancer Res. 1984, 44, 2114–2121. [Google Scholar]
- Oka, K.; Miyamoto, Y.; Matsumura, Y.; Tanaka, S.; Oda, T.; Suzuki, F.; Maeda, H. Enhanced intestinal absorption of a hydrophobic polymer-conjugated protein drug, SMANCS, in an oily formulation. Pharmacol. Res. 1990, 7, 852–855. [Google Scholar]
- Duncan, R. Drug-polymer conjugates: potential for improved chemotherapy. Anti-Cancer Drugs 1992, 3, 175–210. [Google Scholar]
- Putman, D.; Kopecek, J. Polymer conjugates with anticancer activity. Adv. Polym. Sci. 1995, 112, 55–123. [Google Scholar]
- Duncan, R. Polymer conjugates for tumour targeting and intracytoplasmic delivery. The EPR effect as a common gateway? Pharm. Sci. Technol. To. 1999, 2, 441–449. [Google Scholar]
- Vasey, P.A.; Kaye, S.B.; Morrison, R.; Twelves, C.; Wilson, P.; Duncan, R.; Thomson, A.H.; Murray, L.S.; Hilditch, T.E.; Murray, T.; Burtles, S.; Fraier, D.; Frigerio, E.; Cassidy , J. Phase I Clinical and Pharmacokinetic Study of PK1 [N-(2-Hydroxypropyl)methacrylamide Copolymer Doxorubicin]: First Member of a New Class of Chemotherapeutic Agents—Drug-Polymer Conjugates. Clin. Cancer Res. 1999, 5, 83–84. [Google Scholar]
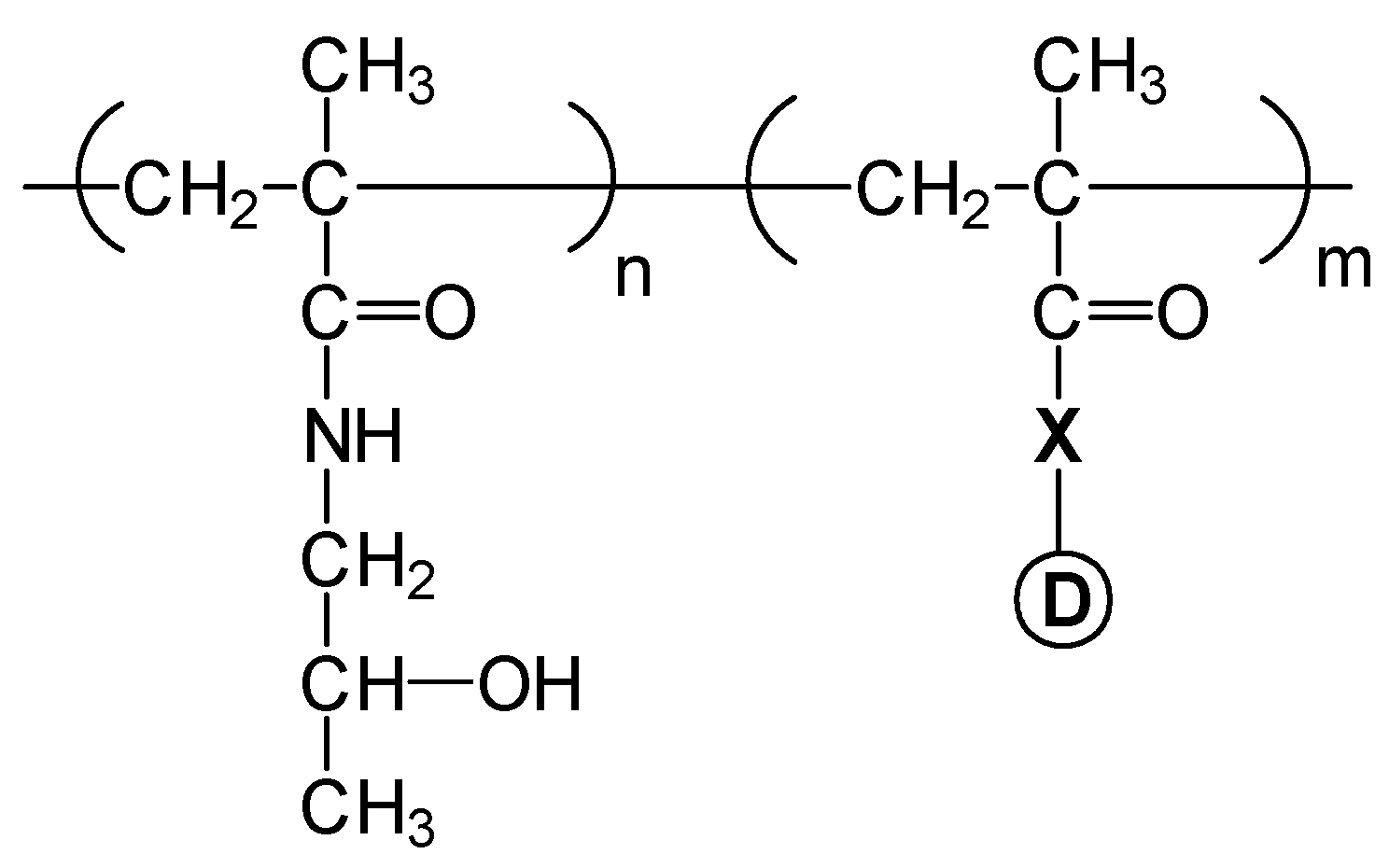
- Elvira, C.; San Román, J. Complexation of polymeric drugs based on polyacrylic chains with aminosalicylic acid side group. J. Mater. Sci.: Mater. Med. 1997, 8, 743–746. [Google Scholar]
- Rodríguez, G.; Gallardo, A.; San Román, J.; Rebuelta, M.; Bermejo, P.; Buján, J.; Bellón, J. M.; Honduvilla, N. G.; Escudero, C. New resorbable polymeric systems with antithrombogenic activity. J. Mater. Sci.: Mater. Med. 1999, 10, 873–878. [Google Scholar]
