Impaired Sulfate Metabolism and Epigenetics: Is There a Link in Autism? †


Abstract
:1. Introduction
2. The Importance of Sulfation
3. Sulfate’s Role in the Developing Brain
3.1. Sulfation of Glycosaminoglycans
3.2. Sulfation of Serotonin
4. Findings of Abnormal Sulfate Metabolism in Autism
4.1. Evidence of Impaired Sulfoconjugation Capacity
4.2. Evidence of Increased Sulfate Excretion: Impairments in the Sodium Sulfate Cotransporter
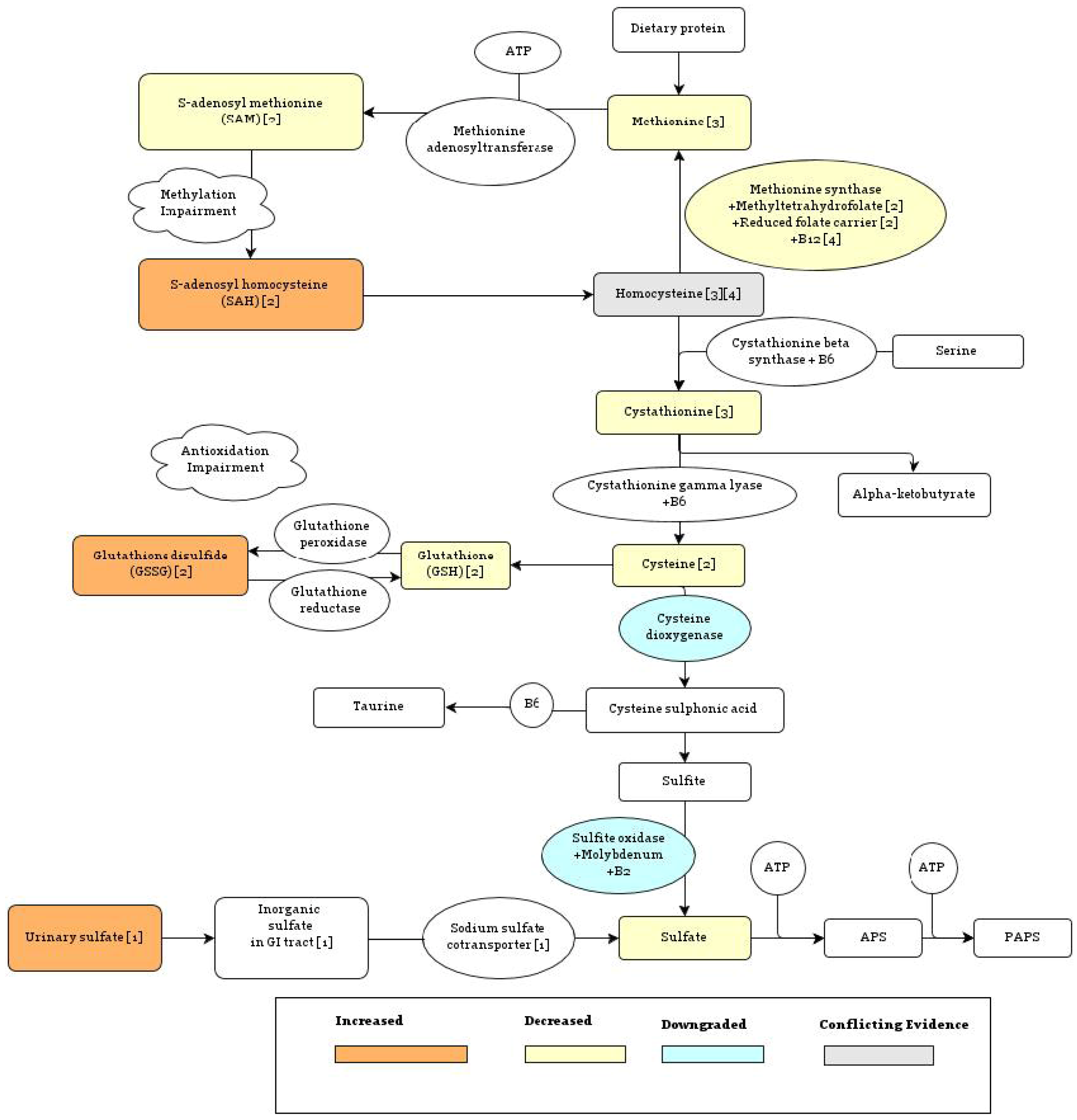
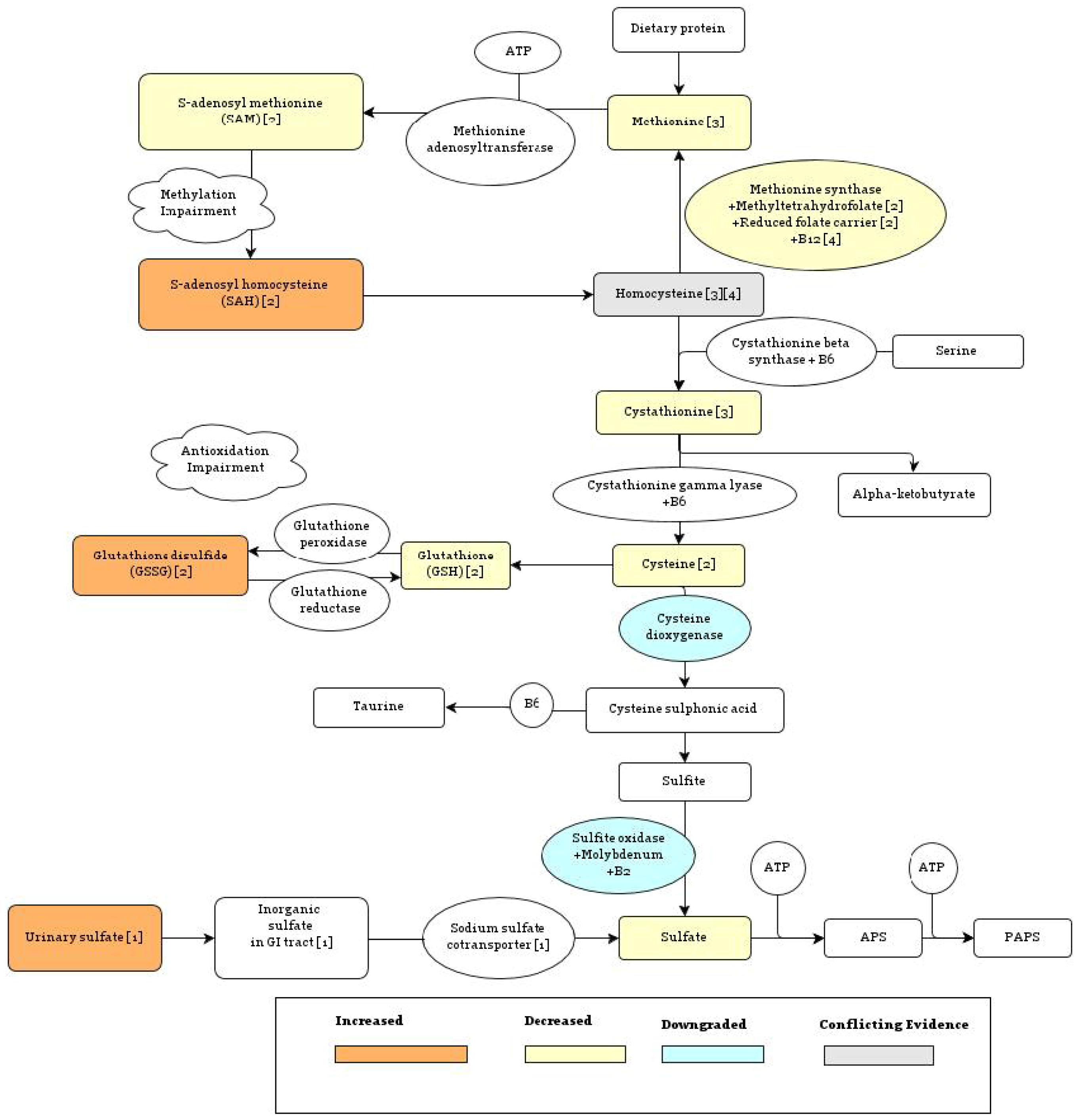{kind=link}
{kind=link}
5. Potential Causes of Abnormal Sulfate Metabolism
5.1. Genetic Factors
5.1.1. Methionine Synthase Reaction
5.1.2. Sulfotransferase Enzymes
5.1.3. Autism Associated Genes with Affinity for Heparan Sulfate
5.2. Environmental Factors
6. Reduced Sulfation Capacity Could Explain Other Symptoms Commonly Associated with Autism
6.1. Gastrointestinal Problems
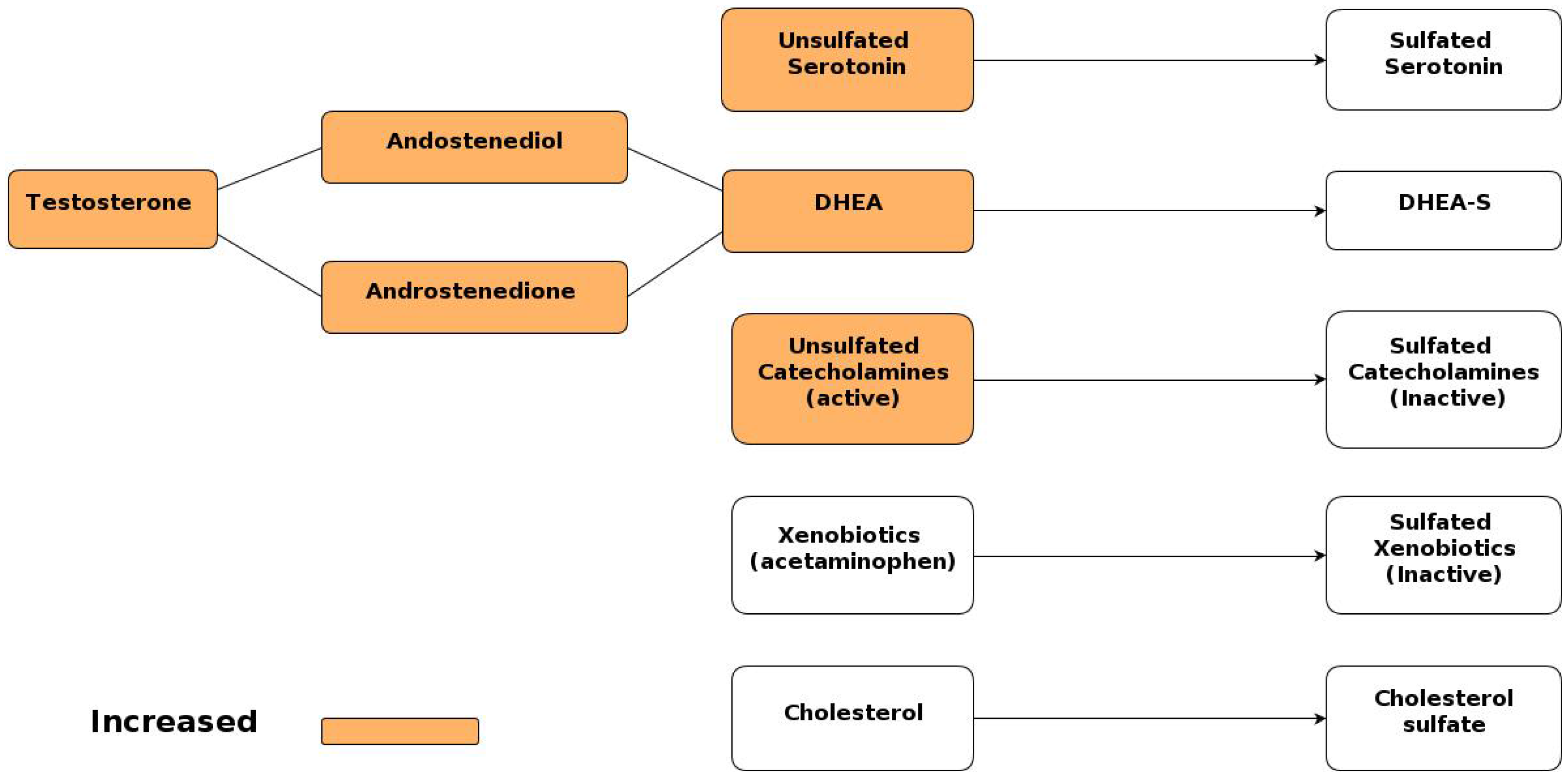
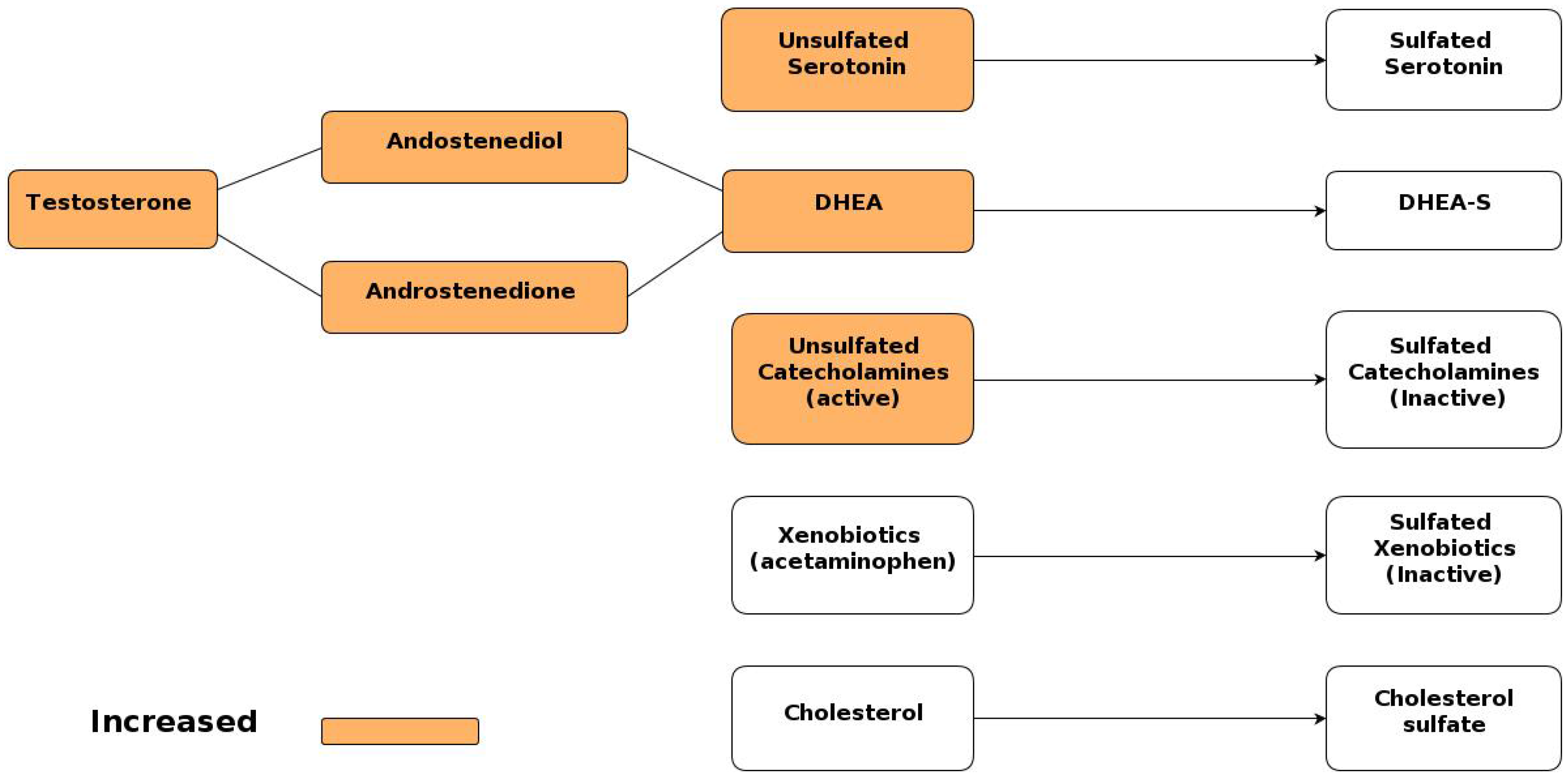
6.2. Increased Androgen Levels
6.3. Inflammation
7. Increasing Sulfate Levels Can Help Alleviate Autistic Symptoms
8. Epigenetics
9. Discussion
10. Conclusion
Acknowledgments
References
- Baio, J. Prevalence of Autism Spectrum Disorders Autism and Developmental Disabilities Monitoring Network, 14 Sites, United States, 2008; Morbidity and Mortality Weekly Report; Centers for Disease Control and Prevention: Atlanta, GA, 2012.
- Betancur, C.; Leboyer, M.; Gillberg, C. Increased rate of twins among affected sibling pairs with autism. Am. J. Hum. Genet. 2002, 70, 1381–1383. [Google Scholar] [CrossRef] [PubMed]
- Launay, J.M.; Ferrari, P.; Haimart, M.; Bursztejn, C.; Tabuteau, F.; Braconnier, A.; Pasques-Bondoux, D.; Luong, C. Serotonin Metabolism and other biochemical parameters in infantile autism: A controlled study of 22 autistic children. Neuropsychobiology 1988, 20, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Laabs, T.; Carulli, D.; Geller, H.M.; Fawcett, J.W. Chondroitin sulfate proteoglycans in neural development and regeneration. Curr. Opin. Neurobiol. 2005, 15, 116–120. [Google Scholar] [CrossRef] [PubMed]
- Al-Yafee, Y.A.; Al-Ayadhi, L.Y.; Haq, S.H.; El-Ansary, A.K. Novel metabolic biomarkers related to sulfur-dependent detoxification pathways in autistic patients of Saudi Arabia. BMC Neurol. 2011, 11, 139. [Google Scholar] [CrossRef] [PubMed]
- Kern, J.K.; Grannemann, B.D.; Trivedi, M.H.; Waring, R.H.; Ramsden, D.B.; Garver, C.R. Abnormal sulfation chemistry in autism. In Trends in Autism Research; Ryaskin, O.T., Ed.; Nova Publishers: Hauppauge, NY, USA, 2004; Chapter XI. [Google Scholar]
- Waring, R.H.; Kovrza, L.V. Sulphur metabolism in autism. J. Nutr. Environ. Med. 2000, 10, 25–32. [Google Scholar] [CrossRef]
- Geiman, T.M.; Muegge, K. DNA methylation in early development. Mol. Reprod. Dev. 2010, 77, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, D.I.; Lott, P.; Korf, I.; LaSalle, J.M. Large-scale methylation domains mark a functional subset of neuronally expressed genes. Genome Res. 2011, 21, 1583–1591. [Google Scholar] [CrossRef] [PubMed]
- Deth, R.; Muratore, C.; Benzecry, J.; Power-Charnitsky, V.-A.; Waly, M. How environmental and genetic factors combine to cause autism: A redox/ methylation hypothesis. Neurotoxicology 2008, 29, 190–201. [Google Scholar] [CrossRef] [PubMed]
- LaSalle, J.M. A genomic point-of-view on environmental factors influencing the human brain methylome. Epigenetics 2001, 6, 862–869. [Google Scholar] [CrossRef]
- Frustaci, A.; Neri, M.; Cesario, A.; Adams, J.B.; Domenici, E.; Dalla-Bernardina, B.; Bonassi, S. Oxidative stress-related biomarkers in autism: systematic review and meta-analyses. Free Radic. Biol. Med. 2012, 52, 2128–2141. [Google Scholar] [CrossRef] [PubMed]
- James, S.J.; Cutler, P.; Melnyk, S.; Jernigan, S.; Janak, L.; Gaylor, D.W.; Neubrander, J.A. Metabolic biomarkers of increased oxidative stress and impaired methylation capacity in children with autism. Am. J. Clin. Nutr. 2004, 80, 1611–1617. [Google Scholar] [PubMed]
