Entropy Variation in the Two-dimensional Phase Transition of Anthracene Adsorbed at the Hg Electrode/Ethylene Glycol Solution Interface

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
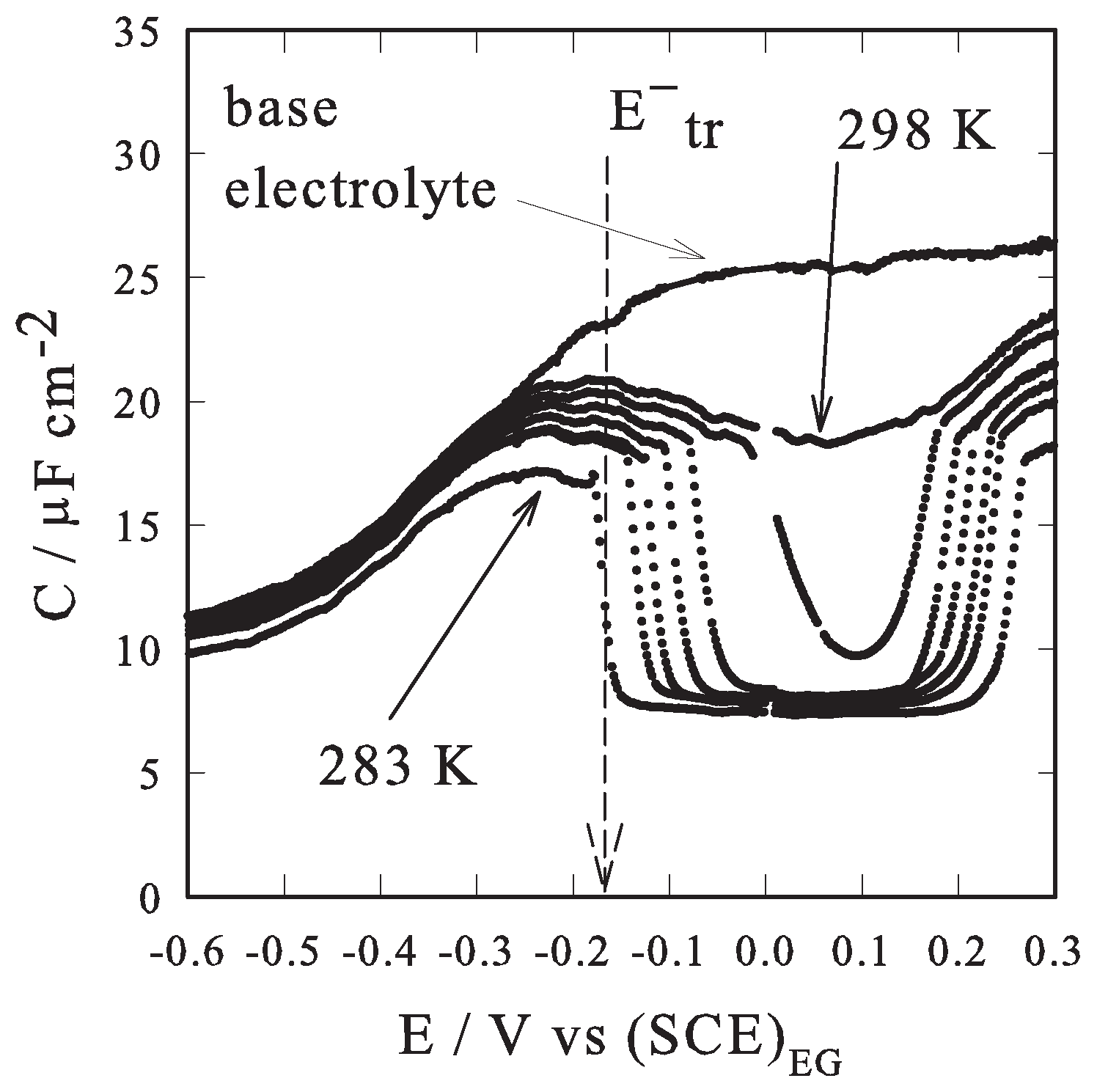
2. Results and Discussion
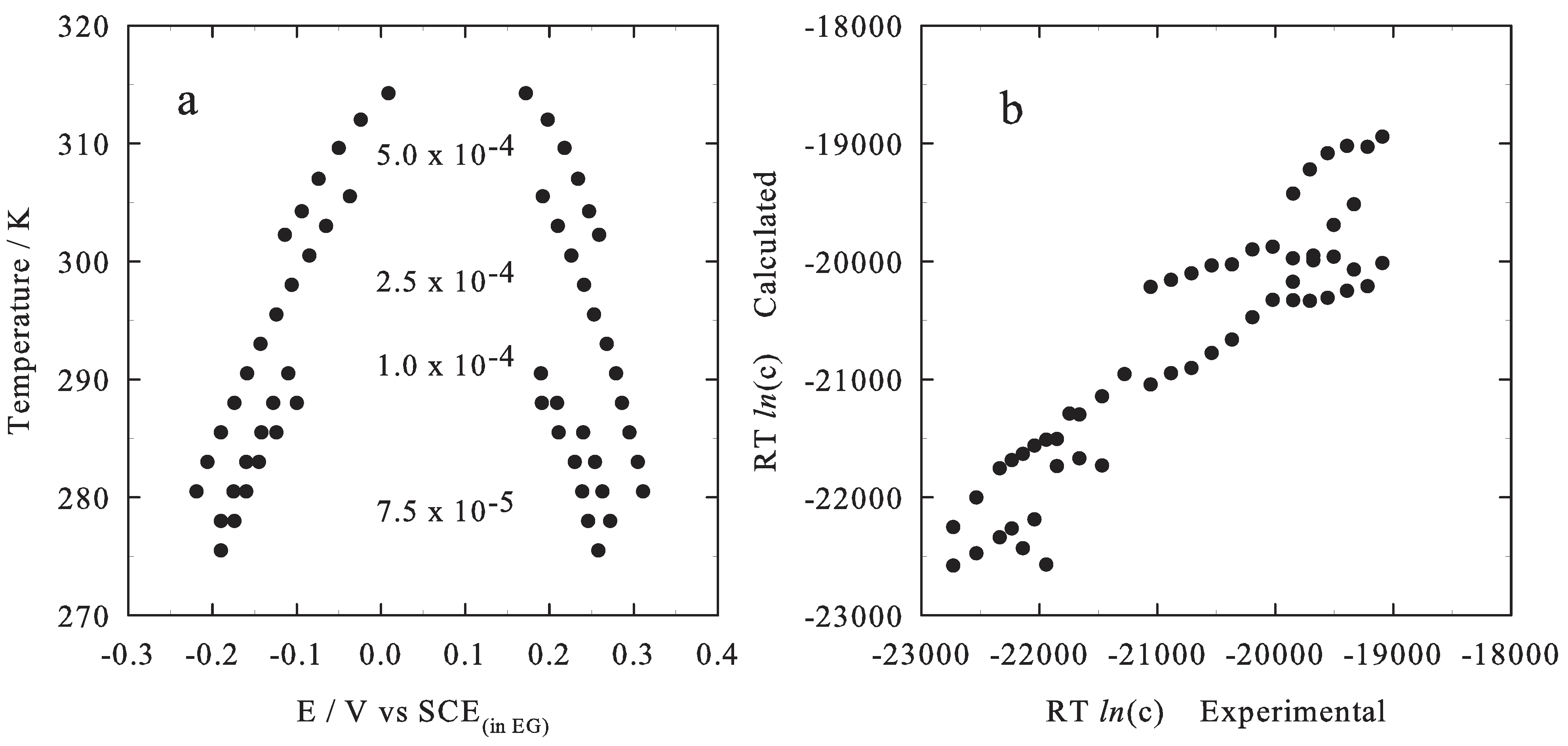
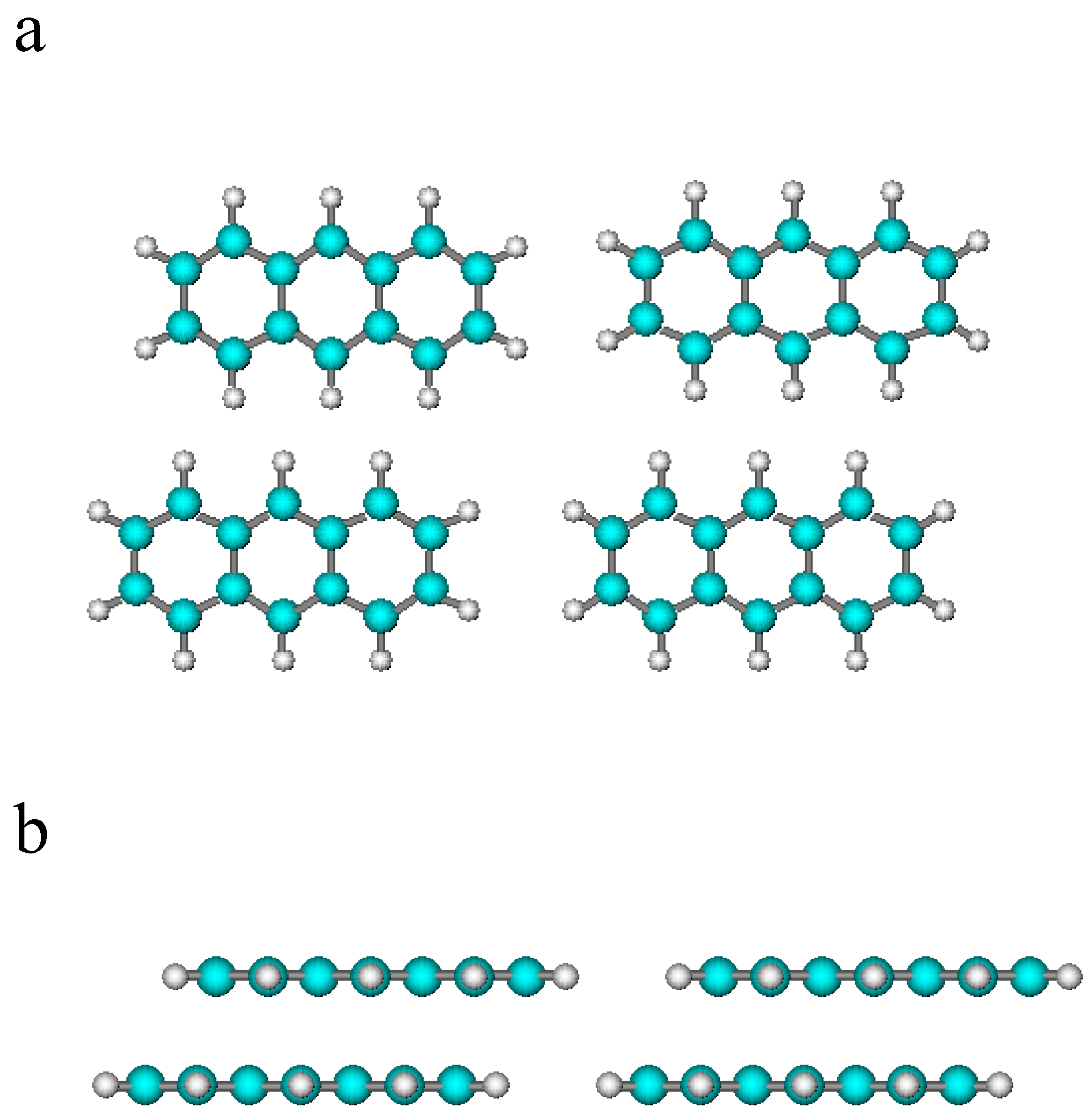
2.1. Theoretical Model
2.2. Results
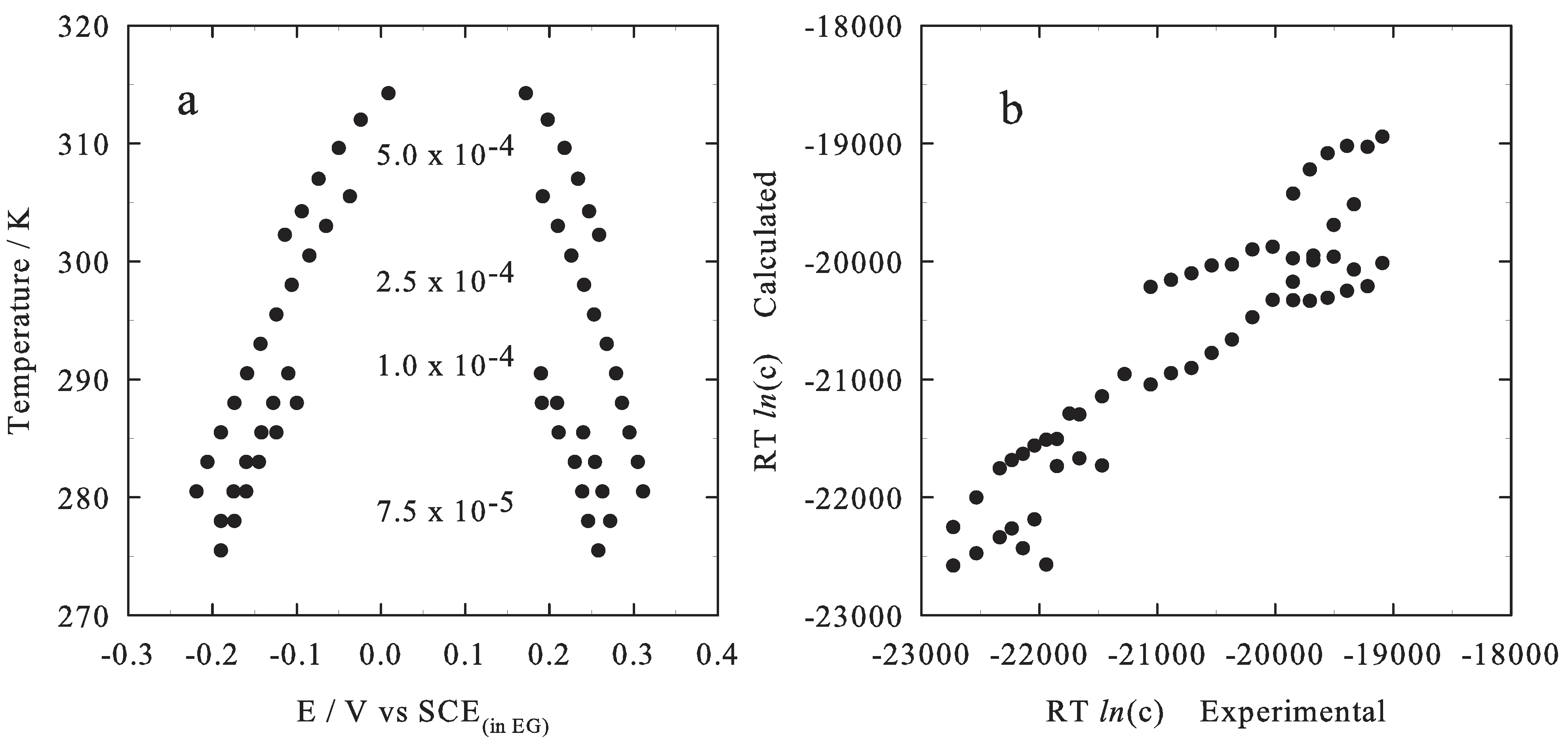
3. Conclusions
4. Experimental Section
References
- Finklea, H.O. Electrochemistry of organized monolayers of thiols and related molecules. In Electroanalytical Chemistry. A Series of Advances; Bard, A.J., Rubinstein, I., Eds.; Marcel Dekker Inc.: New York, NY, USA, 1996; Volume 19, Chapter 3; pp. 110–223. [Google Scholar]
- Vetterl, V. Differentielle kapazitat der elektrolytischen doppelschicht in anwesenheit einiger purin und pyrimidinderivate. Coll. Czech. Chem. Commun. 1966, 31, 2105–2110. [Google Scholar] [CrossRef]
