2.1. Structural Elucidation of the New Compounds
Compound
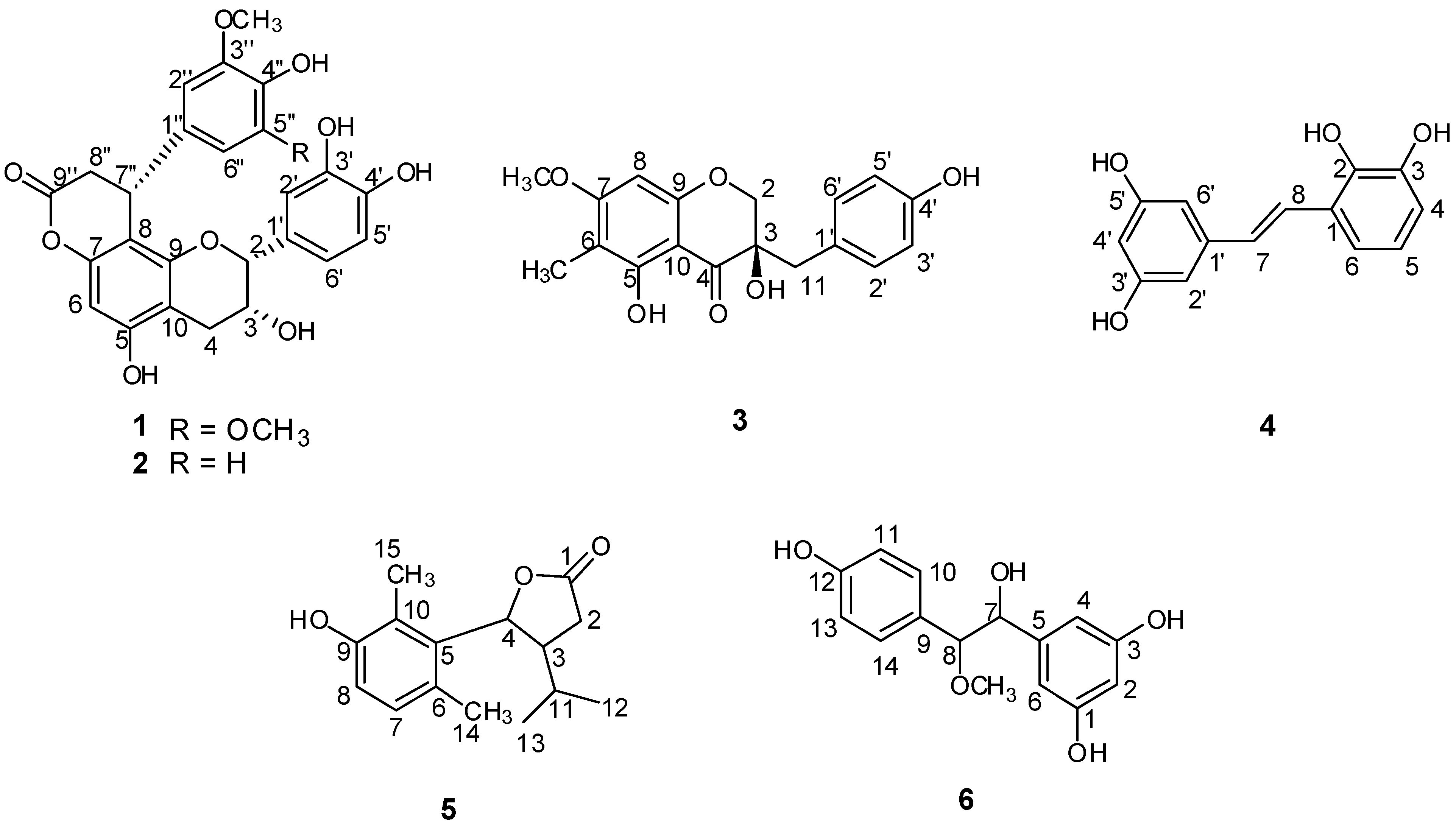
1 (
Figure 1) was obtained as a pale yellow solid. The molecular formula of compound
1, C
26H
24O
10, was concluded from its HRESIMS, which showed a quasi-molecular ion peak at
m/z 495.1299 [M-H]
− (calcd. for C
26H
23O
10 495.1291) in the negative ion mode. Its IR spectrum showed absoptions due to hydroxyl (3,452 cm
−1) and carbonyl (1,751 cm
−1) groups. In the UV spectrum, absorption maxima at 202, 230 and 280 nm were observed. The
1H-NMR and
13C-NMR spectra (
Table 1) showed one ABX coupling system at
δ 7.06 (1H, d,
J = 1.9 Hz),
δ 6.79 (1H, d,
J = 8.1 Hz) and
δ 6.84 (1H, dd,
J = 8.1, 1.9 Hz), two geminal coupling proton signals at
δ 2.92 (1H, dd,
J = 17.2, 4.5 Hz) and
δ 2.85 (1H, dd,
J = 17.2, 1.7 Hz), and three aliphatic carbon signals at
δ 79.8 (C-2),
δ 66.8 (C-3),
δ 29.8 (C-4). These findings revealed the presence of an epicatechin unit. In the
1H-NMR spectrum of
1, an aromatic proton singlet at
δ 6.20 was observed, which suggested substitution at the 6- or 8-position of the A-ring. The presence of one symmetrical 1,3,4,5-tetrasubstituted phenyl group was deduced from the signal at
δ 6.43 (2H, s). The
1H-NMR spectrum also indicated the presence of two methoxy groups at
δ 3.59 (6H, s), a methine proton at
δ 4.60 (1H, br.d,
J = 6.3 Hz), and the methylene protons at
δ 3.12 (1H, dd,
J = 15.9, 7.5 Hz) and
δ 2.91 (1H, dd,
J = 15.9, 1.5 Hz). In addition, resonances attributable to a methine carbon at
δ 36.2 and a methylene carbon at
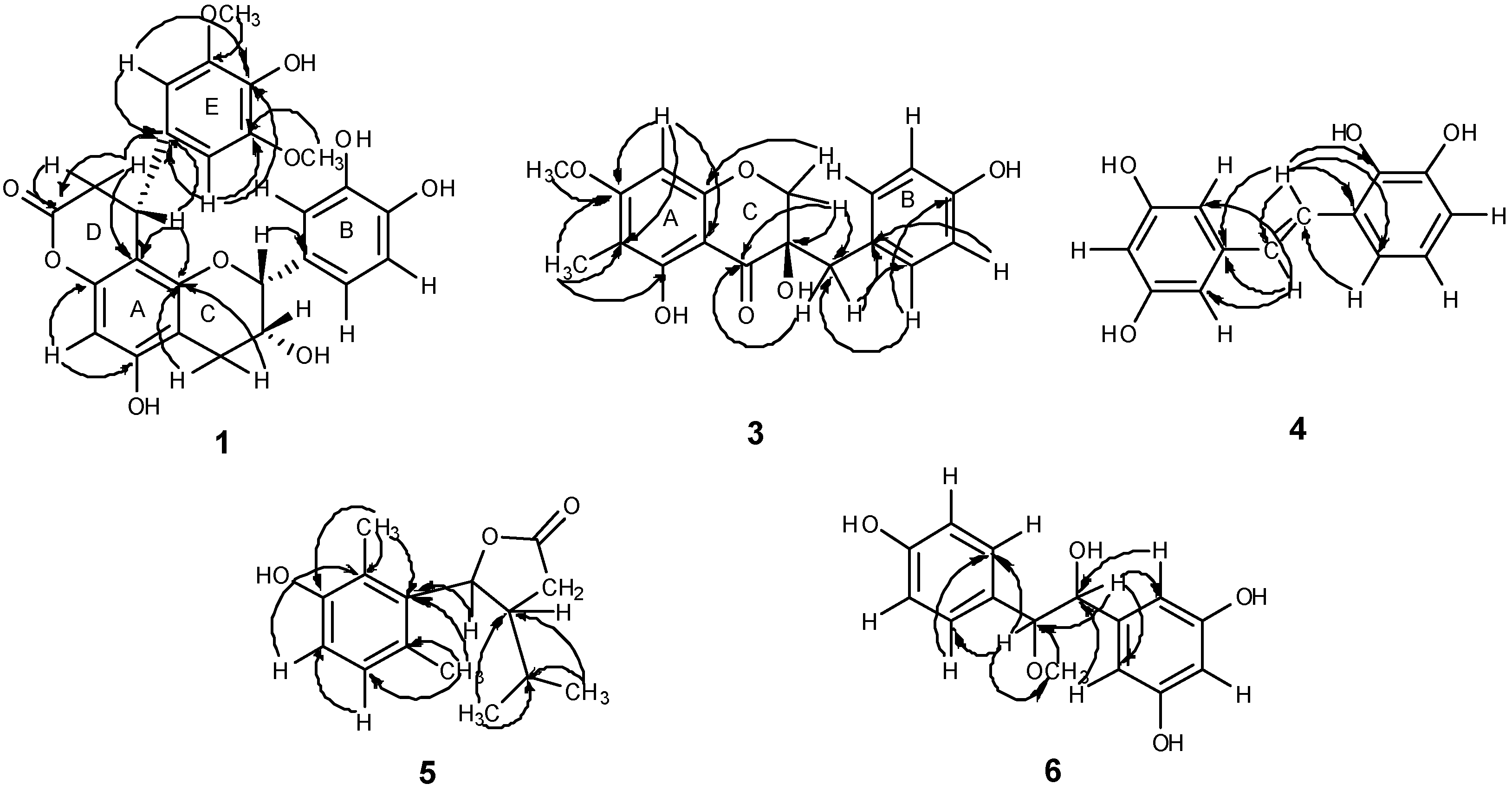
δ 37.5 were observed. The HMBC spectrum (
Figure 3) showed correlations from H-2′′ to C-7′′ (
δ 36.2), H-7′′ to C-1′′ (
δ 135.2) and H-8′′ to C-9′′ (
