The Viral Tegument Protein pp65 Impairs Transcriptional Upregulation of IL-1β by Human Cytomegalovirus through Inhibition of NF-kB Activity

 ,
,  , , , ,
, , , ,  ,
, {kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells and Viruses
2.2. Recombinant Adenoviral Vectors
2.3. Luciferase Assay
2.4. RNA Isolation and Semiquantitative RT-qPCR
2.5. Immunofluorescence Microscopy
2.6. Enzyme-Linked Immunosorbent Assay (ELISA)
2.7. Non-Radioactive Universal EZ-TFA Transcription Factor Assay
2.8. In Vitro Analysis of Caspase-1 and Caspse-8 Activity
2.9. Inhibition of Caspase-1 and Caspase-8 Expression
2.10. Statistical Analysis
3. Results and Discussion
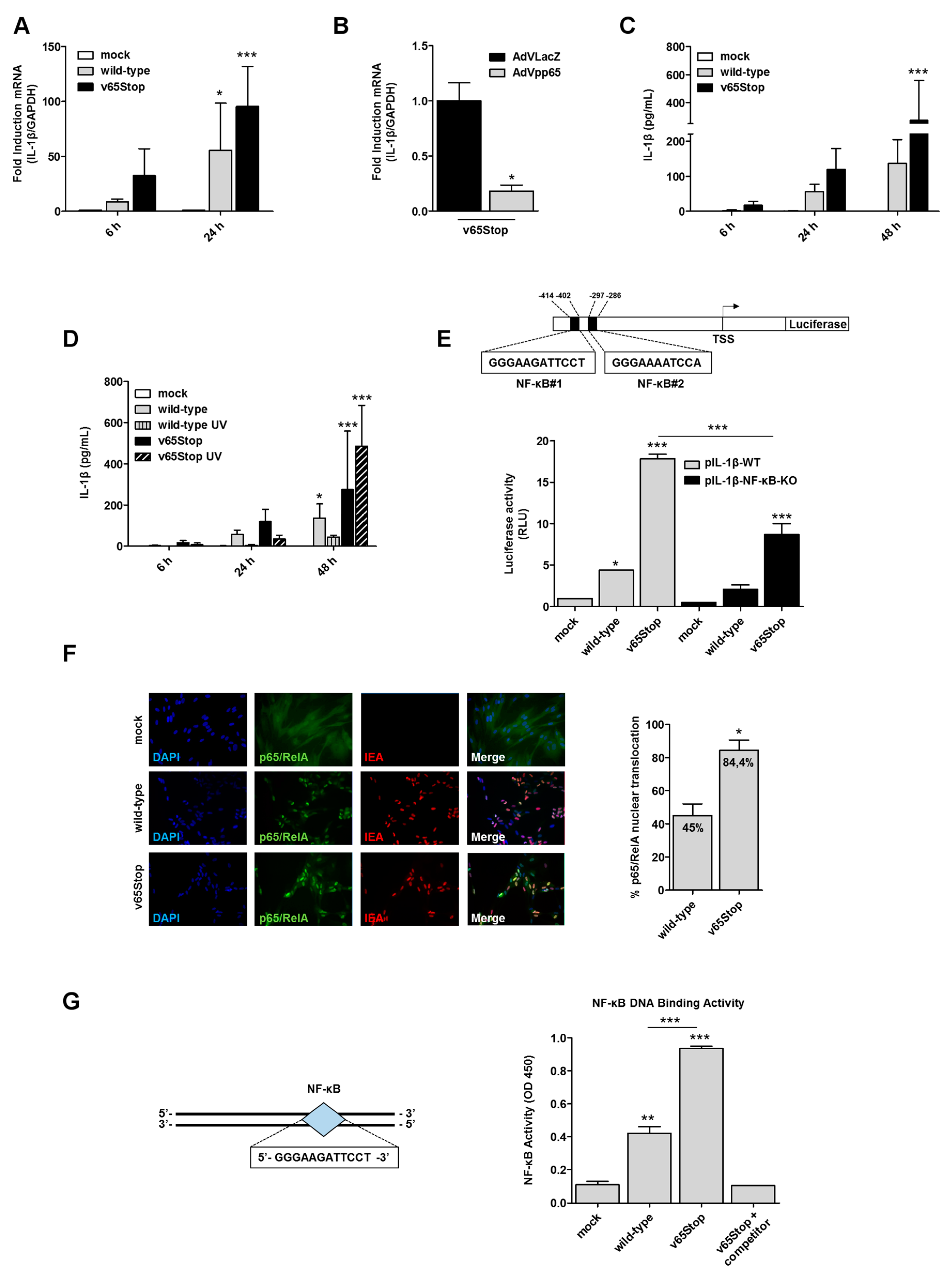
3.1. IL-1β Induction upon HCMV Infection is Inhibited by the HCMV Tegument Protein pp65 in an NF-κB Dependent Manner
3.2. HCMV Primes IL-1β Activation in a Caspase-8 Dependent Manner
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Britt, W.J. Congenital Human Cytomegalovirus Infection and the Enigma of Maternal Immunity. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, P.; Baraniak, I.; Reeves, M. The pathogenesis of human cytomegalovirus. J. Pathol. 2015, 235, 288–297. [Google Scholar] [CrossRef] [PubMed]
- La Rosa, C.; Diamond, D.J. The immune response to human CMV. Future Virol. 2012, 7, 279–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paludan, S.R.; Bowie, A.G.; Horan, K.A.; Fitzgerald, K.A. Recognition of herpesviruses by the innate immune system. Nat. Rev. Immunol. 2011, 11, 143–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luecke, S.; Paludan, S.R. Innate recognition of alphaherpesvirus DNA. Adv. Virus Res. 2015, 92, 63–100. [Google Scholar] [CrossRef] [PubMed]
- Sutterwala, F.S.; Haasken, S.; Cassel, S.L. Mechanism of NLRP3 inflammasome activation. Ann. N. Y. Acad. Sci. 2014, 1319, 82–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yurochko, A.D.; Huang, E.S. Human cytomegalovirus binding to human monocytes induces immunoregulatory gene expression. J. Immunol. 1999, 162, 4806–4816. [Google Scholar] [PubMed]
- Huang, Y.; Liu, L.; Ma, D.; Liao, Y.; Lu, Y.; Huang, H.; Qin, W.; Liu, X.; Fang, F. Human cytomegalovirus triggers the assembly of AIM2 inflammasome in THP-1-derived macrophages. J. Med. Virol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Browne, E.P.; Shenk, T. Human cytomegalovirus UL83-coded pp65 virion protein inhibits antiviral gene expression in infected cells. Proc. Natl. Acad. Sci. USA 2003, 100, 11439–11444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abate, D.A.; Watanabe, S.; Mocarski, E.S. Major human cytomegalovirus structural protein pp65 (ppUL83) prevents interferon response factor 3 activation in the interferon response. J. Virol. 2004, 78, 10995–11006. [Google Scholar] [CrossRef] [PubMed]
