3. Discussion
Transcription factors (Tfs) are useful proteins to evaluate the concept of fuzziness in protein–protein and protein–DNA interactions [
1,
2,
7,
25,
26,
27,
28,
29]. For GCN4 Tf, it has been hypothesized that the binding to DNA occurs sequentially in such a way that a monomer is first assembled into the major groove of DNA, followed by dimerization with a second GCN4 Tf unit [
30]. Generally, mimetics of the GCN4 Tf possess a high degree of randomness, but in the presence of a dimerization motif and their cognate ds DNA, there is a substantial increase of α helical structure due to structure inducing complex formation as a result of the specific binding [
9,
10,
11,
12,
13,
14]. Conformational dynamism and heterogeneity enable context-specific functions to emerge in response to changing environmental conditions and, furthermore, allow a single structural motif to be used in multiple settings [
14]. The sequential interaction pathway is a natural strategy that prevents dimers from being trapped for a relatively long time in non-consensus sequences. In the case of GCN4 mimetics, the dimerization motif (
Figure 1A), which is not involved in the DNA binding interface, is generally conformationally unaffected by binding to the DNA. Thereby, it retains the capability to modulate the interaction [
31]. In general, heterogeneous conformational segments can increase binding affinity. This conformational flexibility and heterogeneity of proteins represent their fuzziness [
1,
2,
3,
4,
5,
6]. The classical framework of protein interactions establishes that there is a deterministic relationship between protein sequence and function. Based on this, a distinguished three-dimensional arrangement of the amino acids is a prerequisite for a given biological activity and is unambiguously encoded in the sequence [
32]. However, protein functions are modulated by different mechanism triggered by different effectors. The effector perturbs one site and thereby leads to altered activity in a second, substrate site [
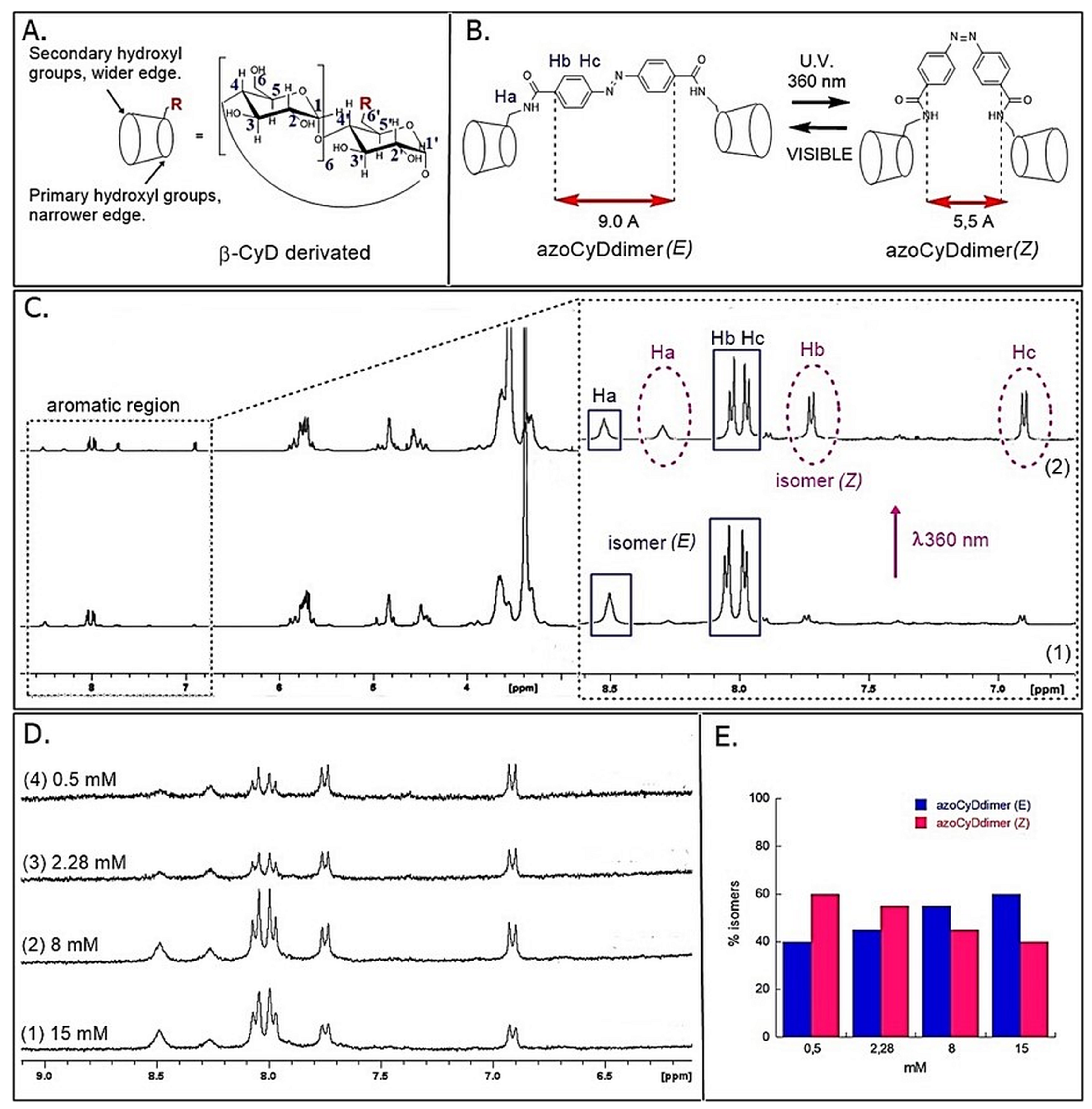
33]. In our system, the effector was the azoCyDdimer, which triggered host–guest interaction of β-CyD of the dimer and two GCN4 peptides containing each an adamantane moiety. The different geometry the azoCyDdimer was previously calculated as 9.1 Å and 6.6 Å for E and Z isomers, respectively [
16]. As confirmed here, the azoCyDdimer exists under two photo-states: the first thermodynamically stable with an E:Z isomer ratio of 95:5 and the second obtained after irradiating with ultraviolet light, E:Z 60:40 ratio. Taking into account, the large geometrical difference between both isomers and the relevance of this distance to form a suitable complex, we had hypothesized that it would be unlikely that both isomers could form a homodimer leading to the same specific dsDNA interaction. Considering that the first and second photo-stationary state contains a major proportion of E isomer 95% and 60%, respectively, it can be anticipated that if the E isomer is the responsible of the tetra-component complex, the specific interaction would be observed under both illumination conditions. However, some differences in binding affinity can be expected. On the contrary, if the Z isomer would give the right geometry for the peptide–DNA interaction, the positive interaction would be observed only mainly in the second photostationary stage where the Z isomer is present in a significant concentration (40%). Nevertheless, if both isomers would contribute equally to the tetra-component complex, similar behavior in all the experiments would be expected.