δ 171.0), indicating the presence of a phenylpropanoid unit. In the HMBC spectrum, correlations of H-8′′ with C-8 (
δ 106.2), C-1′′ (
δ 135.2) and C-9′′ (
δ 171.0) indicated that the phenylpropanoid unit was attached to the C-8 position of the epicatechin, which was further confirmed by the diagnostic
13C-NMR resonances for the C-10, C-6 and C-8 carbons at
δ 105.4, 96.3 and 106.1. It has been established that in the NMR spectra the position of the C-10 chemical shift is distinctive for the location of the lactone function in the A-ring. The C-8 location has the C-10 chemical shift downfield (
δ 105) relative to the C-6 regioisomer (
δ 100) [
12]. For compound
1, the C-10 resonance was in accordance with those of C-8 phenylpropanoid-substituted epicatechins. Furthermore, the
13C-NMR resonance at
δ 171.0 (C-9′′) indicated that the phenylpropanoid unit was fused to the OH group at C-7 of the A-ring of the epicatechin unit through an ester linkage. The HMBC correlation between the methoxy protons at
δ 3.59 (6H, s) and
δ 149.2 (C-3′′, 5′′) confirmed the methoxy groups were linked to C-3′′, 5′′. Thus, compound
1 was very similar to cinchonain Ia, previously isolated from the bark of
Cinchona succirubra, but with additional
O-methyl groups [
13]. The absolute configuration of C-7′′ was defined based on the CD spectrum, which showed negative Cotton effects at 235 and 283 nm and a positive Cotton effect at 259 nm. Hence, the absolute configuration of C-7′′ was determined to be
R [
13]. Consequently, the structure of
1 was elucidated as epicatechin-(7,8-bc)-4
α-(4-hydroxy-3,5-dimethoxyphenyl)-dihydro-2(3
H)-pyranone, and was trivially named smiglabrone A.
Table 1.
1H-NMR (600 MHz) and 13C-NMR (150 MHz) spectra data of compounds 1 and 2 (in CD3OD, J in Hz, δ in ppm).
Table 1.
