Design of a New α-1-C-Alkyl-DAB Derivative Acting as a Pharmacological Chaperone for β-Glucocerebrosidase Using Ligand Docking and Molecular Dynamics Simulation

 ,
,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
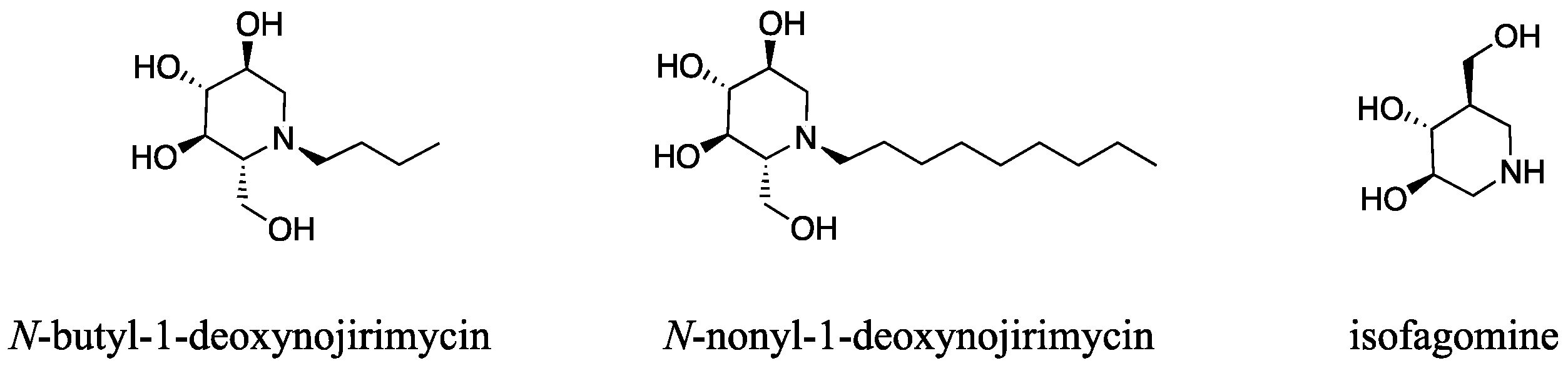
1. Introduction
2. Results
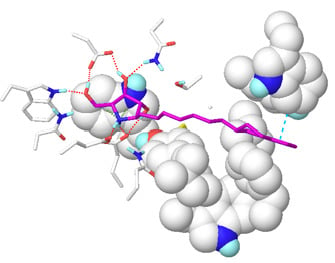
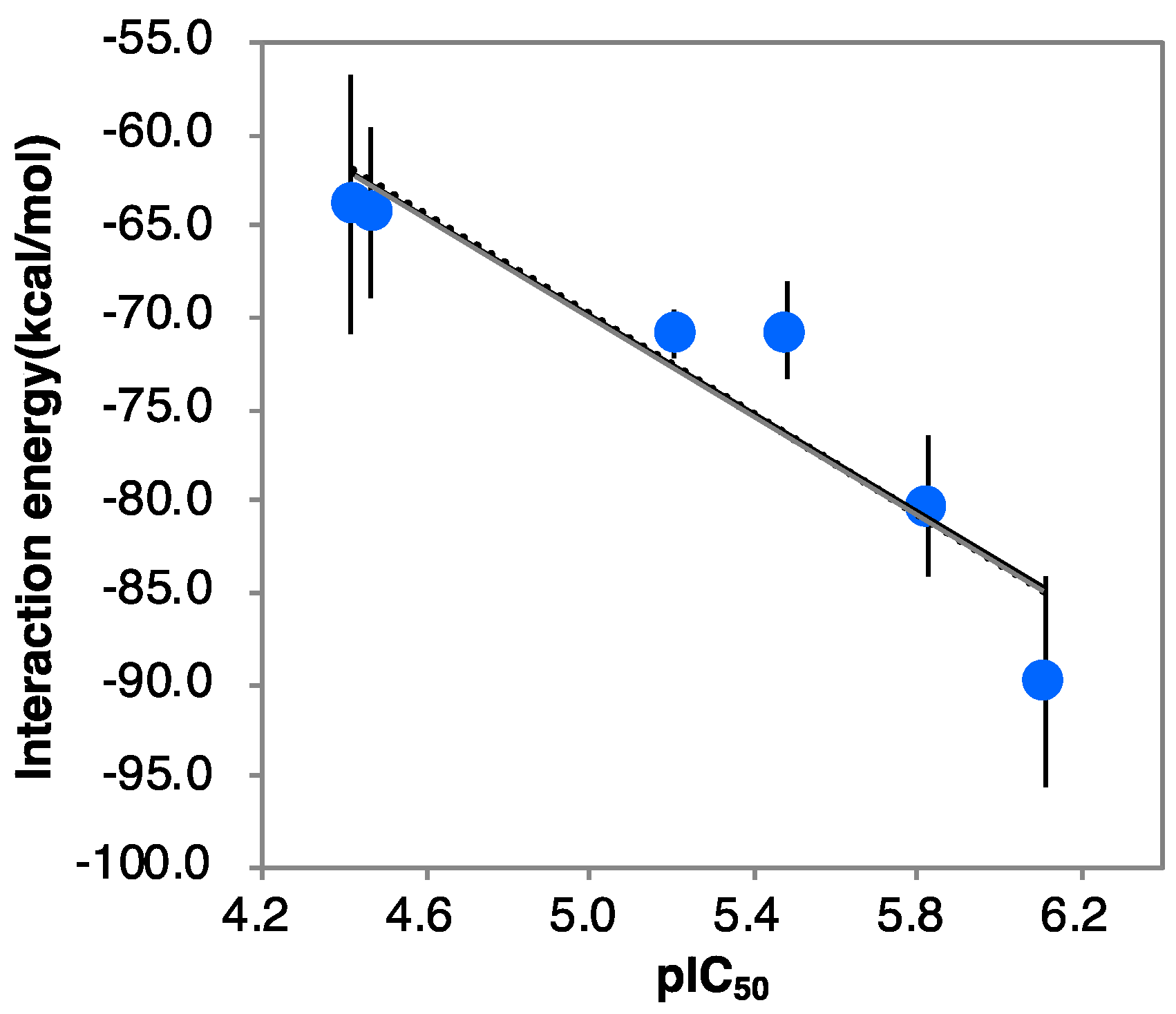
2.1. Docking Results
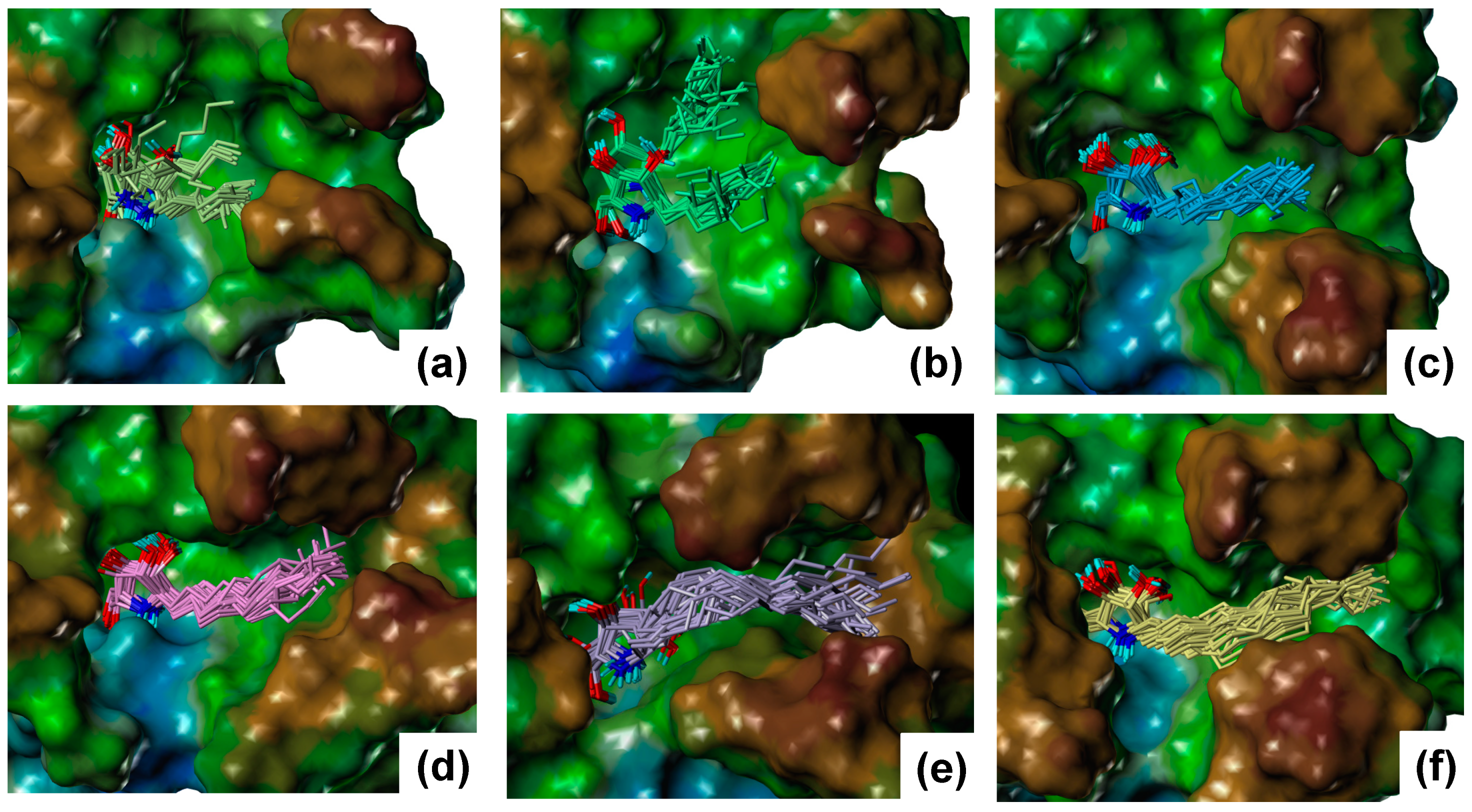
2.2. Results of MD Calculations
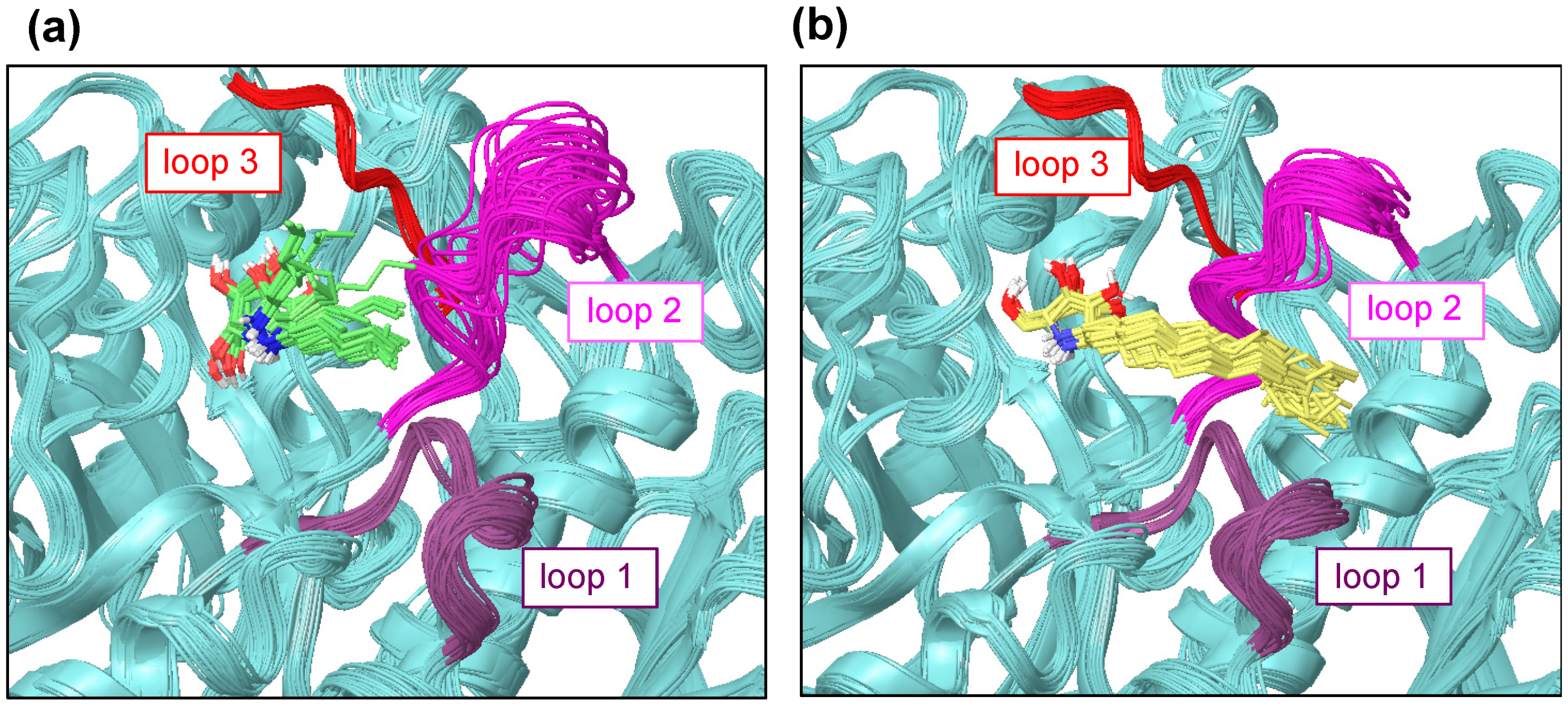
2.3. Fluctuation of Loop 2
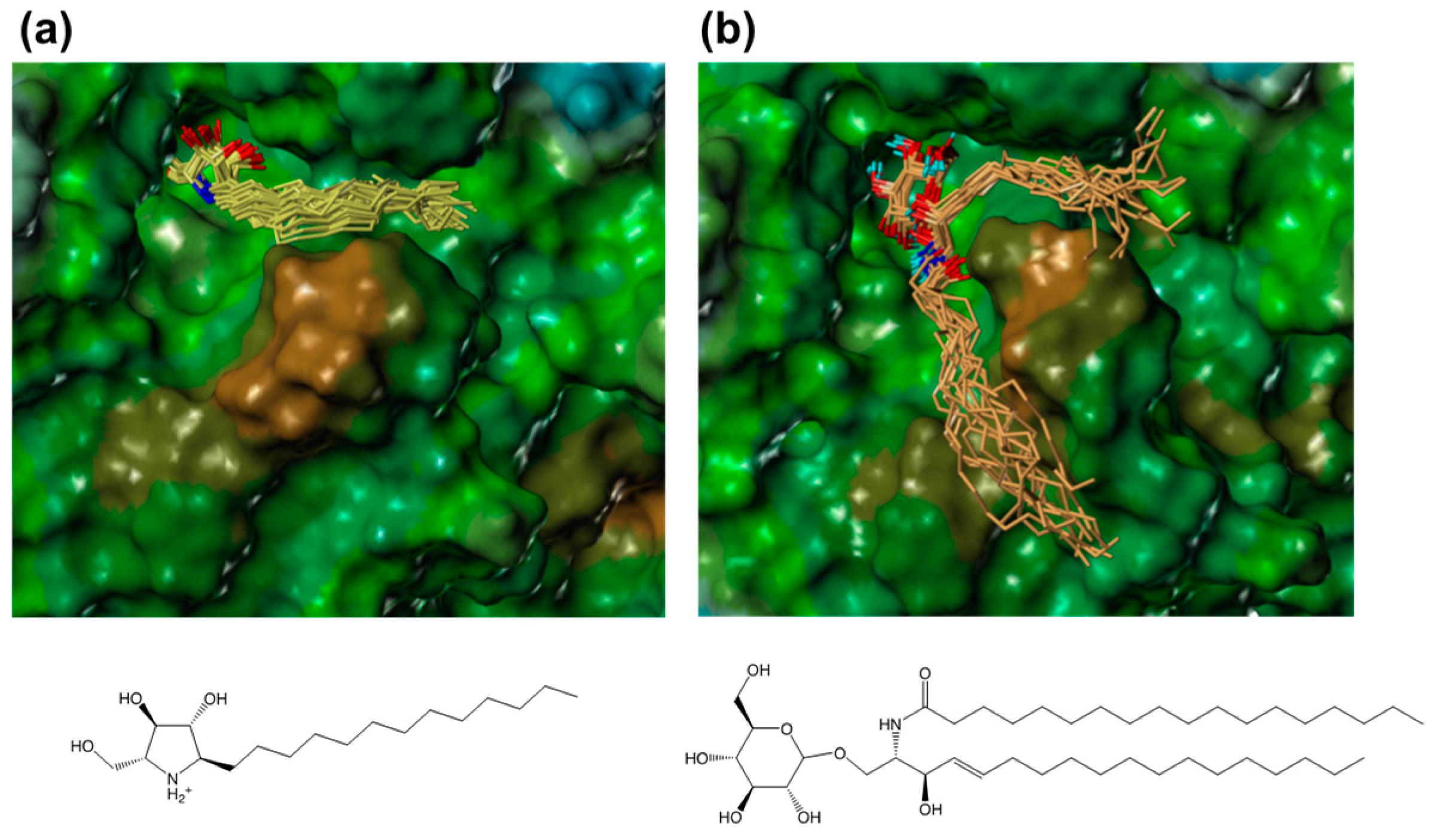
2.4. Modeling of a Complex Structure of β-Glucocerebrosidase with Glucosylceramide
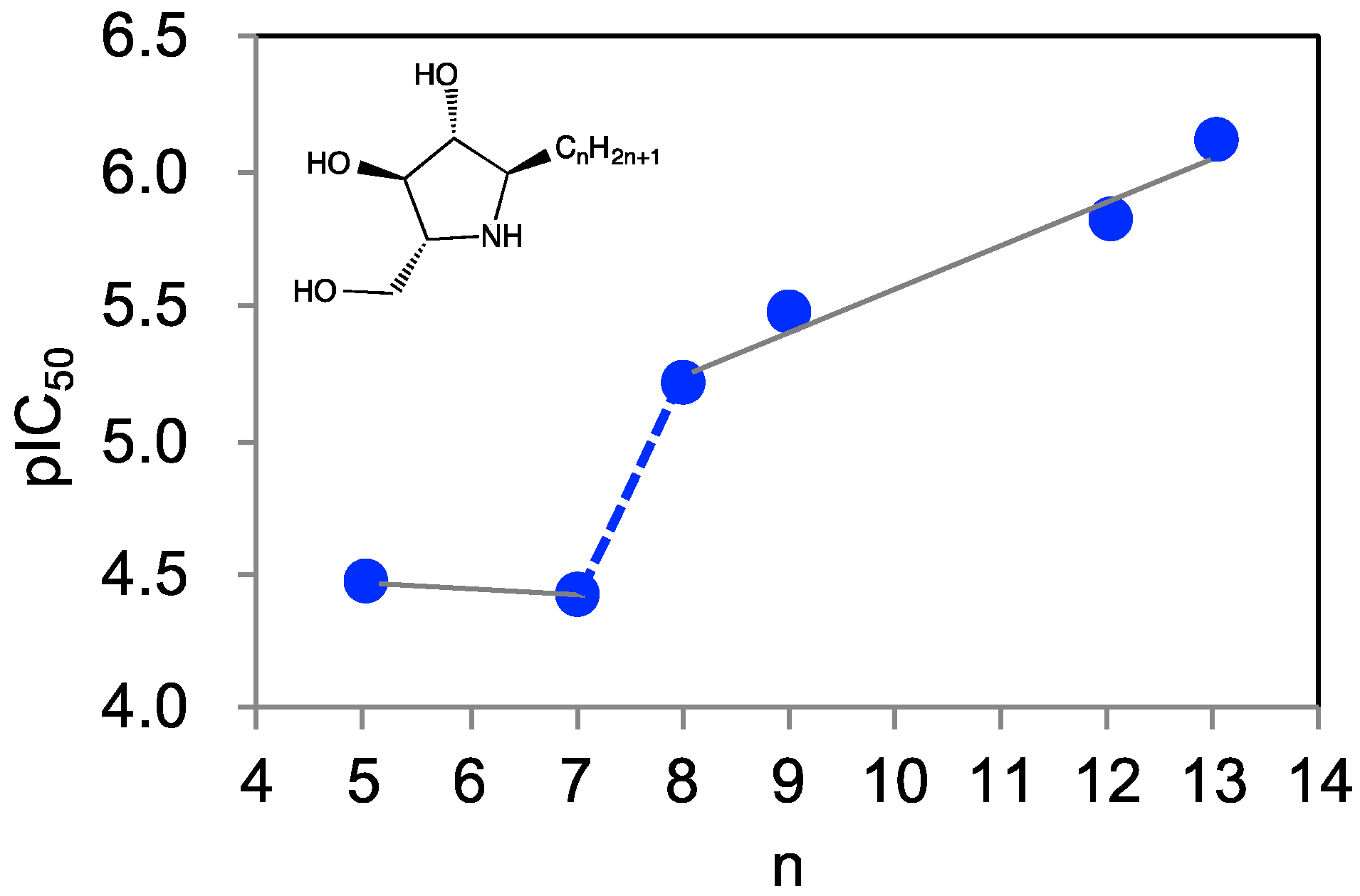
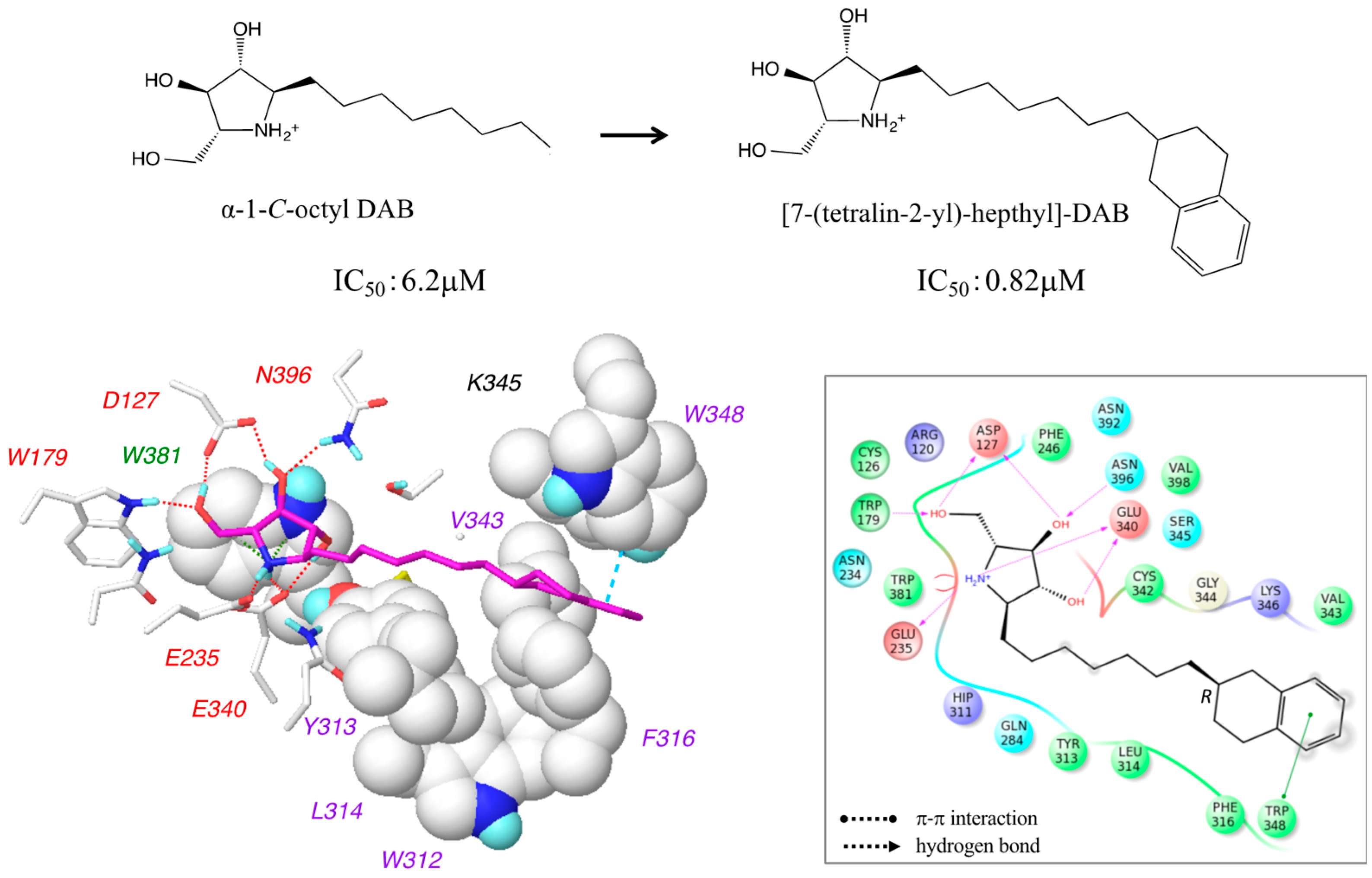
2.5. Design of 7-(Tetralin-2-yl)-heptyl-DAB
3. Discussion
4. Materials and Methods
4.1. Docking Analysis
4.2. MD Calculations
4.3. Enzyme Inhibition Assay
