Genes 2023, 14(2), 322; https://doi.org/10.3390/genes14020322 - 26 Jan 2023
Cited by 1 | Viewed by 2370
Abstract
One of the most important steps in post-translational modifications of collagen type I chains is the hydroxylation of carbon-3 of proline residues by prolyl-3-hydroxylase-1 (P3H1). Genetic variants in P3H1 have been reported to cause autosomal recessive osteogenesis imperfecta (OI) type VIII. Clinical and
[...] Read more.
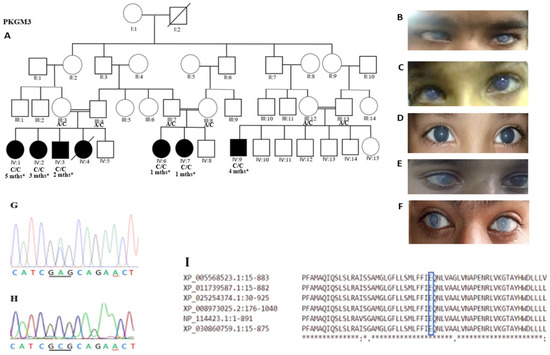
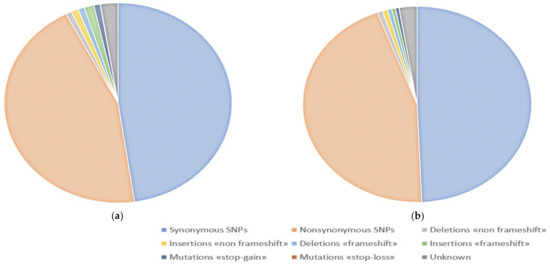
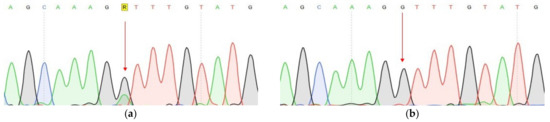
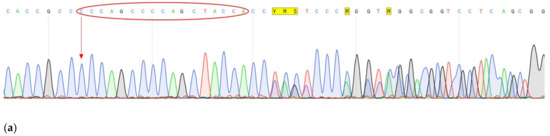
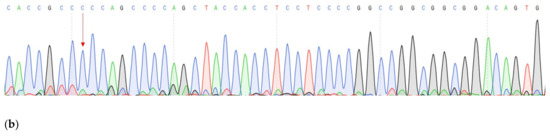
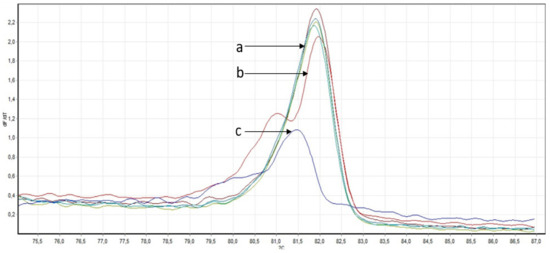
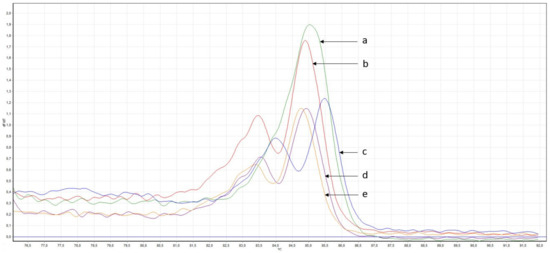
One of the most important steps in post-translational modifications of collagen type I chains is the hydroxylation of carbon-3 of proline residues by prolyl-3-hydroxylase-1 (P3H1). Genetic variants in P3H1 have been reported to cause autosomal recessive osteogenesis imperfecta (OI) type VIII. Clinical and radiographic examinations, whole-exome sequencing (WES), and bioinformatic analysis were performed in 11 Thai children of Karen descent affected by multiple bone fractures. Clinical and radiographic findings in these patients fit OI type VIII. Phenotypic variability is evident. WES identified an intronic homozygous variant (chr1:43212857A > G; NM_022356.4:c.2055 + 86A > G) in P3H1 in all patients, with parents in each patient being heterozygous for the variant. This variant is predicted to generate a new “CAG” splice acceptor sequence, resulting in the incorporation of an extra exon that leads to a frameshift in the final exon and subsequent non-functional P3H1 isoform a. Alternative splicing of P3H1 resulting in the absence of functional P3H1 caused OI type VIII in 11 Thai children of Karen descent. This variant appears to be specific to the Karen population. Our study emphasizes the significance of considering intronic variants.
Full article
(This article belongs to the Section Human Genomics and Genetic Diseases)
►
Show Figures

Figure 1

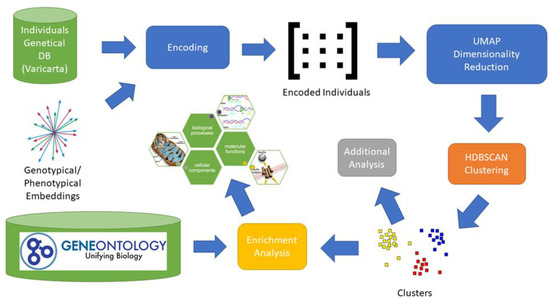



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}