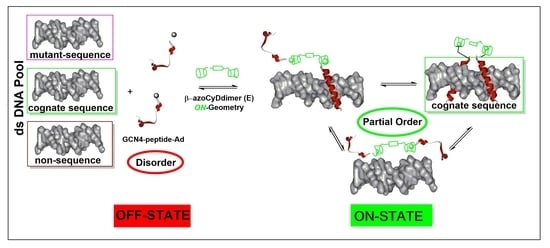
We performed two complementary sets of experiments, EMSA and CD, to evaluate the system. As aforementioned, from EMSA experiments the stoichiometry of the interaction, if any, was identified. Meanwhile, the CD gave information about the transition from disorder-to-order and disorder-to-partial-order in the bound state. In such a case, the value of the molar helicity per residue would let us set a scale of the order obtained for each interaction and connecting them with the stoichiometry obtained in the EMSA experiment. Considering the theory of fuzzy complexes [
1], the disordered-binding elements of GCN4 mimetic may undergo three types of structural transitions upon interaction with the target dsDNA. First, let us consider the disorder-to-order transition to adopt a stable, well-defined conformation in the bound state [
32]. This is also referred to as coupled folding to binding [
34]. Second, upon partner recognition, a transition from disorder-to-partial order could take place in shallow, often hydrophobic binding pockets. In this case, the interface was generated by many redundant contacts and few specificities, as observed by the formation of the complex DNA–Ad30 and DNA–Ad30–azoCyDdimer (E) [
32]. Third, a disorder-to-disorder transition may occur upon binding of the Ad30–azoCyDdimer–Ad30 to the cognate dsDNA; however, in such case, we would not expect an increase of the helical content, but binding should be observed in the EMSA experiment.
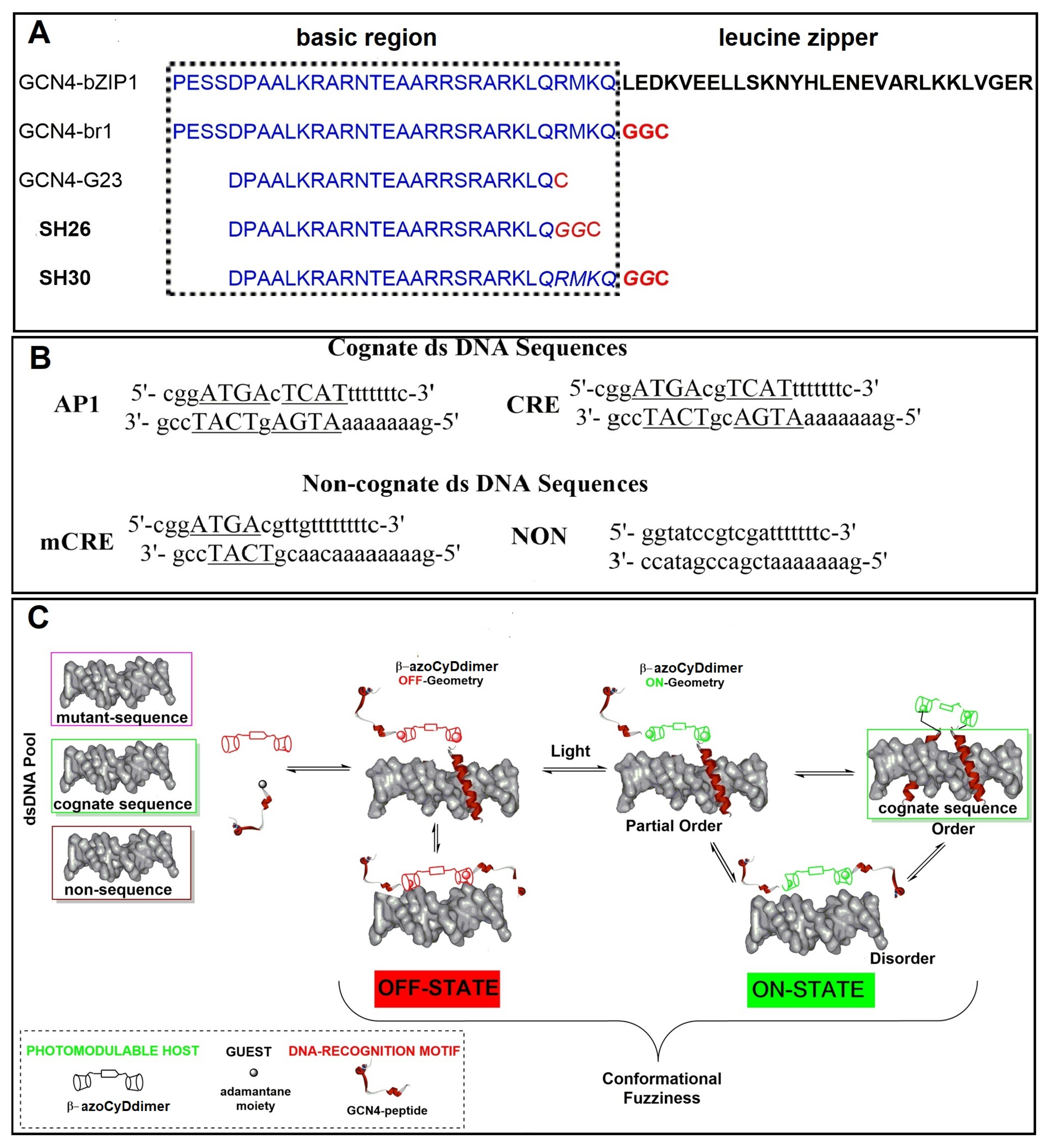
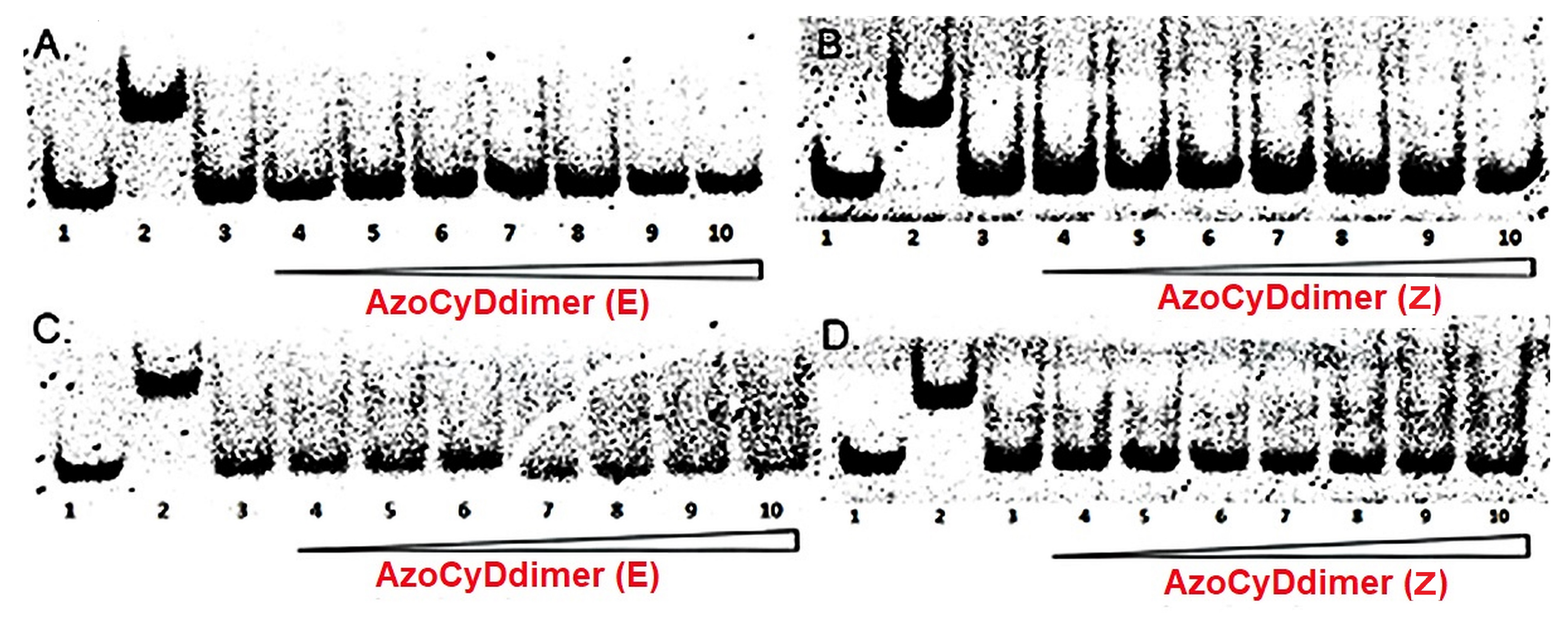
We found that Ad30, but not its shorter form Ad26, was able to form two detectable new interaction shift bands during EMSA experiments. The shifted band detected in the absence of azoCyDdimer has a migration compatible with the monomer of Ad30, which binds to all the dsDNA sequences, inclusive to the random one (NON). Thus, this interaction was considered as unspecific. The second shift band was observed in excess of azoCyDdimer, and it corresponds to the tetra-component complex. This second band of the non-covalent tetra-component complex was observed only in the presence of CRE (′5-..ATGA cg TCAT..