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Liou, B.; Kazimierczuk, A.; Zhang, M.; Scott, C.R.; Hegde, R.S.; Grabowski, G.A. Analyses of variant acid beta-glucosidases: Effects of Gaucher disease mutations. J. Biol. Chem. 2006, 281, 4242–4253. [Google Scholar] [CrossRef] [PubMed]
- Butters, T.D. Gaucher disease. Curr. Opin. Chem. Biol. 2007, 11, 412–418. [Google Scholar] [CrossRef] [PubMed]
- Lieberman, R.L.; D’aquino, J.A.; Ringe, D.; Petsko, G.A. Effects of pH and iminosugar pharmacological chaperones on lysosomal glycosidase structure and stability. Biochemistry 2009, 48, 4816–4827. [Google Scholar] [CrossRef] [PubMed]
- Boyd, R.E.; Lee, G.; Rybczynski, P.; Benjamin, E.R.; Khanna, R.; Wustman, B.A.; Valenzano, K.J. Pharmacological Chaperones as Therapeutics for Lysosomal Storage Diseases. J. Med. Chem. 2013, 56, 2705–2725. [Google Scholar] [CrossRef] [PubMed]
- Trapero, A.; Llebaria, A. Glucocerebrosidase inhibitors for the treatment of Gaucher disease. Future Med. Chem. 2013, 5, 