Microorganisms 2023, 11(1), 124; https://doi.org/10.3390/microorganisms11010124 - 3 Jan 2023
Cited by 57 | Viewed by 10089
Abstract
Animal production is associated with the frequent use of antimicrobial agents for growth promotion and for the prevention, treatment, and control of animal diseases, thus maintaining animal health and productivity. Staphylococcus aureus, in particular methicillin-resistant S. aureus (MRSA), can cause a variety
[...] Read more.
Animal production is associated with the frequent use of antimicrobial agents for growth promotion and for the prevention, treatment, and control of animal diseases, thus maintaining animal health and productivity. Staphylococcus aureus, in particular methicillin-resistant S. aureus (MRSA), can cause a variety of infections from superficial skin and soft tissue infections to life-threatening septicaemia. S. aureus represents a serious public health problem in hospital and community settings, as well as an economic and animal welfare problem. Livestock-associated MRSA (LA-MRSA) was first described associated with the sequence (ST) 398 that was grouped within the clonal complex (CC) 398. Initially, LA-MRSA strains were restricted to CC398, but over the years it has become clear that its diversity is much greater and that it is constantly changing, a trend increasingly associated with multidrug resistance. Therefore, in this review, we aimed to describe the main clonal lineages associated with different production animals, such as swine, cattle, rabbits, and poultry, as well as verify the multidrug resistance associated with each animal species and clonal lineage. Overall, S. aureus ST398 still remains the most common clone among livestock and was reported in rabbits, goats, cattle, pigs, and birds, often together with spa-type t011. Nevertheless, a wide diversity of clonal lineages was reported worldwide in livestock.
Full article
(This article belongs to the Special Issue Pathogens and Antimicrobial Drug Resistance in the Food Chain)
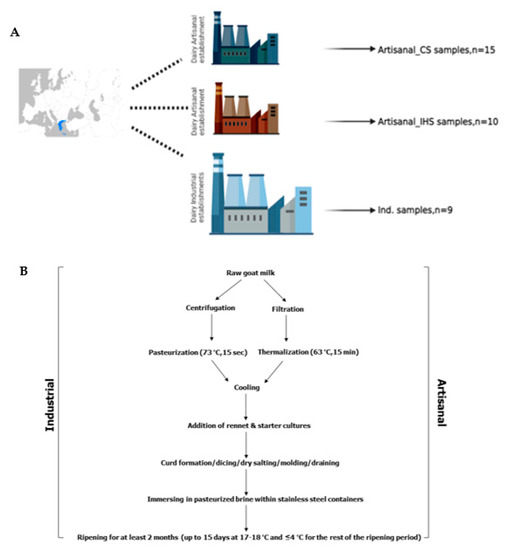

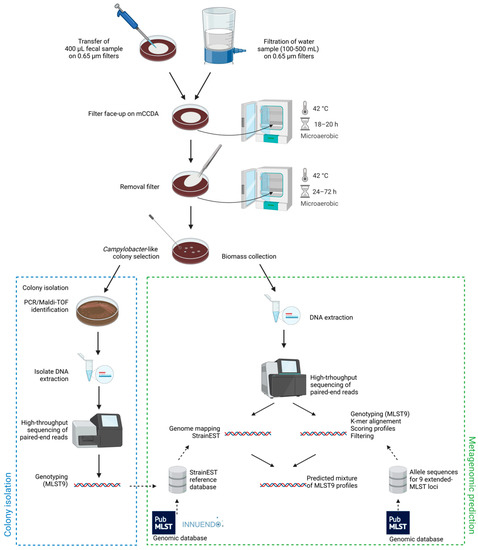
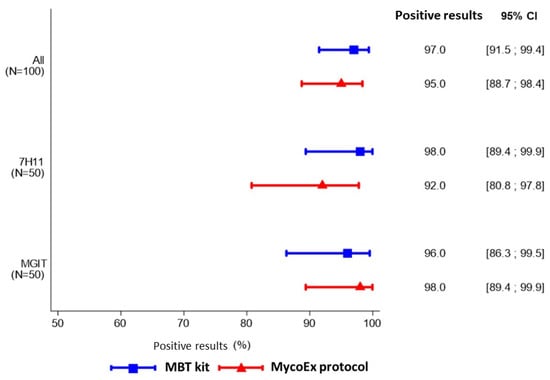
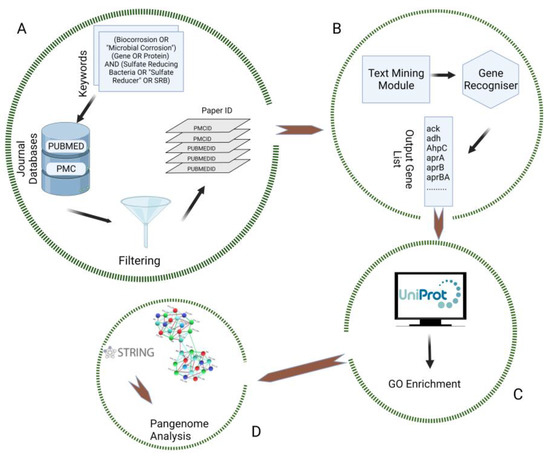



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}