-3′) sequence under both illumination conditions, but not with AP1 (′5-..ATGA c TCAT..-3′), nor with mCRE (’5-..ATGAcg..-3′). The elongation of the sequence in Ad30 by insertion of the affinity enhancing QRMK sequence in before the C-terminal QGGC end, thus leading to the QRMKQGGC sequence (Ad30) instead of QGGC (Ad26), does increase the binding affinity to levels allowing the detection of the interaction of one Ad30 monomer with dsDNA containing the sequences (’5-..ATGAc..-3′). Probably the positive charge of the arginine (R) and lysine (K) groups are stabilizing effects that favor the observed interaction. In general, dynamics of the interaction may vary in a wide range depending on the truncation of the disordered dimerization domain as observed here [
32]. Considering our findings, we hypothesized that the formation of a monomeric complex Ad30–azoCyDdimer and the (’5-..ATGAc..-3′) sequence might favor the subsequent tetra-component complex formation. This can be explained by the fact that when one of the sites is bound to its cognate site receptor, a second site located close-by binds cooperatively, basically because of the lower entropic cost of a (pseudo)-intramolecular interaction [
35]. Considering this concept of multivalency, if the linker connecting the binding and the dimerization domains are flexible, the average distance between the sites is the main factor determining the cooperativity. In such complex, we hypothesized that the QRMKQGGC in Ad30 sequence provides the required flexibility to favor the observed tetra-covalent binding and that this region is responsible for the fuzzy behavior of the mimetic in the bound state.