- Biolatti, M.; Dell’Oste, V.; Pautasso, S.; Gugliesi, F.; von Einem, J.; Krapp, C.; Jakobsen, M.R.; Borgogna, C.; Gariglio, M.; De Andrea, M.; et al. Human Cytomegalovirus Tegument Protein pp65 (pUL83) Dampens Type I Interferon Production by Inactivating the DNA Sensor cGAS without Affecting STING. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Ma, D.; Huang, H.; Lu, Y.; Liao, Y.; Liu, L.; Liu, X.; Fang, F. Interaction between HCMV pUL83 and human AIM2 disrupts the activation of the AIM2 inflammasome. Virol. J. 2017, 14, 34. [Google Scholar] [CrossRef] [PubMed]
- Biolatti, M.; Dell’Oste, V.; De Andrea, M.; Landolfo, S. The human cytomegalovirus tegument protein pp65 (pUL83): A key player in innate immune evasion. New Microbiol. 2018, 41, 87–94. [Google Scholar] [PubMed]
- Li, T.; Chen, J.; Cristea, I.M. Human cytomegalovirus tegument protein pUL83 inhibits IFI16-mediated DNA sensing for immune evasion. Cell Host Microbe 2013, 14, 591–599. [Google Scholar] [CrossRef] [PubMed]
- Gariano, G.R.; Dell’Oste, V.; Bronzini, M.; Gatti, D.; Luganini, A.; De Andrea, M.; Gribaudo, G.; Gariglio, M.; Landolfo, S. The intracellular DNA sensor IFI16 gene acts as restriction factor for human cytomegalovirus replication. PLoS Pathog. 2012, 8, e1002498. [Google Scholar] [CrossRef] [PubMed]
- Chevillotte, M.; Landwehr, S.; Linta, L.; Frascaroli, G.; Lüske, A.; Buser, C.; Mertens, T.; von Einem, J. Major tegument protein pp65 of human cytomegalovirus is required for the incorporation of pUL69 and pUL97 into the virus particle and for viral growth in macrophages. J. Virol. 2009, 83, 2480–2490. [Google Scholar] [CrossRef] [PubMed]
- Biolatti, M.; Dell’Oste, V.; Pautasso, S.; von Einem, J.; Marschall, M.; Plachter, B.; Gariglio, M.; De Andrea, M.; Landolfo, S. Regulatory Interaction between the Cellular Restriction Factor IFI16 and Viral pp65 (pUL83) Modulates Viral Gene Expression and IFI16 Protein Stability. J. Virol. 2016, 90, 8238–8250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baggetta, R.; De Andrea, M.; Gariano, G.R.; Mondini, M.; Rittà, M.; Caposio, P.; Cappello, P.; Giovarelli, M.; Gariglio, M.; Landolfo, S. The interferon-inducible gene IFI16 secretome of endothelial cells drives the early steps of the inflammatory response. Eur. J. Immunol. 