Low-Temperature Spark Plasma Sintering of ZrW2−xMoxO8 Exhibiting Controllable Negative Thermal Expansion

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
3. Results and Discussion
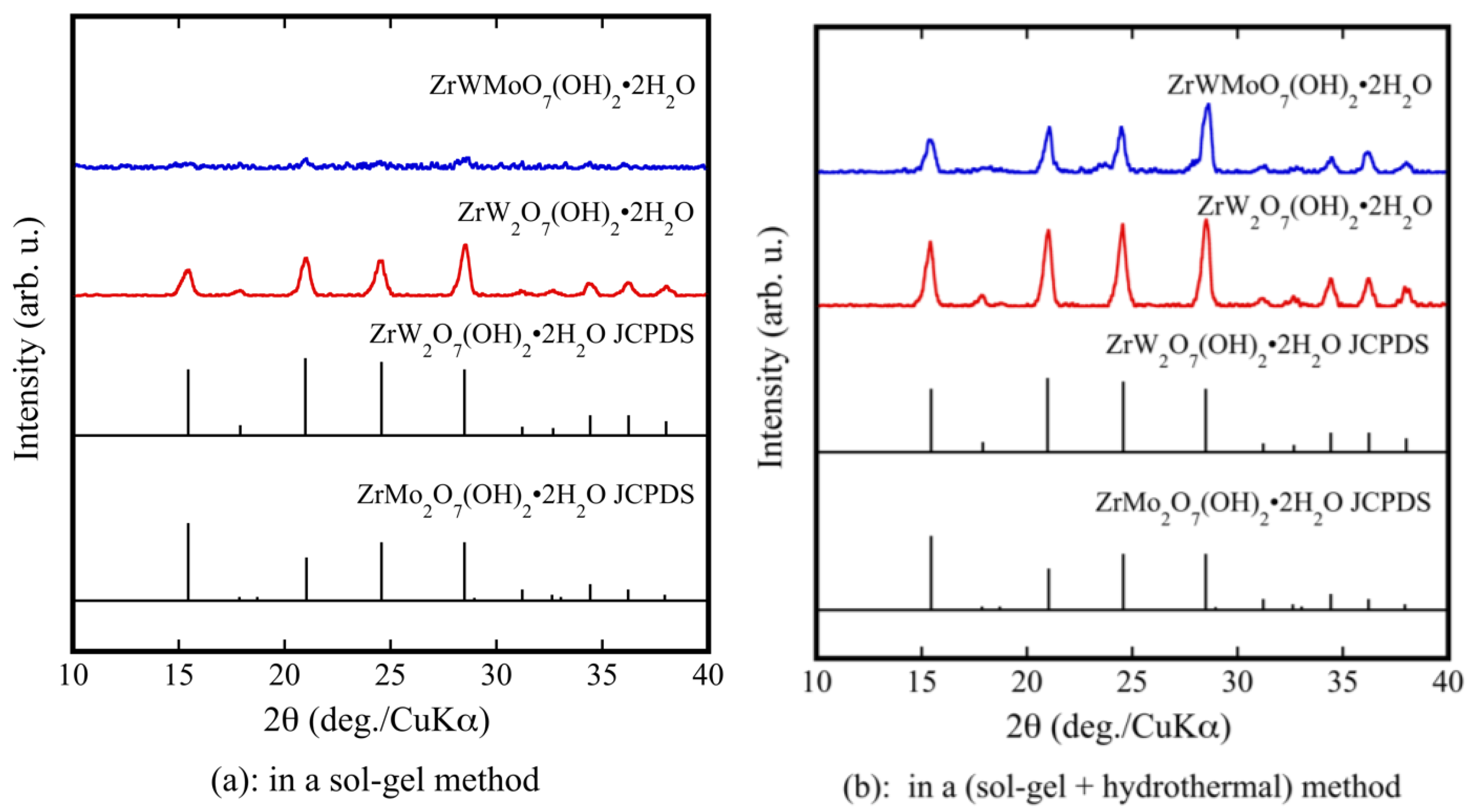
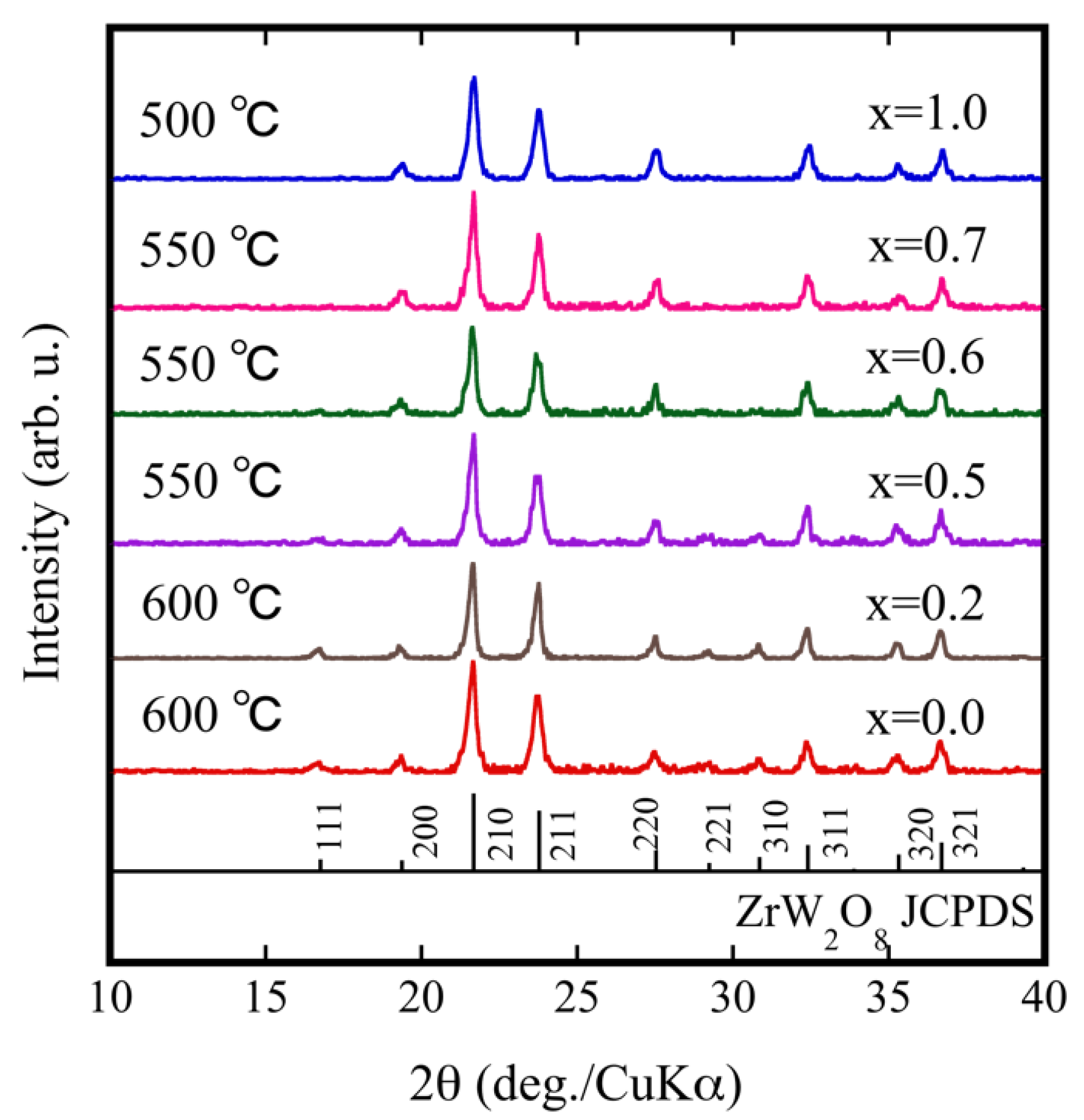
3.1. Fabrication