1
Institute of Human Genetics, Medical Center—University of Freiburg, Faculty of Medicine, University of Freiburg, 79106 Freiburg, Germany
2
Center for Cornification Disorders, Freiburg Center for Rare Diseases, Medical Center, University of Freiburg, 79106 Freiburg, Germany
3
European Reference Networks (ERN Skin), 75015 Paris, France
4
Department of Dermatology, Reference Center for Rare Skin Diseases MAGEC, Saint Louis Hospital AP-HP, 75015 Paris, France
5
Department of Dermatology and Venereology, Muenster University Medical Center, 48149 Muenster, Germany
6
Department of Dermatology, CHU of Bab-El-Oued Algiers, Algiers 16008, Algeria
7
Department of Dermatology and Venereology, Faculty of Medicine and University Hospital, University of Cologne, 50937 Cologne, Germany
8
Clinical Genetics, Karolinska University Hospital, 171 64 Solna, Sweden
9
Institute of Human Genetics, University of Bonn, Medical Faculty & University Hospital Bonn, 53127 Bonn, Germany
10
Department of Dermatology, Venerology und Allergology, University Hospital of Munich, 80337 Munich, Germany
11
Pediatric Skin Center, Dermatology Department, University Children’s Hospital Zurich, 8032 Zurich, Switzerland
12
Synlab Medical Practice for Human Genetics Jena, 07747 Jena, Germany
13
Center for Inflammatory Skin Diseases, Department of Dermatology and Allergy, University Hospital Schleswig-Holstein, Campus Kiel, 24105 Kiel, Germany
14
Pediatric Dermatology Unit, Department of Pathophysiology and Transplantation, Fondazione IRCCS Ca’ Granda—Ospedale Maggiore Policlinico, University of Milan, 20122 Milan, Italy
15
Department of Medical Genetics, Inonu University School of Medicine, 44280 Malatya, Turkey
16
Department of Dermatology and Venereology, Skåne University Hospital, 221 85 Lund, Sweden
add
Show full affiliation list
remove
Hide full affiliation list
Abstract
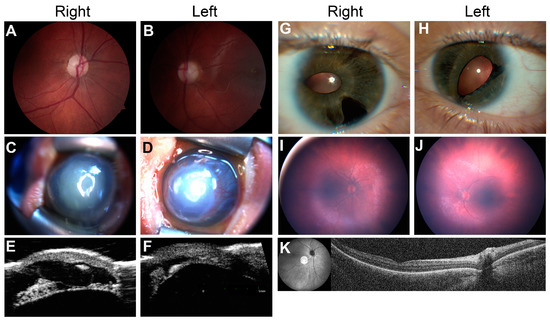
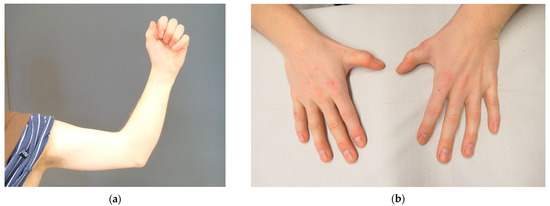
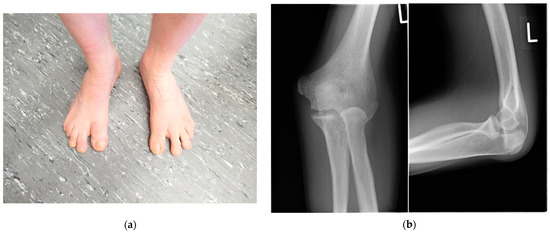
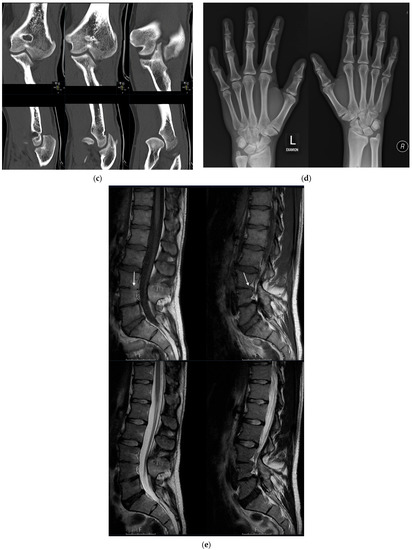
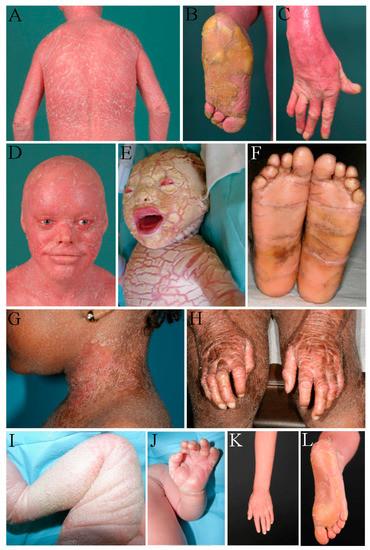
Autosomal recessive congenital ichthyosis (ARCI) is a non-syndromic congenital disorder of cornification characterized by abnormal scaling of the skin. The three major phenotypes are lamellar ichthyosis, congenital ichthyosiform erythroderma, and harlequin ichthyosis. ARCI is caused by biallelic mutations in
ABCA12,
ALOX12B,
[...] Read more.
Autosomal recessive congenital ichthyosis (ARCI) is a non-syndromic congenital disorder of cornification characterized by abnormal scaling of the skin. The three major phenotypes are lamellar ichthyosis, congenital ichthyosiform erythroderma, and harlequin ichthyosis. ARCI is caused by biallelic mutations in
ABCA12,
ALOX12B,
ALOXE3,
CERS3,
CYP4F22,
NIPAL4,
PNPLA1,
SDR9C7,
SULT2B1, and
TGM1. The most severe form of ARCI, harlequin ichthyosis, is caused by mutations in
ABCA12. Mutations in this gene can also lead to congenital ichthyosiform erythroderma or lamellar ichthyosis. We present a large cohort of 64 patients affected with ARCI carrying biallelic mutations in
ABCA12. Our study comprises 34 novel mutations in
ABCA12, expanding the mutational spectrum of
ABCA12-associated ARCI up to 217 mutations. Within these we found the possible mutational hotspots c.4541G>A, p.(Arg1514His) and c.4139A>G, p.(Asn1380Ser). A correlation of the phenotype with the effect of the genetic mutation on protein function is demonstrated. Loss-of-function mutations on both alleles generally result in harlequin ichthyosis, whereas biallelic missense mutations mainly lead to CIE or LI.
Full article
 and Thomas Ben Biddulph 1,*
and Thomas Ben Biddulph 1,* 









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}