- Brosnan, J.T.; Jacobs, R.L.; Stead, L.M.; Brosnan, M.E. Methylation demand: A key determinant of homocysteine metabolism. Acta Biochim. Pol. 2004, 51, 405–413. [Google Scholar] [PubMed]
- James, S.J.; Melnyk, S.; Jernigan, S.; Hubanks, A.; Rose, S.; Gaylor, D.W. Abnormal transmethylation/transsulfuration metabolism and DNA hypomethylation among parents of children with autism. J. Autism Dev. Disord. 2008, 38, 1966–1975. [Google Scholar] [CrossRef] [PubMed]
- Tsitsiou, E.; Sibley, C.P.; D’Souza, S.W.; Catanescu, O.; Jacobsen, D.W.; Glazier, J.D. Homocysteine is transported by the microvillous plasma membrane of human placenta. J. Inherit. Metab. Dis. 2011, 34, 57–65. [Google Scholar] [CrossRef] [PubMed]
- James, S.J.; Melnyk, S.; Pogribna, M.; Pogribny, I.P.; Caudill, M.A. Elevation in S- adenosylhomocysteine and DNA hypomethylation: Potential epigenetic mechanism for homocysteine-related pathology. J. Nutr. 2002, 132, 2361S–2366S. [Google Scholar] [PubMed]
- Caudill, M.A.; Wang, J.C.; Melnyk, S.; Pogribny, I.P.; Jernigan, S.; Collins, M.D.; Santos-Guzman, J.; Swendseid, M.E.; Cogger, E.A.; James, S.J.; et al. Intracellular S-adenosylhomocysteine concentrations predict global DNA hypomethylation in tissues of methyl deficient cystathionine b-synthase heterozygous mice. J. Nutr. 2001, 131, 2811–2818. [Google Scholar]
- Yi, P.; Melnyk, S.; Pogribna, M.; Pogribny, I.P.; Hines, R.J.; James, S.J. Increase in plasma homocysteine associated with parallel increases in plasma S-adenosylhomocysteine and throughout pregnancy predicts fetal homocysteine and birth weight. Clin. Chem. 2000, 50, 1406–1412. [Google Scholar]
- Klaassen, C.D.; Boles, J.W. Sulfation and sulfotransferases 5: The importance of 3’-phosphoadenosine 5’-phosphosulfate (PAPS) in the regulation of sulfation. FASEB J. 1997, 11, 404–418. [Google Scholar] [PubMed]
- Salman, E.D.; Kadlubar, S.A.; Falany, C.N. Expression and localization of cytosolic sulfotransferase (SULT) 1A1 and SULT1A3 in normal human brain. Drug Metab. Dispos. 2009, 37, 706–709. [Google Scholar] [CrossRef] [PubMed]
- Eagle, K. Toxicological effects of red wine, orange juice, and other dietary SULT1A inhibitors via excess catecholamines. Food Chem. Toxicol. 2012, 50, 2243–2249. [Google Scholar] [CrossRef] [PubMed]
- Dooley, T.; Obermoeller, R.; Leiter, E. Mapping of the phenol sulfotransferase gene (STP) to human chromosome 16p12.1-p11.2 and to mouse chromosome 7. Genomics 1993, 18, 440–443. [Google Scholar] [PubMed]
- Kumar, R.A.; KaraMohamed, S.; Sudi, J.; Conrad, D.F.; Brune, C.; Badner, J.A.; Gilliam, T.C.; Nowak, N.J.; Cook, E.H., Jr.; Dobyns, W.B.; et al. Recurrent 16p11.2 microdeletions in autism. Hum. Mol. Genet. 2008, 17, 628–638. [Google Scholar] [CrossRef] [PubMed]
- Weiss, L.; Shen, Y.; Korn, J. Association between microdeletion and microduplication at 16p11.2 and autism. New Engl. J. Med. 2008, 358, 667–675. [Google Scholar] [CrossRef] [PubMed]
- Whiteley, P.; Shattock, P. Biochemical aspects in autism spectrum disorders: updating the opioid-excess theory and presenting new opportunities for biomedical intervention. Expert Opin. Ther. Tar. 2002, 6, 175–183. [Google Scholar] [CrossRef] [PubMed]
- Zamek-Gliszczynski, M.J.; Hoffmaster, K.A.; Nezasa, K.; Tallman, M.N.; Brouwer, K.L. Integration of hepatic drug transporters and phase II metabolizing enzymes: Mechanisms of hepatic excretion of sulfate, glucuronide, and glutathione metabolites. Eur. J. Pharm. Sci. 2006, 27, 447–486. [Google Scholar] [CrossRef] [PubMed]
- Dawson, P. Sulfate in fetal development. Semin. Cell Dev. Biol. 2011, 22, 653–659. [Google Scholar] [CrossRef] [PubMed]
- Seneff, S.; Mascitelli, L.; Davidson, R. Might cholesterol sulfate deficiency contribute to the development of autistic spectrum disorder? Med. Hypotheses 2012, 8, 213–217. [Google Scholar] [CrossRef] [PubMed]
- Lin, B.; Kubushiro, K.; Akiba, Y.; Cui, Y.; Tsukazaki, K.; Nozawa, S.; Iwamori, M. Alteration of acidic lipids in human sera during the course of pregnancy: Characteristic increase in the concentration of cholesterol sulfate. J. Chromatogr. B. Biomed. Sci. Appl. 1997, 704, 99–104. [Google Scholar] [CrossRef]