2010, 40, 2182–2189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dell’Oste, V.; Gatti, D.; Gugliesi, F.; De Andrea, M.; Bawadekar, M.; Lo Cigno, I.; Biolatti, M.; Vallino, M.; Marschall, M.; Gariglio, M.; et al. Innate nuclear sensor IFI16 translocates into the cytoplasm during the early stage of in vitro human cytomegalovirus infection and is entrapped in the egressing virions during the late stage. J. Virol. 2014, 88, 6970–6982. [Google Scholar] [CrossRef] [PubMed]
- Dudding, L.; Haskill, S.; Clark, B.D.; Auron, P.E.; Sporn, S.; Huang, E.S. Cytomegalovirus infection stimulates expression of monocyte-associated mediator genes. J. Immunol. 1989, 143, 3343–3352. [Google Scholar] [PubMed]
- Botero, J.E.; Contreras, A.; Parra, B. Profiling of inflammatory cytokines produced by gingival fibroblasts after human cytomegalovirus infection. Oral Microbiol. Immunol. 2008, 23, 291–298. [Google Scholar] [CrossRef] [PubMed]
- Alcendor, D.J.; Charest, A.M.; Zhu, W.Q.; Vigil, H.E.; Knobel, S.M. Infection and upregulation of proinflammatory cytokines in human brain vascular pericytes by human cytomegalovirus. J. Neuroinflamm. 2012, 9, 95. [Google Scholar] [CrossRef] [PubMed]
- DeMeritt, I.B.; Milford, L.E.; Yurochko, A.D. Activation of the NF-kappaB pathway in human cytomegalovirus-infected cells is necessary for efficient transactivation of the major immediate-early promoter. J. Virol. 2004, 78, 4498–4507. [Google Scholar] [CrossRef] [PubMed]
- Sambucetti, L.C.; Cherrington, J.M.; Wilkinson, G.W.; Mocarski, E.S. NF-kappa B activation of the cytomegalovirus enhancer is mediated by a viral transactivator and by T cell stimulation. EMBO J. 1989, 8, 4251–4258. [Google Scholar] [CrossRef] [PubMed]
- Yurochko, A.D.; Mayo, M.W.; Poma, E.E.; Baldwin, A.S.; Huang, E.S. Induction of the transcription factor Sp1 during human cytomegalovirus infection mediates upregulation of the p65 and p105/p50 NF-kappaB promoters. J. Virol. 1997, 71, 4638–4648. [Google Scholar] [PubMed]
- Cogswell, J.P.; Godlevski, M.M.; Wisely, G.B.; Clay, W.C.; Leesnitzer, L.M.; Ways, J.P.; Gray, J.G. NF-kappa B regulates IL-1 beta transcription through a consensus NF-kappa B binding site and a nonconsensus CRE-like site. J. Immunol. 1994, 153, 712–723. [Google Scholar] [PubMed]