1H-NMR (600 MHz) and 13C-NMR (150 MHz) spectra data of compounds 1 and 2 (in CD3OD, J in Hz, δ in ppm).
| Position | 1 | 2 |
|---|
| δH (J in Hz) | δC | δH (J in Hz) | δC |
|---|
| 2 | 4.81 (1H, s) | 79.8 | 4.82 (1H, s) | 79.8 |
| 3 | 4.26 (1H, m) | 66.8 | 4.25 (1H, m) | 66.7 |
| 4 | 2.85 (1H, dd,
J = 17.2, 1.7) | 29.8 | 2.85 (1H, dd,
J = 17.2, 2.0) | 29.7 |
| | 2.92 (1H, dd,
J = 17.2, 4.5) | | 2.92 (1H, dd,
J = 7.2, 4.4) | |
| 5 | – | 157.4 | – | 157.4 |
| 6 | 6.20 (1H, s) | 96.3 | 6.19 (1H, s) | 96.3 |
| 7 | – | 151.6 | – | 151.7 |
| 8 | – | 106.2 | – | 106.2 |
| 9 | – | 153.6 | – | 153.5 |
| 10 | – | 105.4 | – | 105.3 |
| 1′ | – | 132.0 | – | 132.0 |
| 2′ | 7.06 (1H, d,
J = 1.9) | 115.1 | 7.03 (1H, d,
J = 1.8) | 115.1 |
| 3′ | – | 146.2 | – | 146.1 |
| 4′ | – | 145.9 | – | 145.8 |
| 5′ | 6.79 (1H, d,
J = 8.1) | 116.1 | 6.781 (1H, d,
J = 8.2) | 116.0 |
| 6′ | 6.84 (1H, dd,
J = 8.1, 1.9) | 119.1 | 6.83 (1H, dd,
J = 8.2, 1.8) | 119.1 |
| 1′′ | – | 135.2 | – | 135.8 |
| 2′′ | 6.43 (1H, s) | 105.0 | 6.776 (1H, d,
J = 2.0) | 112.2 |
| 3′′ | – | 149.2 | – | 148.8 |
| 4′′ | – | 135.1 | – | 146.3 |
| 5′′ | – | 149.2 | 6.62 (1H, d,
J = 8.2) | 116.2 |
| 6′′ | 6.43 (1H, s) | 105.0 | 6.50 (1H, dd,
J = 8.2, 2.0) | 119.4 |
| 7′′ | 4.60 (1H, br. d,
J = 6.3) | 36.2 | 4.60 (1H, br. d,
J = 6.3) | 35.7 |
| 8′′ | 2.91 (1H, dd,
J = 15.9, 1.5) | 37.5 | 2.90 (1H, dd,
J = 15.9, 1.5) | 37.8 |
| | 3.12 (1H, dd,
J = 15.9, 7.5) | | 3.10 (1H, dd,
J = 15.9, 7.4) | |
| 9′′ | – | 171.0 | – | 170.9 |
| 3′′-OCH3 | – | – | 3.56 (3H, s) | 56.0 |
| 3′′, 5′′-OCH3 | 3.59 (6H, s) | 56.4 | – | – |
Figure 3.
Main HMBC correlations of compounds 1, 3–6.
Figure 3.
Main HMBC correlations of compounds 1, 3–6.
Compound
2 (
Figure 1) was isolated as a pale yellow solid. The molecular formula of compound
2, C
25H
22O
9, was concluded from its HRESIMS, which showed quasi-molecular ion peaks at
m/z 465.1189 [M−H]
− (calcd. for C
25H
21O
9 465.1186) and 467.1327 [M+H]
+ (calcd. for C
25H
23O
9 467.1342) in the negative and positive modes, respectively. Its IR spectrum showed absorptions due to hydroxyl (3,453 cm
−1) and carbonyl (1,745 cm
−1) groups. UV absorptions at 202, 230 and 280 nm were observed. Detailed
1H,
13C, and HMBC spectra suggested
2 to be a phenylpropanoid-substituted epicatechin quite similar to compound
1, but with differences in the substitution pattern of the E-ring. The
1H-NMR spectrum (
Table 1) showed proton signals at
δ 6.776 (1H, d,
J = 2.0 Hz),
δ 6.62 (1H, d,
J = 8.2 Hz) and
δ 6.50 (1H, dd,
J = 8.2, 2.0 Hz), revealing the presence of a 1,3,4-trisubstituted aromatic ring. A methoxy group at
δ 3.56 (3H, s) was observed. The HMBC spectrum showed correlation from the methoxy protons at
δ 3.56 to
δ 148.8 (C-3′′), indicating that the methoxy group was attached to C-3′′ in the E-ring. To define the absolute configuration of
2 at C-7′′, a circular dichroism investigation was undertaken. The CD spectrum showed a negative Cotton effects at 234 and 284 nm and a positive Cotton effect at 256 nm. Thus, C-7′′ also possessed the
R configuration [
13]. Accordingly, compound
2 was determined as epicatechin-(7,8-bc)-4
α-(3-methoxy-4-hydroxy-phenyl)-dihydro-2(3
H)-pyranone, and trivially named smiglabrone B.
Compound
3 (
Figure 1) was obtained as a pale yellow solid. Its molecular formula was determined as C
18H
18O
6 from the negative-ion HRESIMS with a quasi-molecular ion peak at
m/z 329.1033 [M−H]
− (calcd. for C
18H
17O
6 329.1025). The IR spectrum of
3 showed characteristic absorption bands for hydroxyl (3,450 cm
−1) and carbonyl (1,633 cm
-1) groups. UV absorptions at 216 and 294 nm were observed. The
1H-NMR spectrum (
Table 2) revealed a set of characteristic signals at
δ 4.06 (1H, d,
J = 11.0 Hz) and 3.96 (1H, d,
J = 11.0 Hz) for one methylene linked to an oxygen atom, at
δ 2.90 (1H, d,
J = 14.5 Hz) and 2.86 (1H, d,
J = 14.5 Hz) for one benzylic methylene, indicating that
3 possessed a homoisoflavanone skeleton. A trisubstitution of the A-ring was deduced from the occurence of a one aromatic proton singlet at
δ 6.14, suggesting a substituent either at C-6 or at C-8. Four aromatic protons at
δ 7.05 (2H, d,
J = 8.5 Hz) and 6.70 (2H, d,
J = 8.5 Hz) revealed a 1,4-disubstituted B-ring. In addition, an aromatic methoxyl group at
δ 3.86 (3H, s) and a methyl group at
δ 1.95 (3H, s) were also observed in the
1H-NMR spectrum. The
13C-NMR spectrum (
Table 2) exhibited, in total, eighteen carbon resonances, including one aromatic carbon linked to a carbonyl at
δ 101.8, one alkyl-substituted aromatic carbon at
δ 126.8, one flavanone carbonyl carbon at
δ 200.6, one methylene carbon having an oxygen function at
δ 72.9, one benzylic methylene carbon at
δ 40.8, one methoxyl carbon at
δ 56.5, one methyl carbon at
δ 7.0, and one quaternary carbon having an oxygen function at
δ 73.6. The HMBC correlation of the methoxyl protons at
δ3.86 with C-7 at
δ 167.5 indicated that the linkage position of the methoxyl group was at C-7. The position for the methyl group attaching at C-6 was determined by correlation from methyl protons at
δ1.95 to C-6 (
δ 106.7). The long-range correlation from H-2 to C-9 (
δ 162.7), C-4 (
δ 200.6) and C-11 (
δ 40.8), from H-11 to C-4 (
δ 200.6) and C-2′ (
δ 132.8) further confirmed the speculation of the structure of compound
3 (
Figure 3). The planar structure of compound
3 was identified as 3,5-dihydroxy-7-methoxy-6-methyl-3-(4-hydroxybenzyl)chroman-4-one [
14]. The absolute configuration of C-3 was assessed by optical its rotation (
![Molecules 18 05265 i001]()
+178.72), indicating the
R configuration at C-3 [
15]. Consequently, the structure of compound
3 was identified as (3
R)-3,5-dihydroxy-7-methoxy-6-methyl-3-(4-hydroxybenzyl)chroman-4-one, and trivially named smilachromanone.