- Lee, A.; Beck, L.; Brown, R. Identification of a mammalian brain sulfate transporter. Biochem. Biophys. Res. Commun. 1999, 263, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Blatt, G.J.; Fitzgerald, C.M.; Guptill, J.T.; Booker, A.B.; Kemper, T.L.; Bauman, M.L. Density and distribution of hippocampal neurotransmitter receptors in autism: An autoradiographic study. J. Autism Dev. Disord. 2001, 31, 537–543. [Google Scholar] [CrossRef] [PubMed]
- Fatemi, S.H.; Aldinger, K.A.; Ashwood, P.; Bauman, M.L.; Blaha, C.D.; Blatt, G.J.; Chauhan, A.; Chauhan, V.; Dager, S.R.; Dickson, P.E.; et al. Consensus paper: Pathological role of the cerebellum in autism. Cerebellum 2012, 11, 777–807. [Google Scholar] [PubMed]
- Martin, P.T. Glycobiology of the synapse. Glycobiology 2002, 12, 1R–7R. [Google Scholar] [CrossRef] [PubMed]
- Van Vactor, D.; Wall, D.P.; Johnson, K.G. Heparan sulfate proteoglycans and the emergence of neuronal connectivity. Curr. Opin. Neurobiol. 2006, 16, 40–51. [Google Scholar]
- Yamaguchi, Y. Heparan sulfate proteoglycans in the nervous system: Their diverse roles in neurogenesis, axon guidance, and synaptogenesis. Semin. Cell Dev. Biol. 2001, 12, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Medeiros, G.F.; Mendes, A.; Castro, R.A.; Bau, E.C.; Nader, H.B.; Dietrich, C.P. Distribution of sulfated glycosaminoglycans in the animal kingdom: Widespread occurrence of heparin-like compounds in invertebrates. Biochim. Biophys. Acta 2000, 1475, 287–289. [Google Scholar] [CrossRef]
- Irie, F.; Badie-Mahdavi, H.; Yamaguchi, Y. Autism-like socio-communicative deficits and stereotypies in mice lacking heparan sulfate. Proc. Natl. Acad. Sci. USA 2012, 109, 5052–5056. [Google Scholar] [CrossRef] [PubMed]
- van der Kraan, P.M.; Vitters, E.L.; de Vries, B J.; van den Berg, W.B. High susceptibility of human articular cartilage glycosaminoglycan synthesis to changes in inorganic sulfate availability. J. Orthoped. Res. 1990, 8, 565–571. [Google Scholar]
- Leboyer, M.; Philippe, A.; Bouvard, M.; Guilloud-Bataille, M.; Bondoux, D.; Tabuteau, F.; Feingold, J.; Mouren-Simeoni, M.C.; Launay, J.M. Whole blood serotonin and plasma beta-endorphin in autistic probands and their first-degree relatives. Biol. Psychiat. 1999, 45, 158–163. [Google Scholar] [CrossRef]
- Minderaa, R.B.; Anderson, G.M.; Volkmar, F.R.; Akkerhuis, G.W.; Cohen, D.J. Urinary 5-hydroxyindoleacetic acid and whole blood serotonin and tryptophan in autistic and normal subjects. Biol. Psychiat. 1987, 22, 933–940. [Google Scholar] [CrossRef]
- Whitaker-Azmitia, P.M. Serotonin and brain development: Role in human developmental diseases. Brain Res. Bull. 2001, 56, 479–485. [Google Scholar] [CrossRef]
- Whitaker-Azmitia, P.M. Behavioral and cellular consequences of increasing serotonergic activity during brain development: A role in autism? Int. J. Dev. Neurosci. 2005, 23, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Theoharides, T.C.; Zhang, B. Neuroinflammation, blood-brain barrier, seizures and autism. J. Neuroinflam. 2011, 8, 168. [Google Scholar]
- Green, L.; Fein, D.; Modahl, C.; Feinstein, C.; Waterhouse, L.; Morris, M. Oxytocin and autistic disorder: alterations in peptide forms. Biol. Psychiat. 2001, 50, 609–613. [Google Scholar] [CrossRef]
- Davis, E.; Fennoy, I.; Laraque, D.; Kanem, N.; Brown, G.; Mitchell, J. Autism and developmental abnormalities in children with perinatal cocaine exposure. J. Natl. Med. Assoc. 1988, 84, 315–319. [Google Scholar]
- Aneja, A.; Tierney, E. Cholesterol deficit in autism: Insights from Smith-Lemli-Opitz syndrome. 2008. [Google Scholar] [CrossRef]
- Scanlon, S.M.; Williams, D.C.; Schloss, P. Membrane cholesterol modulates serotonin transporter activity. Biochemistry 2001, 40, 10507–10513. [Google Scholar] [CrossRef] [PubMed]
- Geier, D.A.; Kern, J.K.; Garver, C.R.; Adams, J.B.; Audhya, T.; Geier, M.R. A prospective study of transsulfuration biomarkers in autistic disorders. Neurochem. Res. 2009, 34, 386–393. [Google Scholar] [CrossRef] [PubMed]