- Sridharan, R.; de Levie, R.; Rangarajan, S.K. Two-dimensional phase transitions at electrochemical interfaces. Chem. Phys. Lett. 1987, 142, 43–47. [Google Scholar] [CrossRef]
- De Levie, R. The dynamic double layer: two-dimensional condensation at the Mercury-Water interface. Chem. Rev. 1988, 88, 599–609. [Google Scholar] [CrossRef]
- Donner, C.; Baumgartel, H.; Pohlmann, L.; Retter, U.; Philip, R. Non-faradaic two-dimensional phase transitions on smooth surfaces-an example for time-dependent homogeneous nucleation. Ber. Bunsenges. Phys. Chem. 1996, 100, 403–412. [Google Scholar] [CrossRef]
- Buess-Herman, C. Self-assembled monolayers at electrode metal surfaces. Prog. Surf. Sci. 1994, 46, 335–375. [Google Scholar] [CrossRef]
- Fontanesi, C. Entropy change in the two-dimensional phase transition of adenine adsorbed at the Hg Electrode/Aqueous Solution Interface. J. Chem. Soc., Faraday Trans. 1994, 90, 2925–2930. [Google Scholar] [CrossRef]
- Striegler, H.; Krznaric, D.; Kolb, D.M. Two-dimensional condensation of camphor and its derivatives on Au(111) Electrodes. J. Electroanal. Chem. 2002, 532, 227–235. [Google Scholar] [CrossRef]
- Danckwerts, M.; Li, Y.; Oslonovitch, J.; Pettinger, B.; Krischer, K. Adsorption of camphor on Au(111) and its effects on the electroreduction of periodate (). J. Phys. Chem. B 2004, 108, 14398–14406. [Google Scholar] [CrossRef]
- Tanaka, Y.; Sagara, T. Formation of Viologen Radical Cation condensed phase through two-dimensional molecular organization process on an HOPG Electrode Surface in Binary Viologen Solutions. J. Electroanal. Chem. 2008, 619–620, 65–74. [Google Scholar] [CrossRef]
- Sridharan, R.; De Levie, R.; Rangarajan, S.K. Two-dimensional phase transitions at Electrochemical Interfaces. Chem. Phys. Lett. 1987, 142, 43–46. [Google Scholar] [CrossRef]