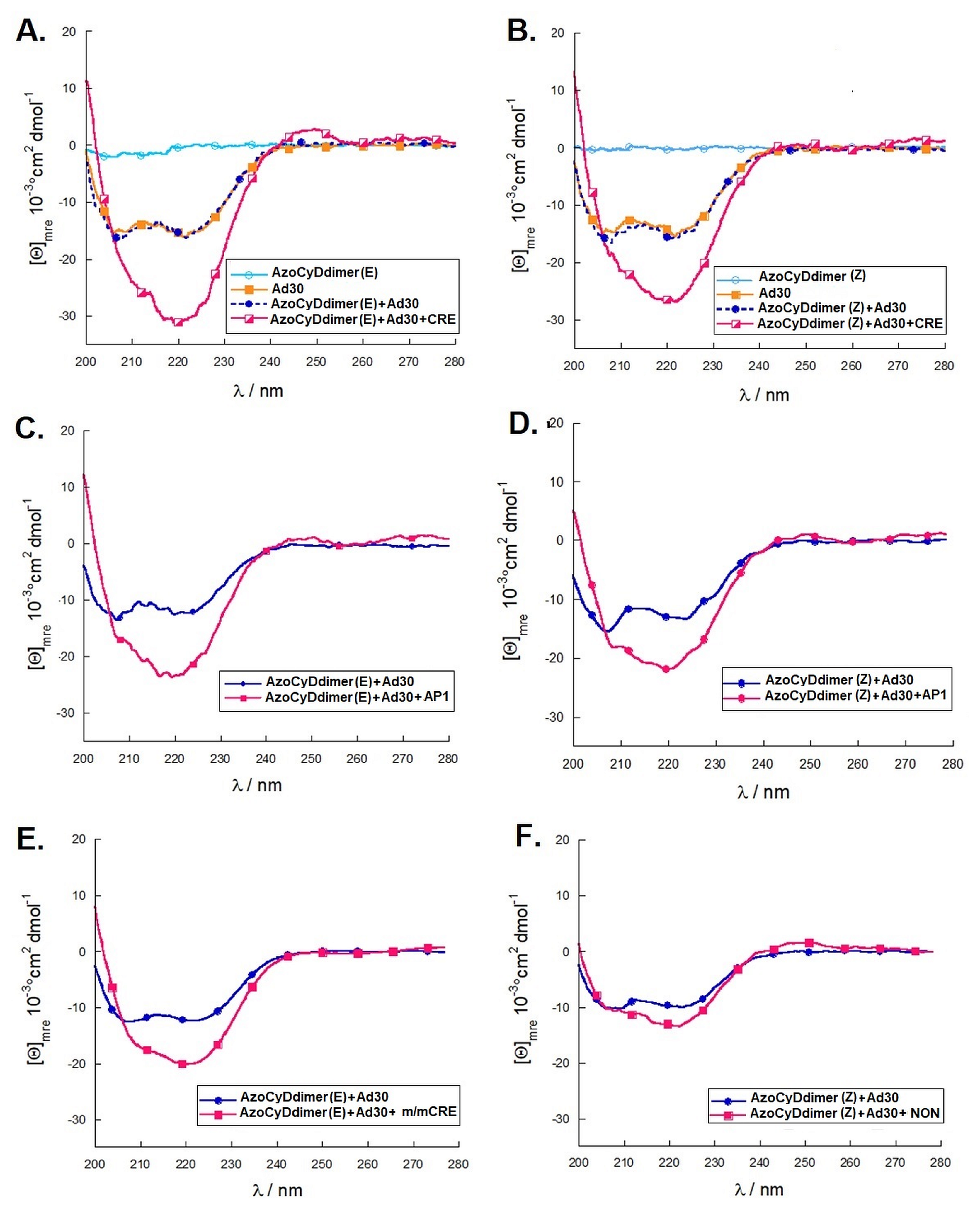
Considering that the specific migration band with CRE was also observed in both photostationary states where the E isomer is 95% and 60%, we hypothesized that the geometry of azoCyDdimer (E) is the adequate combination causing a migration band compatible with the formation of a stable tetra-component complex in the presence of ds CRE. When we evaluated the interaction of Ad30, the azoCyDdimer (E), and the CRE sequence using CD, we observed a significant increase of the helical content compatible with the formation of a tetra-component system, Ad30–azoCyDdimer (E)–Ad30–CRE, similar to those reported by the covalent dimer SS60 and other GCN4 mimetics [
10,
11,
12,
13,
14,
17,
18]. In the case of the second photostationary state (ps) where the ps is E:Z (60:40), a moderate and lower increase of Ad30 helical content compared with the first pss E:Z (95:5) was observed, suggesting that the Z isomer does not contribute to the specific binding.
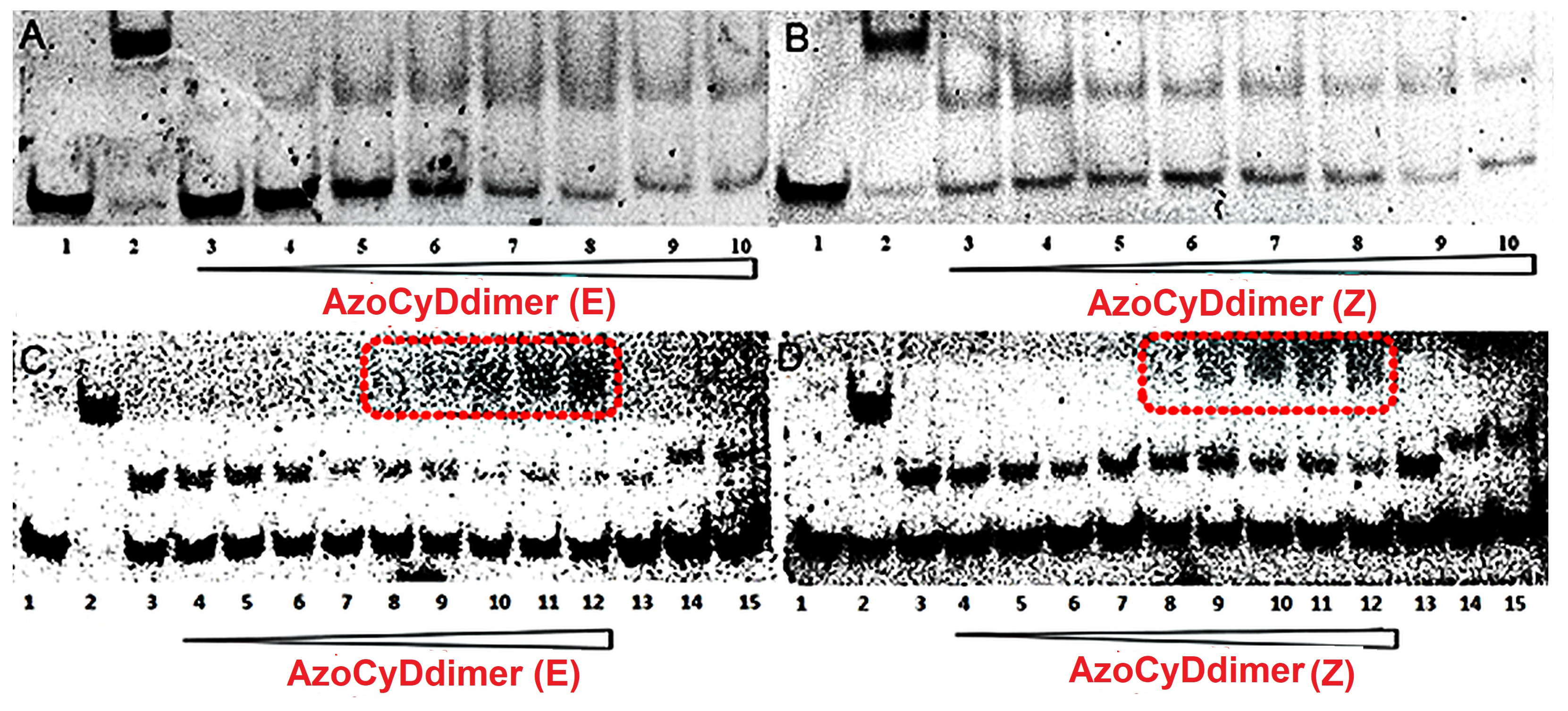
Importantly, for the interaction of Ad30–azoCyDdimer (E) and the mCRE sequence (’5-..ATGAcg..-3′), which contains only one binding site instead of the two necessary (′5-..ATGA cg TCAT..-3′) for the tetra-component formation, a much lower increase of the helical content was observed (
Figure 6E). This is compatible with the interaction of one Ad30–azoCyDdimer (E) or only Ad30 with this sequence, as detected in the EMSA experiment (
Figure 5C,D, lane 15). The interaction with the random sequence (NON,
Figure 6F), which has no recognition motif, showed nearly no interaction, demonstrating that the interaction of the basic region with the phosphate backbone of the dsDNA has at most a minor contribution to the protein–DNA binding. Summarizing the CD experiments, the increase of the helical content upon binding is related to the bound state of Ad30–azoCyDdimer (E) with the different dsDNA as follow: CRE > AP1~ mCRE > NON. After comparison of the increase of helical content (order) of Ad30- in the presence of CRE and the two azoCyDdimer isomers, it is evident that the photo-state (E) contribute more than the (Z). It seems that dimerization interaction between the adamantane and β-cavity of the azoCyDimer occurs at the wider edge of the cyclodextrin (
Figure 2A), instead of its narrow edge, which would increase the binding affinity [