- Pasca, S.P.; Nemes, B.; Vlase, L.; Gagyi, C.E.; Dronca, E.; Miu, A.C.; Dronca, M. High levels of homocysteine and low serum paraoxonase 1 arylesterase activity in children with autism. Life Sci. 2006, 78, 2244–2448. [Google Scholar] [CrossRef] [PubMed]
- Geier, D.A.; Geier, M.R. A clinical and laboratory evaluation of methionine cycle- transsulfuration and androgen pathway markers in children with autistic disorders. Horm. Res. Pediatr. 2006, 66, 182–188. [Google Scholar] [CrossRef] [PubMed]
- Alberti, A.; Pirrone, P.; Elia, M.; Waring, R.H.; Romano, C. Sulphation deficit in ‘low-functioning’ autistic children: a pilot study. Biolog. Psychiat. 1999, 46, 420–424. [Google Scholar] [CrossRef]
- O’Reilly, B.A.; Waring, R.H. Enzyme and sulphur oxidation deficiencies in autistic children with known food/chemical intolerances. J. Orthomol. Med. 1993, 8, 198–200. [Google Scholar]
- Lee, A.; Dawson, P.A.; Markovich, D. NaSi-1 and Sat-1: Structure, function and transcriptional regulation of two genes encoding renal proximal tubular sulfate transporters. Int. J. Biochem. Cell. Biol. 2005, 37, 1350–1356. [Google Scholar] [CrossRef] [PubMed]
- James, S.; Melnyk, S.; Jernigan, S. Metabolic endophenotype and related genotypes are associated with oxidative stress in children with autism. Am. J. Med. Genet. B 2006, 141B, 947–956. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Azeim, S.; Li, X.; Chung, L.W.; Morokuma, K. Zinc-homocysteine binding in cobalamin-dependent methionine synthase and its role in the substrate activation: DFT, ONIOM, and QM/MM molecular dynamics studies. J. Comp. Chem. 2011, 32, 3154–3167. [Google Scholar] [CrossRef] [PubMed]
- Mosharov, E.; Cranford, M.R.; Banerjee, R. The quantitatively important relationship between homocysteine metabolism and glutathione synthesis by the transsulfuration pathway and its regulation by redox changes. Biochemistry 2000, 39, 13005–13011. [Google Scholar] [CrossRef] [PubMed]
- Matherly, L.H. Molecular and cellular biology of the human reduced folate carrier. Prog. Nucleic Acid Res. Mol. Biol. 2001, 67, 131–162. [Google Scholar] [PubMed]
- Yates, Z.; Lucock, M. G80A reduced folate carrier SNP modulates cellular uptake of folate and affords protection against thrombosis via a non homocysteine related mechanism. Life Sci. 2005, 77, 2735–2742. [Google Scholar] [CrossRef] [PubMed]
- James, S.; Melnyk, S. A functional polymorphism in the reduced folate carrier gene and DNA hypomethylation in mothers of children with autism. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2010, 153B, 1209–1220. [Google Scholar] [CrossRef] [PubMed]
- Karatela, R.; Sainani, G. Plasma homocysteine in obese, overweight and normal weight hypertensives and normotensives. Indian Heart J. 2009, 61, 156–159. [Google Scholar] [PubMed]
- Krakowiak, P.; Walker, C.K.; Bremer, A.A.; Baker, A.S.; Ozonoff, S.; Hansen, R.L.; Hertz-Picciotto, I. Maternal metabolic conditions and risk for autism and other neurodevelopmental disorders. Pediatrics 2012, 129, e1121–e1128. [Google Scholar] [CrossRef] [PubMed]
- Seetharam, B.; Bose, S.; Li, N. Cellular import of cobalamin. J. Nutr. 1999, 129, 1761–1764. [Google Scholar] [PubMed]
- Grattan-Smith, P.J.; Wilcken, B.; Procopis, P.G.; Wise, G.A. The neurological syndrome of infantile cobalamin deficiency: Developmental regression and involuntary movements. Mov. Disord. 1997, 12, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Jakubowski, H. The role of paraoxonase 1 in the detoxification of homocysteine thiolactone. Adv. Exp. Med. Biol. 2010, 660, 113–127. [Google Scholar] [PubMed]
- McCully, K.S. Chemical pathology of homocysteine V: thioretinamide, thioretinaco, and cystathionine synthase function in degenerative diseases. Annals Clin. Lab. Sci. 2011, 41, 300–313. [Google Scholar]
- Dufault, R.; Lukiw, W.J.; Crider, R.; Schnoll, R.; Wallinga, D.; Deth, R. A macroepigenetic approach to identify factors responsible for the autism epidemic in the United States. Clin. Epigenet. 2012, 4, 6. [Google Scholar]