573–590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, J.Q. A contradictory treatment for lysosomal storage disorders: Inhibitors enhance mutant enzyme activity. Trends Pharmacol. Sci. 2003, 24, 355–360. [Google Scholar] [CrossRef]
- Sawkar, A.R.; Adamski-Werner, S.L.; Cheng, W.C.; Wong, C.H.; Beutler, E.; Zimmer, K.P.; Kelly, J.W. Gaucher disease-associated glucocerebrosidases show mutation-dependent chemical chaperoning profiles. Chem. Biol. 2005, 12, 1235–1244. [Google Scholar] [CrossRef] [PubMed]
- Sawker, A.R.; Cheng, W.C.; Beautler, E.; Wong, C.H.; Baich, W.E.; Kelly, J.W. Chemical chaperones increase the cellular activity of N370S beta -glucosidase: A therapeutic strategy for Gaucher disease. Proc. Natl. Acad. Sci. USA 2002, 99, 15428–15433. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.H.; Asano, N.; Ishii, S.; Ichikawa, Y.; Fan, J.Q. Hydrophilic iminosugar active-site-specific chaperones increase residual glucocerebrosidase activity in fibroblasts from Gaucher patients. FEBS J. 2006, 273, 4082–4092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Priestman, D.A.; van der Spoel, A.C.; Butters, T.D.; Dwek, R.A.; Platt, F.M. N-Butyldeoxynojirimycin causes weight loss as a result of appetite suppression in lean and obese mice. Diabetes Obes. Metab. 2008, 10, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Ficicioglu, C. Review of miglustat for clinical management in Gaucher disease type 1. Ther. Clin. Risk Manag. 2008, 4, 425–431. [Google Scholar] [CrossRef] [PubMed]
- Khanna, R.; Benjamin, E.R.; Pellegrino, L.; Schilling, A.; Rigat, B.A.; Soska, R.; Nafar, H.; Ranes, B.E.; Feng, J.; Lun, Y.; et al. The Pharmacological Chaperone Isofagomine Increases Activity of the Gaucher Disease L444P Mutant Form of β-Glucosidase. FEBS J. 2010, 277, 1618–1638. [Google Scholar] [CrossRef] [PubMed]