of Dense ZrW2−xMoxO8 Sintered Bodies
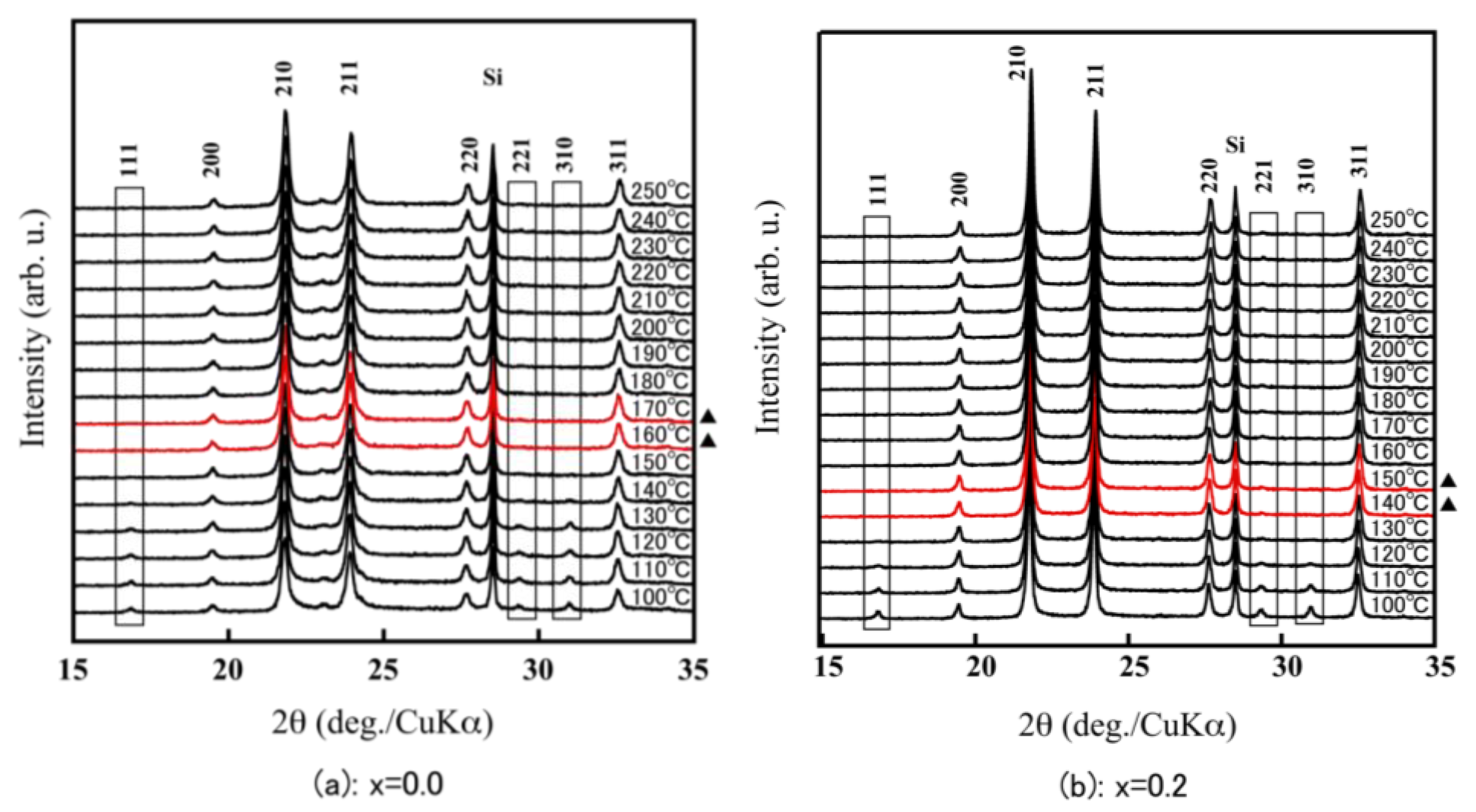