- Ackerman, Z.; Oron-Herman, M.; Pappo, O.; Peleg, E.; Safadi, R.; Schmilovitz-Weiss, H.; Grozovski, M. Hepatic effects of rosiglitazone in rats with the metabolic syndrome. Basic Clin. Pharmacol. Toxicol. 2010, 107, 663–668. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Zhang, H.; Ma, D.; Bucan, M. Common genetic variants on 5p14.1 associate with autism spectrum disorders. Nature 2009, 459, 528–533. [Google Scholar] [PubMed]
- Alarcón, M.; Abrahams, B. Linkage, association, and gene-expression analyses identify CNTNAP2 as an autism-susceptibility gene. Am. J. Hum. Genet. 2008, 82, 150–159. [Google Scholar] [CrossRef] [PubMed]
- Arking, D.E.; Cutler, D.J.; Brune, C.W.; Teslovich, T.M.; West, K.; Ikeda, M.; Rea, A.; Guy, M.; Lin, S.; Cook, E.H.; et al. A common genetic variant in the neurexin superfamily member CNTNAP2 increases familial risk of autism. Am. J. Hum. Genet. 2008, 82, 160–164. [Google Scholar] [PubMed]
- Feng, J.; Schroer, R.; Yan, J.; Song, W.; Yang, C.; Bockholt, A.; Cook, E.H., Jr.; Skinner, C.; Schwartz, C.E.; Sommer, S.S. High frequency of neurexin 1beta signal peptide structural variants in patients with autism. Neurosci. Lett. 2006, 409, 10–13. [Google Scholar] [CrossRef] [PubMed]
- Betancur, C.; Sakurai, T.; Buxbaum, J.D. The emerging role of synaptic cell-adhesion pathways in the pathogenesis of autism spectrum disorders. Trends Neurosci. 2009, 32, 402–412. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, R.; Hansen, R.; Hartiala, J. Prenatal vitamins, one-carbon metabolism gene variants, and risk for autism. Epidemiology 2011, 22, 476–485. [Google Scholar] [CrossRef] [PubMed]
- Bolt, M.J.G.; Liu, W.; Qiao, G.; Kong, J.; Zheng, W.; Krausz, T.; Cs-Szabo, G.; Sitrin, M.D.; Li, Y.C. Critical role of vitamin D in sulfate homeostasis: Regulation of the sodium-sulfate cotransporter by 1,25-dihydroxyvitamin D3. Am. J. Physiol. Endocrinol. Metab. 2004, 287, E744–E749. [Google Scholar] [CrossRef] [PubMed]
- Davidson, R.M.; Seneff, S. The initial common pathway of inflammation, disease, and sudden death. Entropy 2012, 14, 1399–1442. [Google Scholar] [CrossRef]
- Strott, C.A. Cholesterol sulfate in human physiology: What’s it all about? J. Lipid Res. 2003, 44, 1268–1278. [Google Scholar] [CrossRef] [PubMed]
- Rearick, J.I.; Stoner, G.D.; George, M.A.; Jetten, A.M. Cholesterol Sulfate Accumulation in Tumorigenic and Nontumorigenic Rat Esophageal Epithelial Cells: Evidence for Defective Differentiation Control in Tumorigenic Cells. Cancer Res. 1988, 48, 5289–5295. [Google Scholar] [PubMed]
- Grant, W.B.; Soles, C.M. Epidemiologic evidence supporting the role of maternal vitamin D deficiency as a risk factor for the development of infantile autism. Dermatoendocrinol. 2009, 1, 223–238. [Google Scholar] [CrossRef] [PubMed]
- Gardener, H.; Spiegelman, D.; Buka, S.L. Prenatal risk factors for autism: comprehensive meta-analysis. Brit. J. Pschiat. 2009, 195, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Barnevik-Olsson, M.; Gillberg, C.; Fernell, E. Prevalence of autism in children born to Somali parents living in Sweden: A brief report. Dev. Med. Child Neurol. 2008, 50, 598–601. [Google Scholar] [CrossRef] [PubMed]
- Pittas, A.G.; Lau, J.; Hu, F.B.; Dawson-Hughes, B. The role of vitamin D and calcium in type 2 diabetes: A systematic review and meta-analysis. J. Clin. Endocrinol. Metab. 2007, 92, 2017–2029. [Google Scholar] [CrossRef] [PubMed]
- Bodnar, L.M.; Catov, J.M.; Simhan, H.N.; Holick, M.F.; Powers, R.W.; Roberts, J.M. Maternal vitamin D deficiency increases the risk of preeclampsia. J. Clin. Endocrinol. Metab. 2007, 92, 3517–3522. [Google Scholar] [CrossRef] [PubMed]
- Mann, J.R.; McDermott, S.; Bao, H.; Hardin, J.; Gregg, A. Preeclampsia, birth weight, and autism spectrum disorders. J. Autism Dev. Disord. 2010, 40, 548–554. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Klaassen, C.D. Different mechanism of saturation of acetaminophen sulfate conjugation in mice and rats. Toxicol. Appl. Pharmacol. 1996, 139, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Schultz, S.T.; Klonoff-Cohen, H.S.; Wingard, D.L.; Akshoomoff, N.A.; Macera, C.A.; Ji, M. Acetaminophen (paracetamol) use, measles-mumps-rubella vaccination, and autistic disorder: The results of a parent survey. Autism 2008, 12, 293–307. [Google Scholar] [CrossRef] [PubMed]