15]. Previously, it has been shown by molecular dynamic simulations that while the Z azoCyDdimer isomer makes a 1:1 complex with a small organic molecule with two adamantane moieties, through interaction with the narrow edge; the E azoCyDdimer isomer forms supramolecular head to tail complexes through the wider edge [
16]. In the case of the mimetic of GCN4, such supramolecular aggregates are not possible because the peptide has only one binding domain. All our experiments suggested that E conformation provides the required geometry to favor the tetra-component complex, which depends on the dsDNA sequence. On the other hand, we could not discard the formation of the Ad30–azoCyDdimer (Z)–Ad30. Nevertheless, our findings indicate that the geometry of the interaction in the Z conformation is not adequate for DNA recognition.
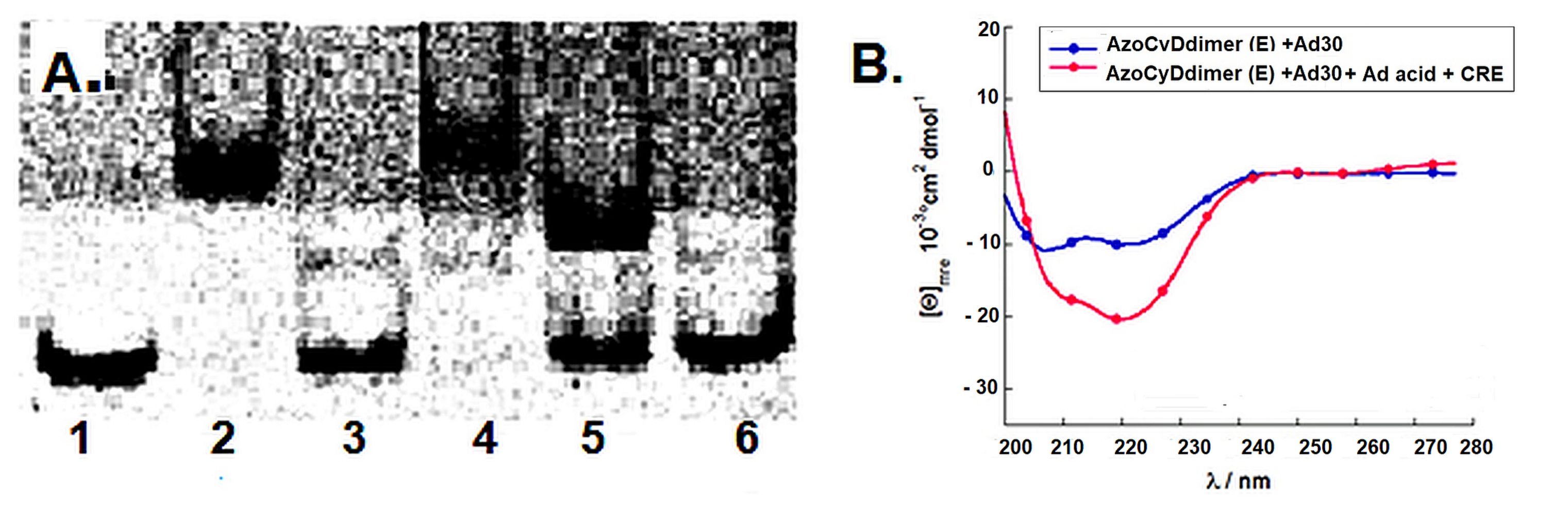
Finally, to confirm that the host–guest interaction between the β-CyD and the Ad moiety is a necessary requisite to recognize CRE sequence specifically, we performed a competitive assay in the presence of 1-adamantane acetic acid. Upon addition of the competitive β-CyD, the only band observed was the unspecific 1:1. A comparable result was obtained by CD upon addition of the competitive β-CyD reaching a helical content value similar to the one of the unspecific binding to mCRE. These final experiments sustain the hypothesis that the azoCyDdimer is necessary to form the homodimeric complex leading to the formation of the tetra-component system in a sequence-specific manner. We had planned to modulate the peptide–DNA interaction by the structural change promoted by the photoisomerization of the azobenzene group located as a linker between the two β-CyDs moieties in the azoCyDdimer. However, as well as for the covalent version of photomodulable GCN4 mimetic reported by Caamaño et al., it was not possible to switch the interaction effectively by changing the azobenzene conformation in situ [
13]. We could not anticipate that the E isomer would be the one that favors the dimerization. Nevertheless, we have the off situation in the absence of azoCyDdimer switching the recognition to
on above a certain azoCyDdimer (E) concentration (
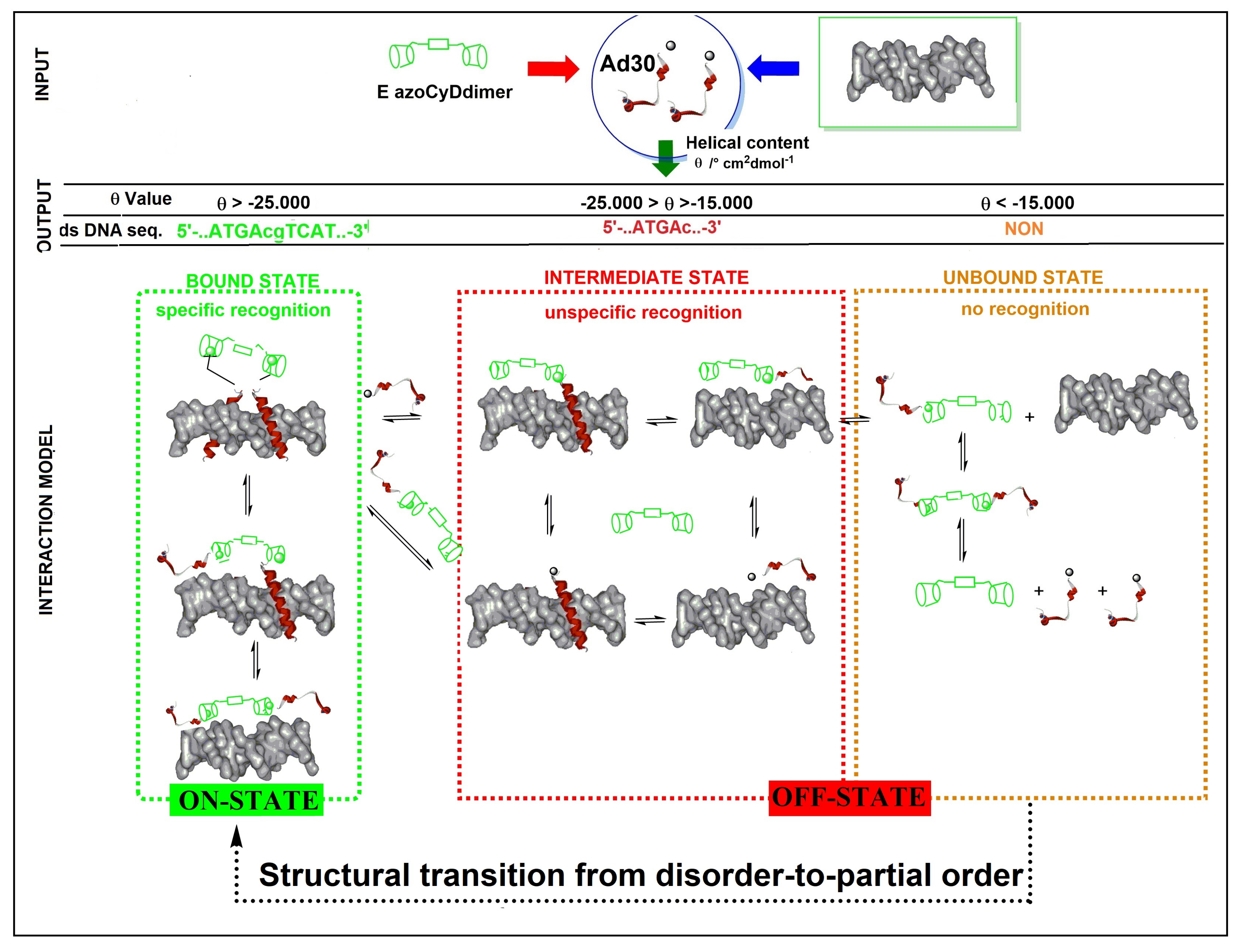