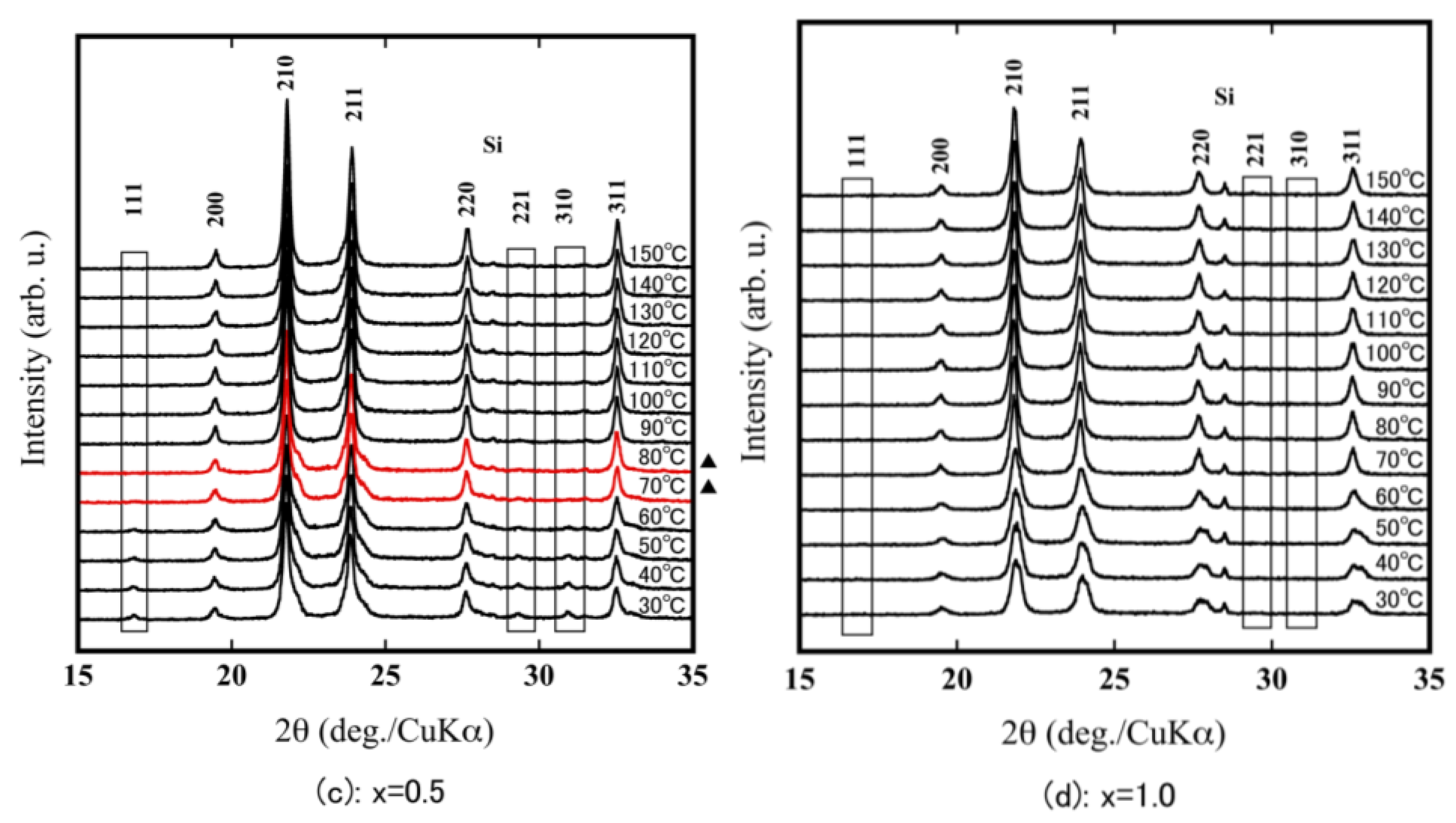
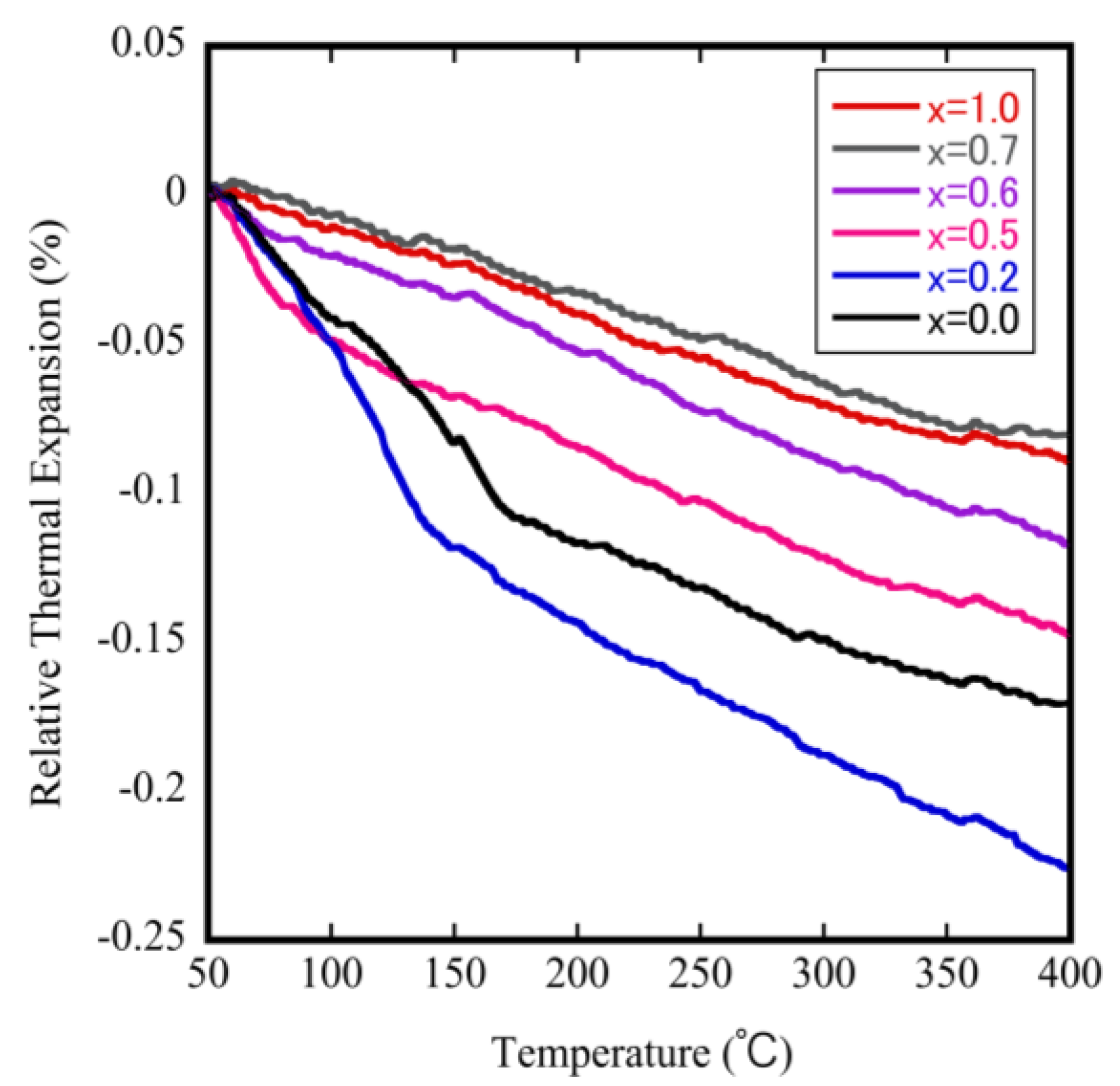
3.2. Evaluation of Thermal