- Waly, M.; Olteanu, H.; Banerjee, R.; Choi, S.W.; Mason, J.B.; Parker, B.S.; Sukumar, S.; Shim, S.; Sharma, A.; Benzecry, J.M.; et al. Activation of methionine synthase by insulin-like growth factor-1 and dopamine: a target for neurodevelopmental toxins and thimerosal. Mol. Psychiatry 2004, 9, 358–370. [Google Scholar] [PubMed]
- Geier, D.A.; Kern, J.K.; Garver, C.R.; Adams, J.B.; Audhya, T.; Nataf, R.; Geier, M.R. Biomarkers of environmental toxicity and susceptibility in autism. J. Neurol. Sci. 2009, 280, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Breitkreutz, R.; Pittack, N.; Nebe, C.T.; Schuster, D.; Brust, J.; Beichert, M.; Hack, V.; Daniel, V.; Edler, L.; Dröge, W. Improvement of immune functions in HIV infection by sulfur supplementation: Two randomized trials. J. Mol. Med. (Berl.). 2000, 78, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Dröge, W.; Breitkreutz, R. Glutathione and immune function. Proc. Nutr. Soc. 2000, 59, 595–600. [Google Scholar] [CrossRef] [PubMed]
- Martensson, J.; Jain, A.; Meister, A. Glutathione is required for intestinal function. Proc. Natl. Acad. Sci. USA 1990, 87, 1715–1719. [Google Scholar] [CrossRef] [PubMed]
- Horvath, K.; Perman, J.A. Autism and gastrointestinal symptoms. Curr. Gastroenterol. Rep. 2002, 4, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Theoharides, T.C.; Doyle, R.; Francis, K.; Conti, P.; Kalogeromitros, D. Novel therapeutic targets for autism. Trends Pharmacol. Sci. 2008, 29, 375–382. [Google Scholar] [CrossRef] [PubMed]
- Furlano, R.I.; Anthony, A.; Day, R.; Brown, A.; McGarvey, L.; Thomson, M.A.; Davies, S.E.; Berelowitz, M.; Forbes, A.; Wakefield, A.J.; et al. Colonic CD8 and γδ T-cell infiltration with epithelial damage in children with autism. J. Pediatr. 2001, 138, 366–372. [Google Scholar]
- Murch, S.H.; MacDonald, T.T.; Walker-Smith, J.A.; Levin, M.; Lionetti, P.; Klein, N.J. Disruption of sulphated glycosaminoglycans in intestinal inflammation. Lancet 1993, 341, 711–714. [Google Scholar]
- Strous, R.D.; Golubchik, P.; Maayan, R.; Mozes, T.; Tuati-Werner, D.; Weizman, A.; Spivak, B. Lowered DHEA-S plasma levels in adult individuals with autistic disorder. Eur. Neuropsychopharmacol. 2005, 15, 305–309. [Google Scholar] [CrossRef] [PubMed]
- Geier, D.A.; Geier, M.R. A prospective assessment of androgen levels in patients with autistic spectrum disorders: Biochemical underpinnings and suggested therapies. Neuro. Endocrinol. Lett. 2007, 28, 565–573. [Google Scholar] [PubMed]
- Geier, D.A.; Kern, J.K.; King, P.G.; Sykes, L.K.; Geier, M.R. An evaluation of the role and treatment of elevated male hormones in autism spectrum disorders. Acta Neurobiol. Exp. (Wars). 2012, 72, 1–17. [Google Scholar] [PubMed]
- Prudova, A.; Albin, M.; Bauman, Z.; Lin, A.; Vitvitsky, V.; Banerjee, R. Testosterone Regulation of Homocysteine metabolism modulates redox status in human prostate cancer cells. Antiox. Redox Sig. 2007, 9, 1875–1882. [Google Scholar] [CrossRef] [PubMed]
- Ashwood, P. The immune response in autism: A new frontier for autism research. J. Leukoc. Biol. 2006, 80, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Blaylock, R.L.; Strunecka, A. Immune-glutamatergic dysfunction as a central mechanism of the autism spectrum disorders. Curr. Med. Chem. 2009, 16, 157–170. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, A.; Chauhan, V. Oxidative stress in autism. Pathophysiology 2006, 13, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Kern, J.K.; Geier, D.A.; Adams, J.B.; Garver, C.R.; Audhya, T.; Geier, M.R. A clinical trial of glutathione supplementation in autism spectrum disorders. Med. Sci. Monit. 2011, 17, CR677–C682. [Google Scholar] [CrossRef] [PubMed]
- Martineau, J.; Barthelemy, C.; Garreau, B.; Lelord, G. Vitamin B6, magnesium, and combined B6-Mg: Therapeutic effects in childhood autism. Biol. Psychiatry 1985, 20, 467–478. [Google Scholar] [CrossRef]
- Mousain-Bosc, M.; Roche, M.; Polge, A.; Pradal-Prat, D.; Rapin, J.; Bali, J.P. Improvement of neurobehavioral disorders in children supplemented with magnesium-vitamin B6. II. Pervasive developmental disorder-autism. Magnesium Res. 2006, 19, 53–62. [Google Scholar]