Table 2.
1H-NMR (500 MHz) and 13C-NMR (125 MHz) spectra data of compound 3 (in CD3OD, J in Hz, δ in ppm).
Table 2.
1H-NMR (500 MHz) and 13C-NMR (125 MHz) spectra data of compound 3 (in CD3OD, J in Hz, δ in ppm).
| Position | δH (J in Hz) | δC |
|---|
| 2 | 4.06 (1H, d,
J = 11.0) | 72.9 |
| | 3.96 (1H, d,
J = 11.0) | – |
| 3 | – | 73.6 |
| 4 | – | 200.6 |
| 5 | – | 161.6 |
| 6 | – | 106.7 |
| 7 | – | 167.5 |
| 8 | 6.14 (1H, s) | 91.8 |
| 9 | – | 162.7 |
| 10 | – | 101.8 |
| 11 | 2.90 (1H, d,
J = 14.5) | 40.8 |
| | 2.86 (1H, d,
J = 14.5) | – |
| 1′ | – | 126.8 |
| 2′, 6′ | 7.05 (2H, d,
J = 8.5) | 132.8 |
| 3′, 5′ | 6.70 (2H, d,
J = 8.5) | 115.9 |
| 4′ | – | 157.5 |
| 7-OCH3 | 3.86 (3H, s) | 56.5 |
| 6-CH3 | 1.95 (3H, s) | 7.0 |
Compound
4 (
Figure 1) was obtained as a brown-yellow powder. Its molecular formula was determined as C
14H
12O
4 by negative-ion HRESIMS with quasi-molecular ion peaks at
m/z 243.0659 [M-H]
− (calcd. for C
14H
11O
4 243.0657). The IR spectrum of
4 showed a characteristic hydroxyl absorption band (3,449 cm
−1). UV absorptions at 220 and 306 nm were observed. The
1H-NMR spectrum (
Table 3) exhibited proton signals at
δ 6.48 (2H, d,
J = 1.8 Hz) and 6.17 (1H, t,
J = 1.8 Hz), suggesting the presence of a 1,3,5-trisubstituted aromatic ring in the molecule. The signals at
δ 7.37 (1H, d,
J = 16.8 Hz) and 6.96 (1H, d,
J = 16.8 Hz) were attributed to a set of
trans-olefinic protons. The remaining protons at
δ 6.69 (1H, dd,
J = 1.8, 7.8 Hz), 6.66 (1H, t,
J = 7.8 Hz) and 7.02 (1H, dd,
J = 1.8, 7.8 Hz) indicated the existence of a 1,2,3-trisubstituted aromatic ring. The
13C-NMR spectrum (
Table 3) exhibited, in total, twelve carbon resonances including two olefinic carbons and ten aromatic carbons. The HMBC spectrum showed correlations from H-7 to C-1′ (
δ 141.6) and C-2′, 6′ (
δ 105.9), indicating that C-7, C-2′ and C-6′ were linked through C-1′; the correlations from H-8 to C-1 (
δ 126.0), C-2 (
δ 144.6) and C-6 (
δ 118.3), suggesting that C-8, C-2 and C-6 were linked through C-1 (
Figure 3). Accordingly, compound
4 was elucidated as (
E)-3-(3,5-dihydroxystyryl)benzene-1,2-diol, and trivially named smiglastilbene.
Table 3.
1H-NMR (600 MHz) and 13C-NMR (150 MHz) spectra data of compound 4 (in CD3OD, J in Hz, δ in ppm).
Table 3.
1H-NMR (600 MHz) and 13C-NMR (150 MHz) spectra data of compound 4 (in CD3OD, J in Hz, δ in ppm).