- Karin, M.; Ben-Neriah, Y. Phosphorylation meets ubiquitination: The control of NF-[kappa]B activity. Annu. Rev. Immunol. 2000, 18, 621–663. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Deng, Y.; He, X.; Chu, J.; Zhou, J.; Zhang, Q.; Guo, W.; Huang, P.; Guan, X.; Tang, Y.; et al. Prenatal inflammation-induced NF-κB dyshomeostasis contributes to renin-angiotensin system over-activity resulting in prenatally programmed hypertension in offspring. Sci. Rep. 2016, 6, 21692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.-S.; Xiang, Y.; Cui, Y.-L.; Lin, K.-M.; Zhang, X.-F. Dietary blue pigments derived from genipin, attenuate inflammation by inhibiting LPS-induced iNOS and COX-2 expression via the NF-κB inactivation. PLoS ONE 2012, 7, e34122. [Google Scholar] [CrossRef] [PubMed]
- Dupaul-Chicoine, J.; Saleh, M. A new path to IL-1β production controlled by caspase-8. Nat. Immunol. 2012, 13, 211–212. [Google Scholar] [CrossRef] [PubMed]
- Gurung, P.; Kanneganti, T.-D. Novel roles for caspase-8 in IL-1β and inflammasome regulation. Am. J. Pathol. 2015, 185, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Gringhuis, S.I.; Kaptein, T.M.; Wevers, B.A.; Theelen, B.; van der Vlist, M.; Boekhout, T.; Geijtenbeek, T.B.H. Dectin-1 is an extracellular pathogen sensor for the induction and processing of IL-1β via a noncanonical caspase-8 inflammasome. Nat. Immunol. 2012, 13, 246–254. [Google Scholar] [CrossRef] [PubMed]
- Skaletskaya, A.; Bartle, L.M.; Chittenden, T.; McCormick, A.L.; Mocarski, E.S.; Goldmacher, V.S. A cytomegalovirus-encoded inhibitor of apoptosis that suppresses caspase-8 activation. Proc. Natl. Acad. Sci. USA 2001, 98, 7829–7834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, G.-C.; Lee, J.H.; Kim, B.Y.; Lee, C.H. Mitochondria-targeted apoptosis in human cytomegalovirus-infected cells. J. Microbiol. Biotechnol. 2013, 23, 1627–1635. [Google Scholar] [CrossRef] [PubMed]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Biolatti, M.; Dell’Oste, V.; Scutera, S.; Gugliesi, F.; Griffante, G.; De Andrea, M.; Musso, T.; Landolfo, S. The Viral Tegument Protein pp65 Impairs Transcriptional Upregulation of IL-1β by Human Cytomegalovirus through Inhibition of NF-kB Activity. Viruses 2018, 10, 567. https://doi.org/10.3390/v10100567
Biolatti M, Dell’Oste V, Scutera S, Gugliesi F, Griffante G, De Andrea M, Musso T, Landolfo S. The Viral Tegument Protein pp65 Impairs Transcriptional Upregulation of IL-1β by Human Cytomegalovirus through Inhibition of NF-kB Activity. Viruses. 2018; 10(10):567. https://doi.org/10.3390/v10100567
Chicago/Turabian StyleBiolatti, Matteo, Valentina Dell’Oste, Sara Scutera, Francesca Gugliesi, Gloria Griffante, Marco De Andrea, Tiziana Musso, and Santo Landolfo. 2018. "The Viral Tegument Protein pp65 Impairs Transcriptional Upregulation of IL-1β by Human Cytomegalovirus through Inhibition of NF-kB Activity" Viruses 10, no. 10: 567. https://doi.org/10.3390/v10100567