- Retter, U. On adsorption according to the Lattice Gas Model (Ising Model). J. Electroanal. Chem. 1987, 236, 21–30. [Google Scholar] [CrossRef]
- Benedetti, L.; Camurri, G.; Fontanesi, C.; Ferrarini, P.; Giovanardi, R. On the 2D phase transition of Adenine adsorbed at the Mercury/Ethylene Glycol Solution Interface. Electrochim. Acta 2004, 36, 1655–1662. [Google Scholar] [CrossRef]
- Tadjeddine, A.; Rahmani, A. Formation of condensed Film at a Mercury-Electrolyte Interface observed by Surface Plasmon Excitation. Electrochim. Acta 1991, 49, 1855–1857. [Google Scholar] [CrossRef]
- In fact, hysteresis occurs because kinetic effects influence the transition potential values, upon selecting the potential scan direction, i.e., positive or negative (the condensed phase formation, from the “parallel” to “perpendicular” disposition of the adsorbate, has a slower kinetics if compared to the reverse process [2,3,4,5,6,7]). In the present work hysteresis effects are negligible when using a 10 mV s−1 potential scan rate, eventually a even lower scan rate, 5 mV s−1, was used in recording capacity vs. potential curves.
© 2010 by the author; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license http://creativecommons.org/licenses/by/3.0/.
Share and Cite
Fontanesi, C. Entropy Variation in the Two-dimensional Phase Transition of Anthracene Adsorbed at the Hg Electrode/Ethylene Glycol Solution Interface. Entropy 2010, 12, 570-577. https://doi.org/10.3390/e12030570
Fontanesi C. Entropy Variation in the Two-dimensional Phase Transition of Anthracene Adsorbed at the Hg Electrode/Ethylene Glycol Solution Interface. Entropy. 2010; 12(3):570-577. https://doi.org/10.3390/e12030570
Chicago/Turabian StyleFontanesi, Claudio. 2010. "Entropy Variation in the Two-dimensional Phase Transition of Anthracene Adsorbed at the Hg Electrode/Ethylene Glycol Solution Interface" Entropy 12, no. 3: 570-577. https://doi.org/10.3390/e12030570