- Allen, L.H. Causes of vitamin B12 and folate deficiency. Food Nutr. Bull. 2008, 29, 20–34. [Google Scholar]
- Reik, W. Stability and flexibility of epigenetic gene regulation in mammalian development. Nature 2007, 447, 425–432. [Google Scholar] [CrossRef] [PubMed]
- Hutnick, L.K.; Golshani, P.; Namihira, M.; Xue, Z.; Matynia, A.; Yang, W.X.; Silva, A.J.; Schweizer, F.E.; Fan, G. DNA hypomethylation restricted to the murine forebrain induces cortical degeneration and impairs postnatal neuronal maturation. Hum. Mol. Genet. 2009, 18, 2875–2888. [Google Scholar] [CrossRef] [PubMed]
- Rinaldi, T.; Kulangara, K.; Antoniello, K.; Markram, H. Elevated NMDA receptor levels and enhanced postsynaptic long-term potentiation induced by prenatal exposure to valproic acid. Proc. Natl. Acad. Sci. USA 2007, 104, 13501–13506. [Google Scholar] [CrossRef] [PubMed]
- Lauri, S.E.; Kaukinen, S.; Kinnunen, T.; Ylinen, A.; Imai, S.; Kaila, K.; Taira, T.; Rauvala, H. Regulatory role and molecular interactions of a cell-surface heparan sulfate proteoglycan (N-syndecan) in hippocampal long-term potentiation. J. Neurosci. 1999, 19, 1226–12235. [Google Scholar] [PubMed]
- Kaksonen, M.; Pavlov, I.; Voikar, V.; Lauri, S.E.; Hienola, A.; Riekki, R.; Lakso, M.; Taira, T.; Rauvala, H. Syndecan-3-deficient mice exhibit enhance LTP and impaired hippocampus-dependent memory. Mol. Cell Neurosci. 2002, 21, 158–172. [Google Scholar] [CrossRef] [PubMed]
- Fan, G.; Martinowich, K.; Chin, M.H.; He, F.; Fouse, S.D.; Hutnick, L.; Hattori, D.; Ge, W.; Shen, Y.; Wu, H.; et al. DNA methylation controls the timing of astrogliogenesis through regulation of JAK-STAT signaling. Development 2005, 132, 3345–3356. [Google Scholar] [PubMed]
- Laurence, J.A.; Fatemi, S.H. Glial fibrillary acidic protein is elevated in superior frontal, parietal and cerebellar cortices of autistic subjects. Cerebellum 2005, 4, 206–210. [Google Scholar] [CrossRef] [PubMed]
- Göttlicher, M.; Minucci, S.; Zhu, P.; Krömer, O.H.; Schimpf, A.; Giavara, S.; Sleeman, J.P.; Lo-Coco, F.; Nervi, C.; Pelicci, P.G.; et al. Valproic acid defines a novel class of HDAC inhibitors inducing differentiation of transformed cells. EMBO J. 2001, 20, 6969–69678. [Google Scholar] [PubMed]
- Woods, R.; Vallero, R.O.; Golub, M.S.; Suarez, J.K.; Ta, T.A.; Yasui, D.H.; Chi, L.H.; Kostyniak, P.J.; Pessah, I.N.; Berman, R.F.; LaSalle, J.M. Long-lived epigenetic interactions between perinatal PBDE exposure and Mecp2308 mutation. Hum. Mol. Genet. 2012, 21, 2399–2411. [Google Scholar] [CrossRef] [PubMed]
- Waterland, R.A.; Jirtle, R.L. Early nutrition, epigenetic changes at transposons and imprinted genes, and enhanced susceptibility to adult chronic diseases. Nutrition 2004, 20, 63–68. [Google Scholar] [CrossRef] [PubMed]
- De Rooij, S.R.; Painter, R.C.; Phillips, D.I.W.; Osmond, C.; Michels, R.P.J.; Godsland, I.F.; Bossuyt, P.M.M.; Bleker, O.P.; Roseboom, T.J. Impaired insulin secretion after prenatal exposure to the Dutch famine. Diabetes Care 2006, 29, 1897–1901. [Google Scholar] [CrossRef] [PubMed]
- Jirtle, R.L.; Skinner, M.K. Environmental epigenomics and disease susceptibility. Nat. Rev. Genet. 2007, 8, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Roberts, L. Controversial from the start. Science 2001, 291, 1182–1188. [Google Scholar] [CrossRef] [PubMed]
- The ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar]
- Biémont, C.; Vieira, C. Genetics: Junk DNA as an evolutionary force. Nature 2006, 443, 521–524. [Google Scholar] [CrossRef] [PubMed]
- Rakyan, V.K.; Chong, S.; Champ, M.E.; Cuthbert, P.C.; Morgan, H.D.; Luu, K.V.K.; Whitelaw, E. Transgenerational inheritance of epigenetic states at the murine AxinFu allele occurs after maternal and paternal transmission. Proc. Natl. Acad. Sci. USA 2003, 100, 2538–2543. [Google Scholar]
- Dolinoy, D.C. The agouti mouse model: An epigenetic biosensor for nutritional and environmental alterations on the fetal epigenome. Nutr. Rev. 2008, 66, S7–S11. [Google Scholar] [CrossRef] [PubMed]