- Brumshtein, B.; Aguilar-Moncayo, M.; Benito, J.M.; García Fernandez, J.M.; Silman, I.; Shaaltiel, Y.; Aviezer, D.; Sussman, J.L.; Futerman, A.H.; Mellet, C.O. Cyclodextrin-mediated crystallization of acid β-glucosidase in complex with amphiphilic bicyclic nojirimycin analogues. Org. Biomol. Chem. 2011, 9, 4160–4167. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Sheth, K.A.; Li, S.; Chang, H.H.; Fan, J.Q. Rational design and synthesis of highly potent beta-glucocerebrosidase inhibitors. Angew. Chem. Int. Ed. 2005, 44, 7450–7453. [Google Scholar] [CrossRef] [PubMed]
- Kato, A.; Nakagome, I.; Sato, K.; Yamamoto, A.; Adachi, I.; Nash, R.J.; Fleet, G.W.J.; Natori, Y.; Watanabe, Y.; Imahori, T.; et al. Docking study and biological evaluation of pyrrolidine-based iminosugars as pharmacological chaperones for Gaucher disease. Org. Biomol. Chem. 2016, 14, 1039–1048. [Google Scholar] [CrossRef] [PubMed]
- Wei, R.R.; Hughes, H.; Boucher, S.; Bird, J.J.; Guziewicz, N.; Van Patten, S.M.; Qiu, H.; Pan, C.Q.; Edmunds, T. X-ray and biochemical analysis of N370S mutant human acid β-glucosidase. J. Biol. Chem. 2011, 286, 299–308. [Google Scholar] [CrossRef] [PubMed]