| Position | δH (J in Hz) | δC |
|---|
| 1 | – | 126.0 |
| 2 | – | 144.6 |
| 3 | – | 146.5 |
| 4 | 6.69 (1H, dd,
J = 7.8, 1.8) | 114.9 |
| 5 | 6.66 (1H, t,
J = 7.8) | 120.5 |
| 6 | 7.02 (1H, dd,
J = 7.8, 1.8) | 118.3 |
| 7 | 6.96 (1H, d,
J = 16.8) | 129.5 |
| 8 | 7.37 (1H, d,
J = 16.8) | 124.7 |
| 1′ | – | 141.6 |
| 2′, 6′ | 6.48 (2H, d,
J = 1.8) | 105.9 |
| 3′, 5′ | – | 159.7 |
| 4′ | 6.17 (1H, t,
J = 1.8) | 102.8 |
Compound
5 (
Figure 1) was obtained as a white powder. Its molecular formula was determined as C
15H
20O
3 by HREIMS with a molecular ion peak at
m/z 248.1415 [M]
+ (calcd. for C
15H
20O
3 248.1412). The IR spectrum of
5 showed characteristic absorption bands for hydroxyl (3,357 cm
−1) and carbonyl (1,747 cm
−1) groups. UV absorptions at 201 and 289 nm were observed. The
1H-NMR spectrum (
Table 4) displayed four methyl groups at
δ 0.76 (3H, d,
J = 6.6 Hz), 0.98 (3H, d,
J = 6.6 Hz), 2.30 (3H, s), 2.22 (3H, s), a proton signal at
δ 5.72 (1H, d,
J = 9.0 Hz) attached to the oxygenated carbon, and two methenyl proton signals at
δ 2.75 (1H, m) and 1.76 (1H, m). In addition, a pair of vicinal coupled aromatic proton signals at
δ 6.85 (1H, d,
J = 8.4 Hz), 6.69 (1H, d,
J = 8.4 Hz) indicated the presence of one 1,2,3,4-tetrasubstituted phenyl ring. The
13C-NMR spectrum (
Table 4) exhibited one quaternary carbon signal attached to the O-atom at
δ 179.6; four other quaternary carbons in the benzene ring at
δ 156.2, 136.3, 128.9 and 125.2. Among these signals, the lower field carbon signal at
δ 156.2 indicated that this C-atom was connected to the O-atom. The
13C-NMR spectrum also gave two tertiary carbons in the benzene ring at
δ 130.7 and 116.2, one methylene carbon at
δ 33.8, three methenyl carbons at
δ 49.5, 85.1 and 31.3, four methyl carbons at
δ 21.9, 19.9, 20.8 and 13.0. From the above information it was possible to deduce the presence of dihydrofuran in the molecule. The HMBC spectrum showed correlations from H-15 to C-5 (
δ 136.3), C-9 (
δ 156.2) and C-10 (
δ 125.2), from H-14 to C-5 (
δ 136.3), C-6 (
δ 128.9) and C-7 (
δ 130.7), thus indicating that two methyl groups are placed at C-10 and C-6, respectively. The HMBC correlations from H-12 and H-13 to C-11 (
δ 31.3) and C-3 (
δ 49.5) indicated that the presence of an isopropyl at C-3; the correlation from
δ 5.72 (1H, d,
J = 9.0 Hz, H-4) to C-5 (
δ 136.3) suggested that the dihydrofuran was connected to C-5 in the benzene ring (
Figure 3). The relative configuration of H-3 and H-4 was determined as
trans by the 9.0 Hz coupling constant between H-3 and H-4. Irradiation of H-4 caused NOE enhancements of H-11, H-12 and H-13 in the 1D NOE spectra and indicated that isopropyl was positioned in the same plane with H-4. Thus, the NOE result further confirmed the
trans conformation of H-4 and H-3. Consequently, compound
5 was elucidated as 5-(3-hydroxy-2,6-dimethylphenyl)-4-isopropyldihydrofuran-2(3
H)-one, trivially named smiglactone.
Table 4.
1H-NMR (600 MHz) and 13C-NMR (150 MHz) spectra data of compound 5 (in CD3OD, J in Hz, δ in ppm).
Table 4.
1H-NMR (600 MHz) and 13C-NMR (150 MHz) spectra data of compound 5 (in CD3OD, J in Hz, δ in ppm).
| Position | δH (J in Hz) | δC |
|---|
| 1 | – | 179.6 |
| 2 | 2.65 (2H, m) | 33.8 |
| 3 | 2.75 (1H, m) | 49.5 |
| 4 | 5.72 (1H, d,
J = 9.0) | 85.1 |
| 5 | – | 136.3 |
| 6 | – | 128.9 |
| 7 | 6.85 (1H, d,
J = 8.4) | 130.7 |
| 8 | 6.69 (1H, d,
J = 8.4) | 116.2 |
| 9 | – | 156.2 |
| 10 | – | 125.2 |
| 11 | 1.76 (1H, m) | 31.3 |
| 12 | 0.76 (3H, d,
J = 6.6) | 21.9 |
| 13 | 0.98 (3H, d,
J = 6.6) | 19.9 |
| 14 | 2.30 (3H, s) | 20.8 |
| 15 | 2.22 (3H, s) | 13.0 |
Compound
6 (
Figure 1) was obtained as a pale yellow solid. Its molecular formula was determined as C
15H
16O
5 by negative-ion HRESIMS with quasi-molecular ion peaks at
m/z 311.06917 [M+Cl]
−, 338.08813 [M+NO
3]
− and 587.16893 [2M+Cl]− (calcd. for C
15H
16O
5Cl 311.06921, C
15H
16O
5NO
3 338.08835 and C
30H
32O
10Cl 587.16829, respectively). The IR spectrum of
6 showed a characteristic hydroxyl absorption band (3,336 cm
−1). UV absorptions at 206, 225 and 278 nm was observed. The
1H-NMR spectrum (
Table 5) showed proton signals at
δ 7.30 (2H, d,
J = 8.4 Hz) and 7.06 (2H, d,
J = 8.4 Hz), revealing the presence of a 1,4-disubstituted aromatic ring, and the signals at
δ 6.88 (1H, t,
J = 2.4 Hz) and 6.97 (2H, d,
J = 2.4 Hz) indicating the presence of a 1,3,5-trisubstituted aromatic ring. The
1H-NMR spectrum of
6 also indicated the presence of four hydroxyl groups at
δ 11.24 (2H, br. s), 11.38 (1H, br. s) and 3.59 (1H, br. s), and one methoxy group at
δ 3.28 (3H, s). The
13C-NMR spectrum (
Table 5) exhibited, in total, eleven carbon resonances involving eight aromatic carbons, a methoxy carbon, and two oxygenated carbons. In the HMBC spectrum, the correlations from methoxy proton at
δ 3.28 (3H, s) to C-8 (
δ 89.5), and from H-8 to methoxy carbon at
δ 56.8 suggested that the methoxy group was connected with C-8. The correlations from H-8 to C-10, 14 (
δ 129.9), H-14 to C-10 (
δ 129.9) indicated that C-8, C-10 and C-14 were linked through C-9; the correlations from H-7 to C-4, 6 (
δ 107.1), H-6 to C-7 (
δ 78.7) and H-4 to C-7 (
δ 78.7) suggested that C-7, C-4 and C-6 were linked through C-5 (
Figure 3). The large coupling constant between H-7 and H-8 (
J = 7.8 Hz) suggested a
threo conformation of C-7/C-8 [
16,
17]. Thus, the structure of compound
6 was determined as
threo-5-[1-hydroxy-2-(4-hydroxyphenyl)-2-methoxyethyl] benzene-1,3-diol, trivially named smiglabrol.