- Miltenberger, R.; Mynatt, R.; Wilkinson, J.; Woychik, R. The role of the agouti gene in the yellow obese syndrome. J. Nutr. 1997, 127, S1902–S1907. [Google Scholar]
- Bonilla, C.; Boxill, L.A.; Donald, S.A.; Williams, T.; Sylvester, N.; Parra, E.J.; Dios, S.; Norton, H.L.; Shriver, M.D.; Kittles, R.A. The 8818G allele of the agouti signaling protein (ASIP) gene is ancestral and is associated with darker skin color in African Americans. Hum. Genet. 2005, 116, 402–406. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, J.B.; Chen, Y.; Jiang, N.; Beasley, B.E.; Salerno, J.C.; Ghosh, D.K. Inhibition of nitric oxide synthase by cobalamins and cobinamides. Free Radic. Biol. Med. 2009, 46, 1626–1632. [Google Scholar] [CrossRef] [PubMed]
- Fulton, D.; Gratton, J.-P.; Sessa, W.C. Post-translational control of endothelial nitric oxide synthase: Why isn’t calcium/calmodulin enough? J. Pharmacol. Exp. Ther. 2001, 299, 818–824. [Google Scholar] [PubMed]
- Sögüt, S.S.; Zoroglu, S.S.; Özyurt, H.; Ylmaz, H.R.; Ozugurlu, F.; Sivasli, E.; Yetkin, O.; Yanik, M.; Tutkun, H.; Savas, H.A.; et al. Changes in nitric oxide levels and antioxidant enzyme activities may have a role in the pathophysiological mechanisms involved in autism. Clin. Chim. Acta 2003, 331, 111–117. [Google Scholar] [PubMed]
- Akyol, O.; Zoroglu, S.S.; Armutcu, F.; Sahin, S.; Gurel, A. Nitric Oxide as a Physiopathological Factor in Neuropsychiatric Disorders. In Vivo 2004, 18, 377–390. [Google Scholar] [PubMed]
- Chen, Y.; Huang, C.; Zhou, T.; Chen, G. Genistein induction of human sulfotransferases in HepG2 and Caco-2 cells. Basic Clin. Pharmacol. Toxicol. 2008, 103, 553–559. [Google Scholar] [CrossRef] [PubMed]
- Asaba, H.; Hosoya, K.; Takanaga, H.; Ohtsuki, S.; Tamura, E.; Takizawa, T.; Terasaki, T. Blood-brain barrier is involved in the efflux transport of a neuroactive steroid, dehydroepiandrosterone sulfate, via organic anion transporting polypeptide 2. J. Neurochem. 2000, 75, 1907–1916. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.Y.; Roh, D.H.; Seo, H.S.; Kang, S.Y.; Han, H.J.; Beitz, A.J.; Lee, J.H. Intrathecal injection of the neurosteroid, DHEAS, produces mechanical allodynia in mice: Involvement of spinal sigma-1 and GABAA receptors. Br. J. Pharmacol. 2009, 157, 666–773. [Google Scholar]
- Chisari, M.; Wu, K.; Zorumski, C.F.; Mennerick, S. Hydrophobic anions potently and uncompetitively antagonize GABA(A) receptor function in the absence of a conventional binding site. Br. J. Pharmacol. 2011, 164, 667–680. [Google Scholar] [CrossRef] [PubMed]
- Menold, M.M.; Shao, Y.; Wolpert, C.M.; Donnelly, S.L.; Raiford, K.L.; Martin, E.R.; Ravan, S.A.; Abramson, R.K.; Wright, H.H.; Delong, G.R.; et al. Association analysis of chromosome 15 GABAA receptor subunit genes in autistic disorder. J. Neurogenet. 2001, 15, 245–259. [Google Scholar] [PubMed]
- Goin-Kochel, R.P.; Porter, A.E.; Peters, S.U.; Shinawi, M.; Sahoo, T.; Beaudet, A.L. The MTHFR 677C-T polymorphism and behaviors in children with autism: Exploratory genotypephenotype correlations. Autism Res. 2009, 2, 98–108. [Google Scholar] [CrossRef] [PubMed]
- Frye, R.E.; Sequeira, J.M.; Quadros, E.V.; James, S.J.; Rossignol, D.A. Cerebral folate receptor autoantibodies in autism spectrum disorder. Mol. Psychiatr. 2012. [Google Scholar] [CrossRef] [PubMed]
© 2012 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Hartzell, S.; Seneff, S. Impaired Sulfate Metabolism and Epigenetics: Is There a Link in Autism? Entropy 2012, 14, 1953-1977. https://doi.org/10.3390/e14101953
Hartzell S, Seneff S. Impaired Sulfate Metabolism and Epigenetics: Is There a Link in Autism? Entropy. 2012; 14(10):1953-1977. https://doi.org/10.3390/e14101953
Chicago/Turabian StyleHartzell, Samantha, and Stephanie Seneff. 2012. "Impaired Sulfate Metabolism and Epigenetics: Is There a Link in Autism?" Entropy 14, no. 10: 1953-1977. https://doi.org/10.3390/e14101953
APA StyleHartzell, S., & Seneff, S. (2012). Impaired Sulfate Metabolism and Epigenetics: Is There a Link in Autism? Entropy, 14(10), 1953-1977. https://doi.org/10.3390/e14101953