Figure 8). About the mechanism of interaction, we hypothesized that recognition occurs through a sequential mechanism where the monomer Ad30 probably forming a 1:1 complex with the azoCyDdimer recognizes the sequence (’5-..ATGAc..-3′) Initially with high affinity followed by the cooperative union of a second monomer promoted by the host–guest dimerization motif of azoCyDdimer in the E conformation. This second stage is stabilized by the specific interaction with CRE sequences (′5-..ATGA cg TCAT..-3′). During this interaction, different fuzzy conformers are possible that favor the specific interaction with CRE by a transition from disorder-to-partial order of the four components of the complex (
Figure 8). For the others dsDNA that lack of the complete binding sequence in mCRE or only one base as a connector between the binding site in AP1, let to the formation of bi-or tri-component complexes. As described before, Tompa et al. have proposed a categorization for fuzzy complexes, as static or dynamic [
1]. Considering that in the bound state, most of the Ad30 is order, it is possible to describe the interaction by the polymorphism model of static fuzziness [
1]. In such model, one part of the molecule makes the contacts for the interaction whereas its dimerization domain adopts several distinct conformations, and establish the right geometry to favor dimerization and binding to the DNA with the complete recognition sequence in CRE. We hypothesized that the QRMKQGGC region of Ad30 might adopt multiple unrelated conformations to stabilize the interaction with the larger azoCyDdimer (E) instead of the compact (Z) isomer, thus favoring dimerization that contributes to CRE recognition. Probably, this structural variability limits an unfavorable decrease in entropy accompanying complex formation, which enables the combination of rapid and thermodynamically favorable binding. Truncation of this region led to no-binding as observed for Ad26, which validates our hypothesis. In conclusion, the present report is the first example of a GCN4 mimetic that forms a specific non-covalent tetra-component system with the cognate binding sequence only in the presence of an external ligand in one of two possible conformations, working as an off–on switch. Moreover, we demonstrated that chemically modified mimetics of GCN4 are suitable minimalist models to investigate conformational fuzziness in protein–DNA interactions, opening the opportunity to investigate biomolecular interaction by implementing fuzzy logic sets as proposed by Gentili [
36].