Table 5.
1H-NMR (600 MHz) and 13C-NMR (150 MHz) spectra data of compound 6 (in C5D5N, J in Hz, δ in ppm).
Table 5.
1H-NMR (600 MHz) and 13C-NMR (150 MHz) spectra data of compound 6 (in C5D5N, J in Hz, δ in ppm).
| Position | δH (J in Hz) | δC |
|---|
| 1, 3 | – | 159.7 |
| 2 | 6.88 (1H, t,
J = 2.4) | 102.9 |
| 4, 6 | 6.97 (2H, d,
J = 2.4) | 107.1 |
| 5 | – | 145.4 |
| 7 | 5.13 (1H, d,
J = 7.8) | 78.7 |
| 8 | 4.56 (1H, d,
J = 7.8) | 89.5 |
| 9 | – | 130.0 |
| 10, 14 | 7.30 (2H, d,
J = 8.4) | 129.9 |
| 11, 13 | 7.06 (2H, d,
J = 8.4) | 115.7 |
| 12 | – | 158.4 |
| 1, 3-OH | 11.24 (2H, br. s) | – |
| 7-OH | 3.59 (1H, br. s) | – |
| 8-OCH3 | 3.28 (3H, s) | 56.7 |
| 12-OH | 11.38 (1H, br. s) | – |
2.2. Structural Elucidation of the Known Isolates
Compound
43 (
Figure 2) was isolated as a pale yellow powder. Its molecular formula was determined as C
21H
20O
7 from the negative-ion HRESIMS with a quasi-molecular ion peak at
m/z 383.1133 [M−H]
− (calcd. for C
21H
19O
7 383.1131) and the positive negative-ion HRESIMS at
m/z 407.1103 [M+Na]
+ (calcd. for C
21H
20O
7Na 407.1107). The IR spectrum of
43 showed characteristic absorption bands for hydroxyl (3,448 cm
−1) and carbonyl (1,686 cm
−1) groups. UV absorptions at 210, 228 and 310 nm were observed. The
1H-NMR spectrum exhibited signals of two
trans-double bonds at
δ 7.57 (1H, d,
J = 16.2 Hz), 6.39 (1H, d,
J = 16.2 Hz), 7.540 (1H, d,
J = 15.6 Hz) and 6.38 (1H, d,
J = 15.6 Hz). Eight aromatic protons at
δ 7.541 (2H, d,
J = 8.4 Hz), 6.77 (2H, d,
J = 8.4 Hz), 7.53 (2H, d,
J = 8.4Hz) and 6.76 (2H, d,
J = 8.4 Hz), revealing the presence of two 1,4-disubstituted aromatic rings. Additionally, an acylated methine proton at
δ 5.11 (1H, m), acylated methylene protons at
δ 4.40 (1H, dd,
J = 11.4, 3.0 Hz) and 4.25 (1H, dd,
J = 12.0, 6.6 Hz), and hydroxymethyl protons at 3.60 (2H, t) suggested the presence of a 1,2-diacylglycerol moiety. The
13C-NMR spectrum showed two ester carbonyl carbon signals at
δ 166.41 and 166.19, four olefinic carbons at
δ 114.03, 145.21, 113.68 and 145.16, one oxymethine carbon at
δ 72.13, and one oxymethylene carbon at
δ 62.54. The HMBC correlation of H-1, H-6 with
δ 166.19 (C-4), H-3′ with
δ 166.41 (C-1′) further confirmed the presence of two non-equivalent
p-coumarate moieties. The HMBC spectrum showed correlations from H-1, H-3 to
δ 72.13 (C-2), H-1 to
δ 59.67 (C-3), indicating that the hydroxymethyl and acylated methylene were linked through C-2. Thus, the planar structure of
43 was determined as 1,2-
O-di-
trans-
p-coumaroylglycerol, that was reported as being isolated from stromata of
Epichloe typhina on
Phleum pretense [
18]. To determine the absolute configuration of
43, the exciton-coupled circular dichroism (ECCD) technique was applied [
19,
20]. The CD of
43 (
Figure 4) exhibited a positive split between the two chromophores of the
p-coumarate coupled with π→π
* transition (287 nm, Δε -11.12; 326 nm, Δε +14.09), indicating that the transition dipole moments of the two chromophores were oriented in a clockwise manner. This positive CD shows that the electric transition dipole of the
p-coumarate chromophores constitute positive chirality. Thus the absolute configuration of the chiral center in
43 was deduced as 2
S. Accordingly, compound
43 was elucidated as (2
E, 2'
E)-[(
S)-3-hydroxypropane-1,2-diyl] bis[3-(4-hydroxyphenyl)acrylate], and the compound was trivially named smiglycerol.
Figure 4.
CD and UV spectra of compound 43.
Figure 4.
CD and UV spectra of compound 43.
