Genes 2021, 12(10), 1512; https://doi.org/10.3390/genes12101512 - 26 Sep 2021
Cited by 14 | Viewed by 4027
Abstract
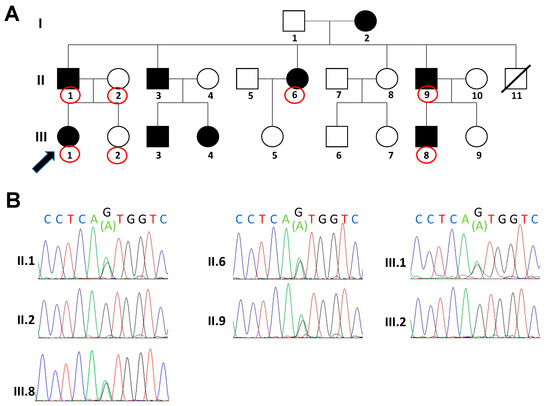
Knobloch syndrome is an inherited disorder characterized by high myopia, retinal detachment, and occipital defects. Disease-causing mutations have been identified in the COL18A1 gene. This study aimed to investigate novel variants of COL18A1 in Knobloch syndrome and describe the associated phenotypes in Chinese
[...] Read more.
Knobloch syndrome is an inherited disorder characterized by high myopia, retinal detachment, and occipital defects. Disease-causing mutations have been identified in the COL18A1 gene. This study aimed to investigate novel variants of COL18A1 in Knobloch syndrome and describe the associated phenotypes in Chinese patients. We reported six patients with Knobloch syndrome from four unrelated families in whom we identified five novel COL18A1 mutations. Clinical examination showed that all probands presented with high myopia, chorioretinal atrophy, and macular defects; one exhibited rhegmatogenous retinal detachment in one eye. Occipital defects were detected in one patient.
Full article
(This article belongs to the Section Genetic Diagnosis)
►
Show Figures

Figure 1




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}