4. Materials and Methods
4.1. Peptide Synthesis
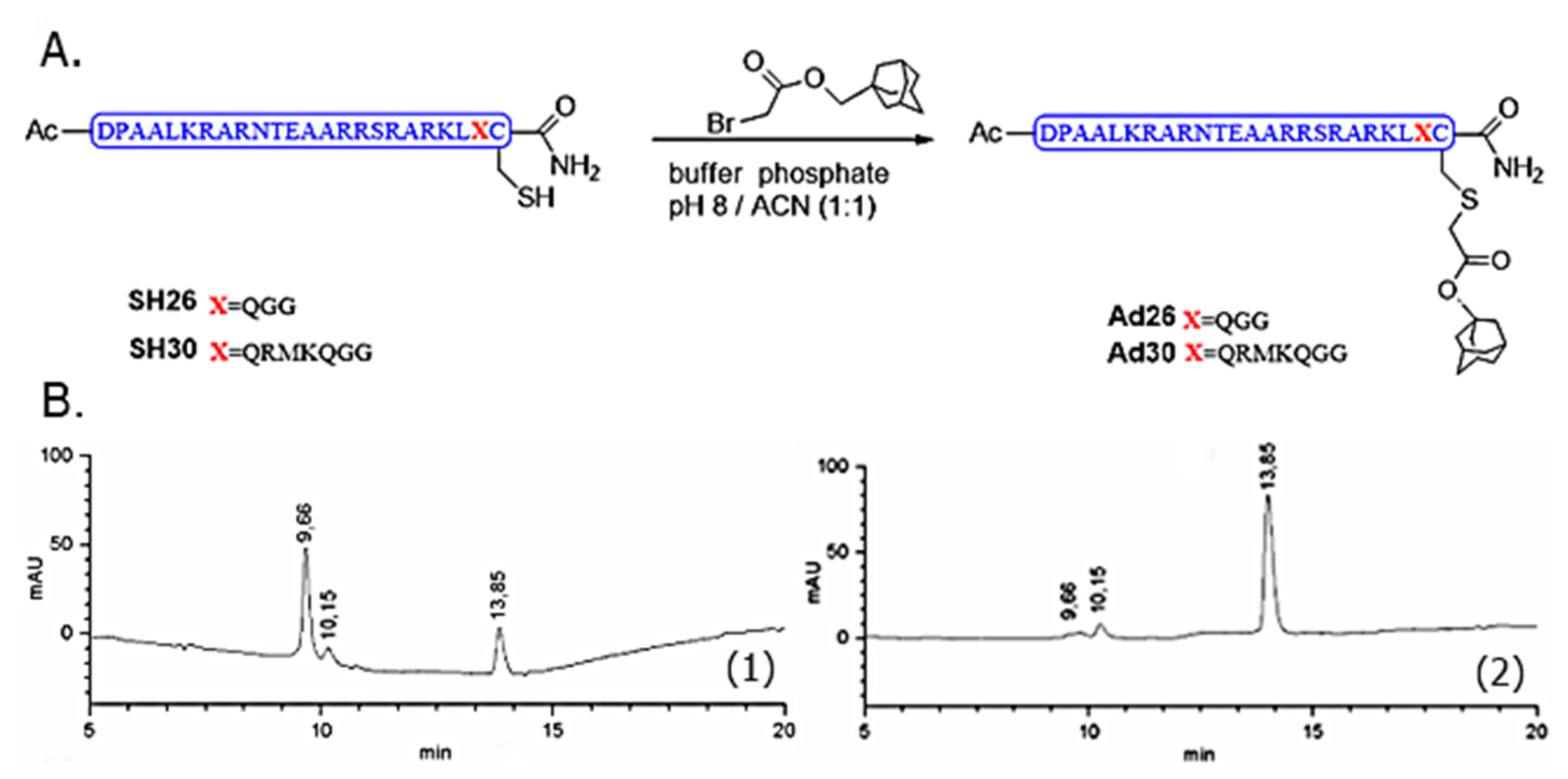
Disulfide dimers SS52 and SS60 were synthesized from commercial peptides SH26 and SH30, to have reference standards to study their interactions with DNA [
15]. Both products were purified by semi-preparative reverse phase HPLC (Waters, Milford, MA, USA) and then lyophilized. The characterization was carried out by MALDI–TOF (Bruker Daltonik, Bremen, Germany), SS52: (M + H) calc. C
234H
414N
98O
74S
2 5786, found: 5788.5 (43%yield). SS60: (M + H)
+ calc. C
278H
498N
116O
80S
4 6873.95, found: 6875.36 (50% yield).
For the synthesis of Ad26 [
15] and Ad30, on both deoxygenated solutions of SH26 (1.9 mg, 6.2 × 10
−4 mmol) and SH30 (2.0 mg, 5.82 × 10
−4 mmol) in potassium phosphate buffer (150 μL, 100 mM, pH = 8.0) and CH
3CN 50 μL were added 4 equiv of bromo acetyldamantane in CH
3CN (0.79 mg, 9 μL). The mixtures were stirred at room temperature for 3 h under N
2 and checked by RP-LC–MS (Agilent 1100, Santa Clara, CA, USA),. The new compounds were purified by semi-preparative RP-HPLC and then lyophilized, identified by mass spectrometry as the alkylated peptides. Once purified and lyophilized, Ad26 (59%) and Ad30 were obtained in 59% and 66% yield, respectively. MALDI–TOF for Ad26, C
130H
229N
49O
37S
1: calc. (M + H)
+ 3101.36, found 3102.81; and for Ad30, C
152H
272N
58O
42S
2: calc. (M + H)
+ 3646.04, found 3647.08.