Based on their 1D and 2D NMR, CD and MS spectroscopic data and comparison of the data with those reported in the literature, fifty-seven known compounds were identified. These included thirteen flavanones: taxifolin (
7) [
7], naringenin (
8), dihydrokaempferol (
9) [
21], sakuranetin (
10) [
22], isoastilbin (
11) [
23], astilbin (
12) [
23], neoastilbin (
13) [
23], neoisoastilbin (
14) [
23], engeletin (
15) [
21], arthromerin B (
16) [
23,
24], sinensin (
17) [
23,
25], (2
R, 3
R)-taxifolin 3′-
O-
β-D-glucopyranoside (
18), (2
S,3
S)-glucodistylin (
19) [
26], four flavanes: (-)-epicatechin (
20) [
13], (+)-catechin (
21) [
12], cinchonain Ib (
22) [
13], cinchonain Ia (
23) [
13], four flavonoids: apigenin (
24), quercetin (
25), luteolin (
26), myricetin (
27), one chalcone, kukulkanin B (
28) [
27], two aurones: 4,4′,6-trihydroxyaurone (
29) [
28], aureusidin (
30) [
29], six lignans: (-)-secoisolariciresinol (
31) [
30], 4-ketopinoresinol (
32) [
31], 1,4-bis(4-hydroxy-3,5-dimethoxyphenyl)-2,3-bis(hydroxymethyl)-1,4-butanediol (named smiglabranol by us for being utilized conveniently in future) (
33) [
32], (+)-lyoniresinol (
34) [
33], kompasinol A (
35) [
34], aiphanol (
36) [
35], three stilbenes:
trans-resveratrol (
37) [
36],
trans-piceid (
38) [
36], piceatannol (
39) [
36], six phenylpropanoids:
trans-caffeic acid (
40), 5-
O-caffeoylshikimic acid (
41) [
21], 3-
O-
p-coumaroylshikimic acid (
42) [
37], (2
S)-1,2-
O-di-
trans-
p-coumaroylglycerol (named smiglycerol) (
43) [
18], juncusyl ester B (
44) [
38], 1-
O-
p-coumaroylglycerol (
45) [
39], nine phenolics: vanillin (
46),
p-hydroxy-benzaldehyde (
47), acetovanillone (
48), (+)-scytalone (
49) [
40], glucosyringic acid (
50), protocatechuic acid (
51), 3-methoxygallic acid (
52), vanillic acid 1-
O-
β-D-glucopyranosyl ester (
53), hydroxytyrosol (
54), one triterpene, acetyl-11-keto-
β-boswellic acid (
55), three steroids: stigmasterol (
56),
β-sitosterol (
57), daucosterol (
58), and five other compounds: smilagenin (
59), 5-hydroxymaltol (
60), 5-hydroxyuridine (
61), 2-methylbutanedioic acid-4-ethyl ester (
62), isoselachoceric acid (
63) (see
Figure 2). Isolation of twenty-seven compounds (
10,
17,
26–
36,
42,
43,
45,
46,
48–
50,
52–
55,
60–
62) from the genus
Smilax is reported here for the first time and fourteen compounds (
9,
15,
16,
19,
21–
24,
39,
40,
44,
47,
51,
63) were obtained from the rhizomes of
S. glabra for the first time.
During our investigations, four phenylpropanoid-substituted epicatechins (compounds
1,
2,
22,
23), which were a class of flavan-3-ols substituted at the A ring with a C
6-C
3 unit, were obtained. To our knowledge, these have never been previously reported in
S. glabra. Pharmacological studies revealed that compounds
22 and
23possessed antioxidant [
10], antifungal and antiviral [
11] activities. This is the first report of the presence in
Smilax of chalcone
28, which was previously isolated from
Mimosa tenuefolia [
27]. Compounds with this skeleton were found to have antioxidant [
41] and antitumor [
42] activities. The two aurones, namely compounds
29 and
30, were found to possess antiviral [
28] and antioxidant [
29] activities, respectively. Their isolation from the genus
Smilax is reported here for the first time. Compound
36 represents a stilbenolignan skeleton in which a stilbene moiety is linked with a phenylpropane unit through a dioxane bridge. It was previously reported from
Aiphanes aculeate and exhibited significant inhibitory activities against cyclooxygenases-1 and -2 [
35]. Compound
35 is also a stilbenolignan, which was isolated from
Koompassia malaccensis [
34] and
Syagrus romanzoffiana and had anti-
α-glucosidase activity [
43]. This is first report of the presence of compounds with this type of skeleton in
Smilax plants.
On the basis of previous phytochemical studies and our investigations, flavonoids are the main constituents in the rhizomes of
S. glabra, and especially flavanones were relatively more abundant than other kinds of flavonoids. It was reported that the contents of the five flavonoids were in the range of 0.0290–1.06, 0.0128–0.0543, 1.53–11.3, 0.449–13.7 and 0.552–4.837 mg/g crude drugs for taxifolin, neoastilbin, astilbin, neoisoastilbin and isoastilbin, respectively [
8]. In addition, quantitative analysis showed that the content of engeletin was in the range of 0.14–3.1 mg/g crude drug [
44]. Previous phytochemical investigations showed that fourteen flavonoids were isolated the rhizomes of
S. glabra, which included ten flavanones: taxifolin, astilbin, neoastilbin, isoastilbin, neoisoastilbin, (2
R,3
R)-taxifolin-3′-
O-
β-D-glucopyranoside, isoengelitin, naringenin, engeletin, smitilbin [
45,
46,
47], two flavonoids: quercetin, quercetin 4′-
O-
β-D-glucoside [
48], one isoflavone, 7,6′-dihydroxy-3'-methoxy-isoflavone [
46], and one flavane, (-)-epicatechin [
49].
2.3. Antimicrobial Activity
The MIC obtained in the antimicrobial assessment of S. glabra extracts demonstrated considerable activity in the ethanolic extract, ethyl acetate fraction and n-butanol fraction against S. aureus ATCC6538 (50 μg/mL). The ethyl acetate fraction and water fraction showed activity against C. albicans SC5314 and S. aureus ATCC6538 with MIC values of 200 μg/mL. This is the first report on the antifungal properties of S. glabra.
Thirty compounds were tested for their antimicrobial activity (
Table 6). The results demonstrated that seventeen of these compounds were found to have antibacterial activity against Gram-positive bacteria and ten compounds displayed activity against the tested fungus, while eight compounds were found to be antibacterial against Gram-negative bacteria. As for the four flavan-3-ols (
Figure S1, Table S1),
1,
20 and
22 demonstrated antimicrobial properties, whereas
21 was inactive in the concentration range tested. Compound
1 exhibited activity against all tested microorganisms, which was stronger against
C. albicans SC5314 with a MIC value of 0.146 mM and weaker against the Gram-positive and Gram-negative bacteria where the MIC values were in the range of 0.302-0.604 mM. Compound
22 showed activity against methicillin-resistant
S. aureus and
S. aureus ATCC6538 with a MIC value of 0.0801 mM and activity against
C. albicans SC5314 and
E. faecalis with MIC valued of 0.160 mM. Compound
20 was only found to be active against methicillin-resistant
S. aureus,
S. aureus ATCC6538 and
E. faecalis. Compounds
1,
20 and
22 all possessed epicatechin units, while
21 was identified as catechin. The result suggested that the antimicrobial activity was influenced by the stereochemistry at C2 and C3.