- Gallardo, A.; San Román, J. Synthesis and characterization of a new poly(methacrylamide) bearing side groups with biomedical interest. Polymer 1993, 34, 394–400. [Google Scholar]
- Ortiz, C.; Vázquez, B.; San Román, J. Synthesis, characterization and properties of polyacrylic systems derived from vitamin E. Polymer 1998, 39, 4107–4114. [Google Scholar]
- Gallardo, A.; Parejo, C.; San Román, J. NSAIDs bound to methacrylic carriers: microstructural characterization and in vitro release analysis. J. Control. Rel. 2001, 71, 127–140. [Google Scholar]
- Okano, T. Biorelared polymers and gels; Okano, T., Ed.; Academic Press: San Diego, USA, 1998. [Google Scholar]
- Chen, G.H.; Hoffman, A.S. A new temperature and pH-responsive copolymer for possible use in protein conjugation. Macromol. Chem. Phys. 1995, 196, 1251–1259. [Google Scholar]
- Hoffman, A.S. Intelligent polymers in medicine and biotechnology. Macromol. Symp. 1995, 98, 645–664. [Google Scholar] [CrossRef]
© 2005 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
Elvira, C.; Gallardo, A.; Roman, J.; Cifuentes, A. Covalent Polymer-Drug Conjugates. Molecules 2005, 10, 114-125. https://doi.org/10.3390/10010114
Elvira C, Gallardo A, Roman J, Cifuentes A. Covalent Polymer-Drug Conjugates. Molecules. 2005; 10(1):114-125. https://doi.org/10.3390/10010114
Chicago/Turabian StyleElvira, C., A. Gallardo, J. Roman, and A. Cifuentes. 2005. "Covalent Polymer-Drug Conjugates" Molecules 10, no. 1: 114-125. https://doi.org/10.3390/10010114
APA StyleElvira, C., Gallardo, A., Roman, J., & Cifuentes, A. (2005). Covalent Polymer-Drug Conjugates. Molecules, 10(1), 114-125. https://doi.org/10.3390/10010114