4.2. Synthesis and Characterization of azoCyDdimer
4,4′-bis (carboxy) azobenzene (0.036 g, 0.132 mmol) was dissolved in dry DMF (2 mL), HATU in DMF was added (0.10 g, 0.317 mmol) and 0.183 mL DIPEA (0.136 g, 1.056 mmol), was stirred for 5 min at room temperature, and to this solution was added β-CyD-NH2 (0.300 g, 0.264 mmol) dissolved in 2 mL of DMF, the resulting mixture was stirred for 3 h under nitrogen atmosphere. The mixture was poured over a container with ice-cold acetone and a precipitate appeared. The solid was separated by filtration and then dissolved in DMF to be purified by silica gel chromatography column. (CH3CN–H2O–NH4OH 14:0:0.5/4:10:0.5). 1H-NMR (500 MHz (Avance II 500, Bruker, Germany), DMSO-d6) δ (ppm): 3.3–4.33 (m, 42H ov 2 H2O), 4.33–4.56 (br.s, 12H), 4.96 (br.s, 14H), 4.82 (br.s, 7H), 5.7–5.82 (m, OH, 27H), 7.98 (4 H, d, J = 8.6 Hz), 8.04 (4 H, d, J = 8.6 Hz), 8.50 (2 H, br. s). 13C-NMR (125.75 MHz, DMSO-d6) δ (ppm): 41.8 (CH2), 59.7 (CH2), 71.9 (CH), 72.3 (CH), 73.0 (CH), 81.3 (CH), 81.9 (CH), 84.1 (CH), 101.64 (CH), 101.9 (CH), 122.2 (CH), 128.4 (CH), 137.4 (C), 153.7 (C), 166.2 (C). MALDI–TOF (M + H)+: calcd. for C98H148N4O70 2500.8, (M + Na)+: calc. 2523.58, found 2523.53, (M + K)+: calc. 2539.98, found 2539.96.
H-NMR Photoisomerization Experiment
Four independent photoisomerization experiments were performed using different initial concentrations of azoCyDdimer: 15 mM, 8 mM, 2.28 mM, and 0.5 mM. Dilutions were made from a 15 mM stock solution with an isomer ratio of 95:5 (E:Z). The stock solution and the dilutions were irradiated at 360 nm for 20 min, and then the corresponding 1HRMN spectra were acquired (AMX 300, Bruker, Germany), protecting the solution from the visible light. Switching experiments were performed with an 8 W mercury arc lamp with filter of 360 nm from Pleuger, Antwerp, Brussels.
4.3. Annealing of dsDNA
Oligonucleotides were purchased from Thermo Fisher Scientific GmbH on a 0.2 mmol scale as freeze-dried solids. After solving in H2O milliQ, their concentrations were measured by ultraviolet absorption at 260 nm with a BioRad SmartSpec Plus Spectrophotometer. Absorbance was measured twice, and concentrations were calculated applying Lambert–Beer’s equation. The molar extinction coefficients of single-strand oligonucleotides were calculated by using the following formula ε(260 nm) = {(8.8 × #T) + (7.3 × #C) + (11.7 × #G) + (15.4 × #A)} × 0.9 × 103 M−1 cm−1, where #A, #T, #C, #G stand for the number of each type of bases in the DNA strand. Oligonucleotides were annealed by heating an equimolar mixture of the two complementary single-strand DNAs to 90 °C for 2 min and then cooling slowly to room temperature (12 h).
4.4. Electrophoretic Shift binding Assays
EMSA was performed with a BioRad Mini Protean gel system, powered by an electrophoresis power supplies PowerPac Basic model, maximum power 150 V, frequency 50.60 Hz at 140 V (constant V). The binding reactions were performed over 30 min in a binding mixture (20 or 40 μL) containing 18 mm tris(hydroxymethyl)aminomethane (Tris; pH 7.5), 90 mm KCl, 1.8 mm MgCl
2, 1.8 mm EDTA, 9%glycerol, 0.11 mgmL
−1 bovine serum albumin (BSA), and 2.2% NP-40 (nonidet-P40). Products were resolved by PAGE by using a 10% nondenaturing polyacrylamide gel and 0.5XTBE buffer solution (44.5 mm Tris, 44.5 mm boric acid,1 mm EDTA, pH 8) and analyzed by staining with SyBrGold (Molecular Probes: 5 mL in 50 mL of 1XTBE) for 10 min and visualized with fluorescence. Ad26 working concentration was 200 nM, 50 nM of DNA, and for azoCyDdimer was used from 0 to 100 equivalents of the E and Z dimer, respectively, was selected (stock solution 0.74 mM in H
2O, ratio E:Z determined by RP-HPLC,
Supplementary Materials Figure S5). The order of addition was azoCyDdimer, Ad26 (pre-incubation for 10 min at 4 °C), and then the corresponding DNA. Duplicates of independent experiments were performed. For the competitive binding assay, we added 200 equiv of 1-adamantane acetic acid after the addition of azoCyDdimer.
4.5. Circular Dichroism Spectroscopy
CD experiments were performed on a Jasco spectrometer (Jasco 715, Tokyo, Japan) with 1 mm path-length cell [
17]. Samples in 10 mM phosphate buffer (pH 7.0) and 100 mM NaCl contained 10 or 5 µM of peptide and 5 µM oligonucleotides (double-stranded) in the absence or presence of 1 equiv of azoCyDdimer at 4 °C (stock solution 0.74 mM in H
2O, ratio E:Z determined by RP-HPLC, see
Supplementary Materials Figure S5). Every sample was incubated for 5 min before registering. Duplicates of independent experiments were performed. The CD spectra of the peptides (when measured in the presence of DNA) were calculated as the difference between the spectrum of the peptide/DNA mixture and the measured spectrum of the respective dsDNA oligonucleotide. For the competitive binding assay, we added 200 equiv of 1-adamantane acetic acid after the addition of azoCyDdimer. Smoothing of the signals was performed by using the software KaleidaGraph.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}