A similar situation happens with respect to some of the investigated flavanones. Compound
7 was inactive and its flavanonol rhamnoside isomers (compounds
12,
13 and
14) showed antimicrobial activity except for
11 (
Figure S2, Table S2). Among them, compound
14, with 2
S,3
R configuration, presented the best activity and showed inhibitory effects toward all of the tested microorganisms. This indicates that the stereochemistry at C2 and C3 of taxifolin largely governs the potency of inhibition, and controls the steric configuration of the rhamnose moiety. Taxifolin (
7), astilbin (
12), neoastilbin (
13) and isoastilbin (
11) have been found to display antimicrobial activity against
Streptococcus sobrinus with MIC values in the 0.500–0.740 mM range. Among these compounds, neoastilbin showed an especially potent GTase inhibitory activity, which suggested that the antimicrobial activity was related to stereochemistry [
7]. To some extent, our results mirror the previous study [
7].
Compound
9 and its glycoside (compound
17) demonstrated activity against methicillin-resistant
S. aureus and
S. aureus ATCC6538, whereas
15 and
16 did not show inhibitory effects toward these microorganisms. This implied that for the C ring, hydroxylation at position 3 improved the activity of flavanones (
Figure S3, Table S3). Compound
10 was found to be antibacterial against methicillin-resistant
S. aureus,
S. aureus ATCC6538 and
E. faecalis with MIC values of 0.524, 0.524 and 1.05 mM. The homoisoflavanone
3 showed antibacterial activity against
S. aureus ATCC6538, with a MIC value of 0.303 mM, and weaker activity against
C. albicans SC5314, methicillin-resistant
S. aureus and
E. faecalis with a uniform MIC of 0.605 mM.
All the investigated stibenes (compounds
4,
37–
39) displayed antimicrobial activity against the microorganisms tested (
Figure S4, Figure S5, Table S4). Compound
37 showed antibacterial activity against methicillin-resistant
S. aureus,
S. aureus ATCC6538 and
E. faecalis with MIC values of 0.159, 0.0794 and 0.159 mM, respectively. It appeared that compound
38 which was the glycosylated version of resveratrol (
37) had much weaker activity than its parent, which was reported to have antibacterial activity against
Streptococcus mutans and
Streptococcus sanguis with MIC values of 0.219 and 0.110 mM, respectively, but piceid (
38) did not inhibit microbial growth [
50].The structures of compound
4 and
39 were quite similar, except for the position of the hydroxyl, while the activity of
4 was better than
39. They displayed inhibitory activity toward
S. aureus ATCC6538 and methicillin-resistant
S. aureus with MIC values of 0.205 and 0.409 mM, respectively. We infer that the hydroxyl position may influence antimicrobial effect, and the activity of 2,3-hydroxyl-substituted compounds was better than that of 3,4-hydroxyl-substituted compounds. The stibenolignan
36 showed antimicrobial activity against all the microorganisms tested. We suppose that the presence of a stilbene moiety in compound
36contributes to the antimicrobial effect.
Table 6.
Minimum inhibitory concentrations (MICs, mM) of the selected compounds obtained from the rhizomes of S. glabra.
Table 6.
Minimum inhibitory concentrations (MICs, mM) of the selected compounds obtained from the rhizomes of S. glabra.
| Compound | EC | PA | KP | MRSA | SA | EF | CA |
|---|
| 1 | 0.604 | 0.604 | 0.604 | 0.302 | 0.302 | 0.302 | 0.146 |
| 3 | >1.21 | >1.21 | >1.21 | 0.605 | 0.303 | 0.605 | 0.605 |
| 4 | 1.64 | 1.64 | 1.64 | 0.409 | 0.205 | 0.819 | 0.819 |
| 9 | >2.10 | >2.10 | >2.10 | 2.10 | 2.10 | >2.10 | >2.10 |
| 10 | >2.10 | >2.10 | >2.10 | 0.524 | 0.524 | 1.05 | >2.10 |
| 11 | >1.33 | >1.33 | >1.33 | >1.33 | >1.33 | >1.33 | >1.33 |
| 12 | >1.33 | >1.33 | >1.33 | >1.33 | >1.33 | >1.33 | 0.666 |
| 13 | >1.33 | >1.33 | >1.33 | 1.33 | >1.33 | >1.33 | >1.33 |
| 14 | 1.33 | 1.33 | 1.33 | 1.33 | 0.666 | 1.33 | 1.33 |
| 15 | >1.38 | >1.38 | >1.38 | >1.38 | >1.38 | >1.38 | >1.38 |
| 16 | >1.33 | >1.33 | >1.33 | >1.33 | >1.33 | >1.33 | >1.33 |
| 17 | >1.33 | >1.33 | >1.33 | 1.33 | 1.33 | 1.33 | >1.33 |
| 20 | >2.07 | >2.07 | >2.07 | 1.03 | 0.517 | 1.03 | >2.07 |
| 22 | >1.33 | 0.663 | >1.33 | 0.0801 | 0.0801 | 0.160 | 0.160 |
| 33 | >0.880 | >0.880 | >0.880 | >0.880 | >0.880 | >0.880 | >0.880 |
| 36 | 0.663 | 0.663 | 0.663 | 1.33 | 0.663 | 1.33 | 0.332 |
| 37 | >2.63 | >2.63 | >2.63 | 0.159 | 0.0794 | 0.159 | 0.657 |
| 38 | >1.54 | >1.54 | >1.54 | 0.768 | 0.768 | 1.54 | >1.54 |
| 39 | 1.64 | 1.64 | 1.64 | 0.409 | 0.205 | >1.64 | 0.819 |
| 44 | 2.52 | 2.52 | 2.52 | 0.630 | 0.630 | 1.26 | 0.630 |
| 49 | 3.09 | 3.09 | 3.09 | 3.09 | 1.55 | 3.09 | 3.09 |
| 51 | >2.60 | >2.60 | >2.60 | >2.60 | >2.60 | >2.60 | >2.60 |
| Cipro | 0.00302 | 0.00302 | 0.00604 | | | | |
| Van | | | | 0.000690 | 0.000690 | 0.00138 | |
| Keto | | | | | | | 0.0000301 |



{kind=link}
{kind=link}
{kind=link}
{kind=link}

 +178.72), indicating the R configuration at C-3 [15]. Consequently, the structure of compound 3 was identified as (3R)-3,5-dihydroxy-7-methoxy-6-methyl-3-(4-hydroxybenzyl)chroman-4-one, and trivially named smilachromanone.
+178.72), indicating the R configuration at C-3 [15]. Consequently, the structure of compound 3 was identified as (3R)-3,5-dihydroxy-7-methoxy-6-methyl-3-(4-hydroxybenzyl)chroman-4-one, and trivially named smilachromanone.