- Lieberman, R.L.; Wustman, B.A.; Huertas, P.; Powe, A.C., Jr.; Pine, C.W.; Khanna, R.; Schlossmacher, M.G.; Ringe, D.; Petsko, G.A. Structure of acid beta-glucosidase with pharmacological chaperone provides insight into Gaucher disease. Nat. Chem. Biol. 2007, 3, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Kato, A.; Nakagome, I.; Nakagawa, S.; Koike, Y.; Nash, R.J.; Adachi, I.; Hirono, S. Docking and SAR studies of calystegines: Binding orientation and influence on pharmacological chaperone effects for Gaucher’s disease. Bioorg. Med. Chem. 2014, 22, 2435–2441. [Google Scholar] [CrossRef] [PubMed]
- Sherman, W.; Day, T.; Jacobson, M.P.; Friesner, R.A.; Farid, R. Novel procedure for modeling ligand/receptor induced fit effects. J. Med. Chem. 2006, 49, 534–553. [Google Scholar] [CrossRef] [PubMed]
- Shivakumar, D.; Williams, J.; Wu, Y.; Damm, W.; Shelley, J.; Sherman, W. Prediction of Absolute Solvation Free Energies using Molecular Dynamics Free Energy Perturbation and the OPLS Force Field. J. Chem. Theory Comput. 2010, 6, 1509–1519. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Mohanty, U.; Noehre, J.; Sawyer, T.K.; Sherman, W.; Krilov, G. Probing the alpha-helical structural stability of stapled p53 peptides: Molecular dynamics simulations and analysis. Chem. Biol. Drug Des. 2010, 75, 348–359. [Google Scholar] [CrossRef] [PubMed]
- Bowers, K.J.; Chow, E.; Xu, H.; Dror, R.O.; Eastwood, M.P.; Gregersen, B.A.; Klepeis, J.L.; Kolossvary, I.; Moraesm, M.A.; Sacerdotim, F.D.; et al. Scalable algorithms for molecular dynamics simulations on commodity clusters. Presented at the 2006 ACM/IEEE conference on Supercomputing, Tampa, FL, USA, 11–17 November 2006. [Google Scholar]
Sample Availability: Not available. |








© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nakagome, I.; Kato, A.; Yamaotsu, N.; Yoshida, T.; Ozawa, S.-i.; Adachi, I.; Hirono, S. Design of a New α-1-C-Alkyl-DAB Derivative Acting as a Pharmacological Chaperone for β-Glucocerebrosidase Using Ligand Docking and Molecular Dynamics Simulation. Molecules 2018, 23, 2683. https://doi.org/10.3390/molecules23102683
Nakagome I, Kato A, Yamaotsu N, Yoshida T, Ozawa S-i, Adachi I, Hirono S. Design of a New α-1-C-Alkyl-DAB Derivative Acting as a Pharmacological Chaperone for β-Glucocerebrosidase Using Ligand Docking and Molecular Dynamics Simulation. Molecules. 2018; 23(10):2683. https://doi.org/10.3390/molecules23102683
Chicago/Turabian StyleNakagome, Izumi, Atsushi Kato, Noriyuki Yamaotsu, Tomoki Yoshida, Shin-ichiro Ozawa, Isao Adachi, and Shuichi Hirono. 2018. "Design of a New α-1-C-Alkyl-DAB Derivative Acting as a Pharmacological Chaperone for β-Glucocerebrosidase Using Ligand Docking and Molecular Dynamics Simulation" Molecules 23, no. 10: 2683. https://doi.org/10.3390/molecules23102683