Genes 2022, 13(8), 1380; https://doi.org/10.3390/genes13081380 - 1 Aug 2022
Cited by 2 | Viewed by 1921
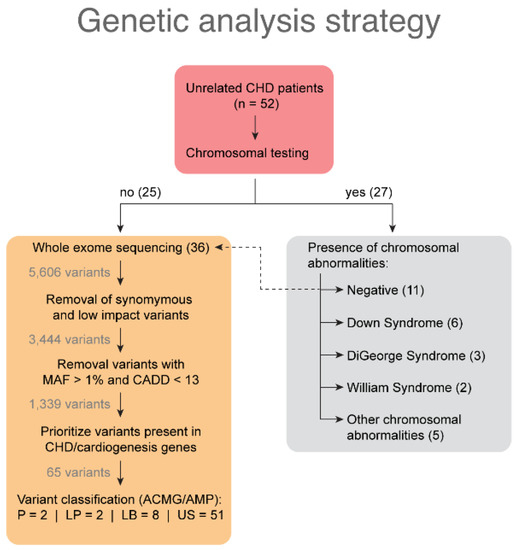
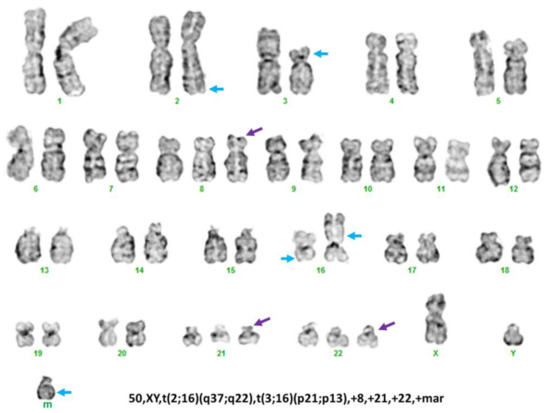
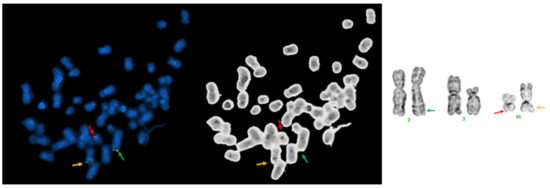
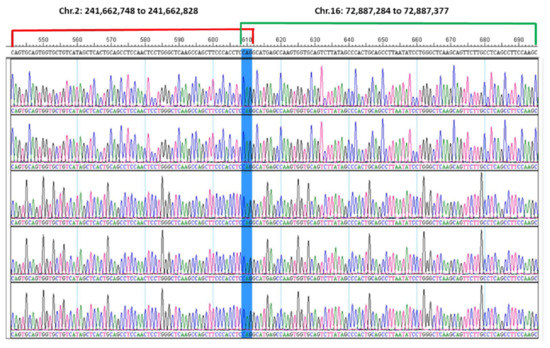
Abstract
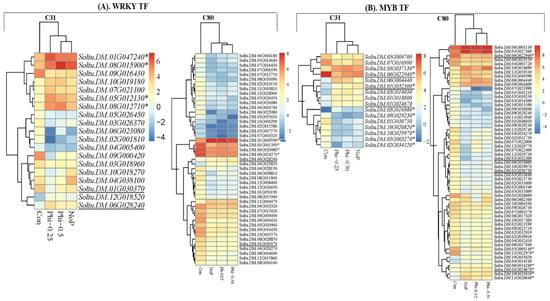
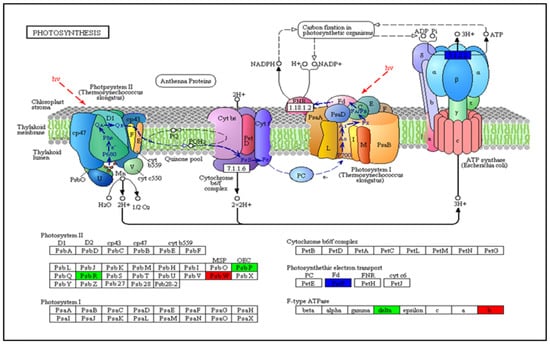
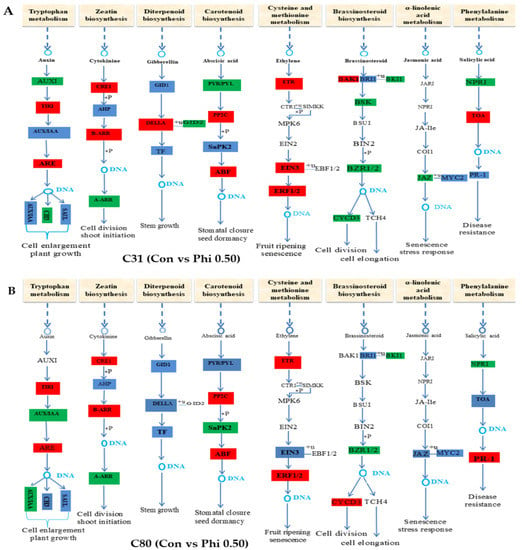
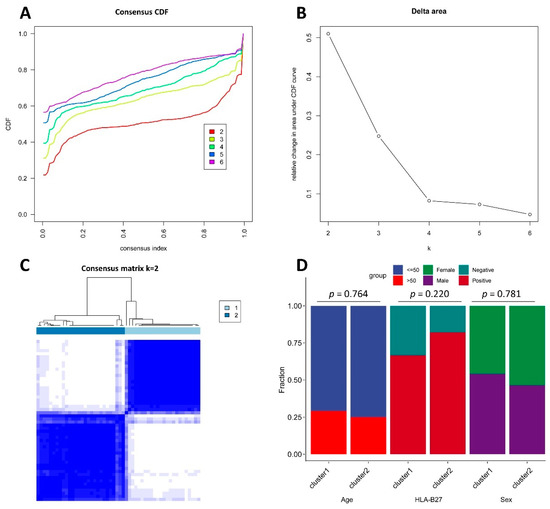
Now more than ever, the understanding of the genetics and evolution of the gene mechanisms and the networks of different molecular pathways acting on plant abiotic stress tolerance has an important role in the finding of new solutions and approaches mitigating the effects
[...] Read more.
Now more than ever, the understanding of the genetics and evolution of the gene mechanisms and the networks of different molecular pathways acting on plant abiotic stress tolerance has an important role in the finding of new solutions and approaches mitigating the effects of global climate changes, thus contributing to a correct equilibrium among human needs, food security and human health and wellbeing [...]
Full article
(This article belongs to the Special Issue Genetics and Evolution of Abiotic Stress Tolerance in Plants)






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}