Expansion Properties of ZrW2−xMoxO8 Sintered Bodies
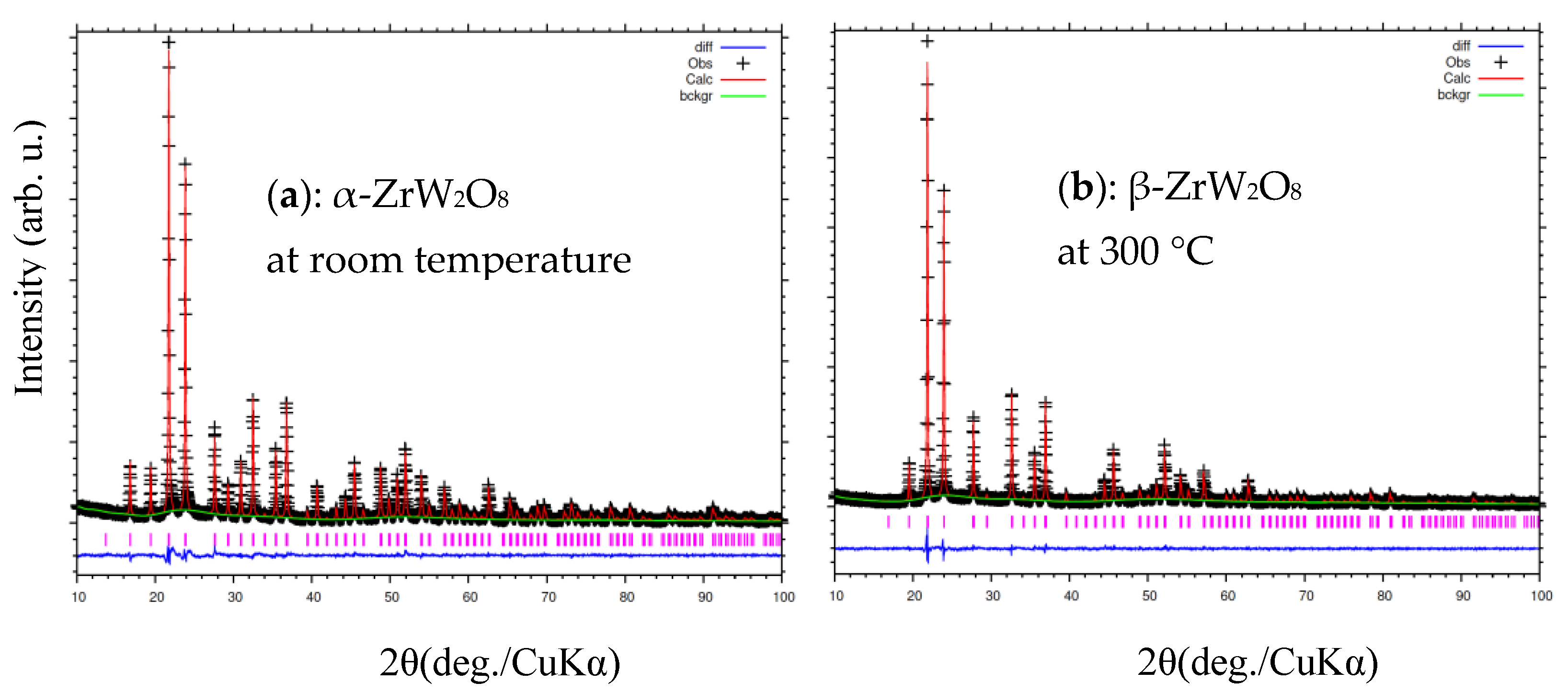
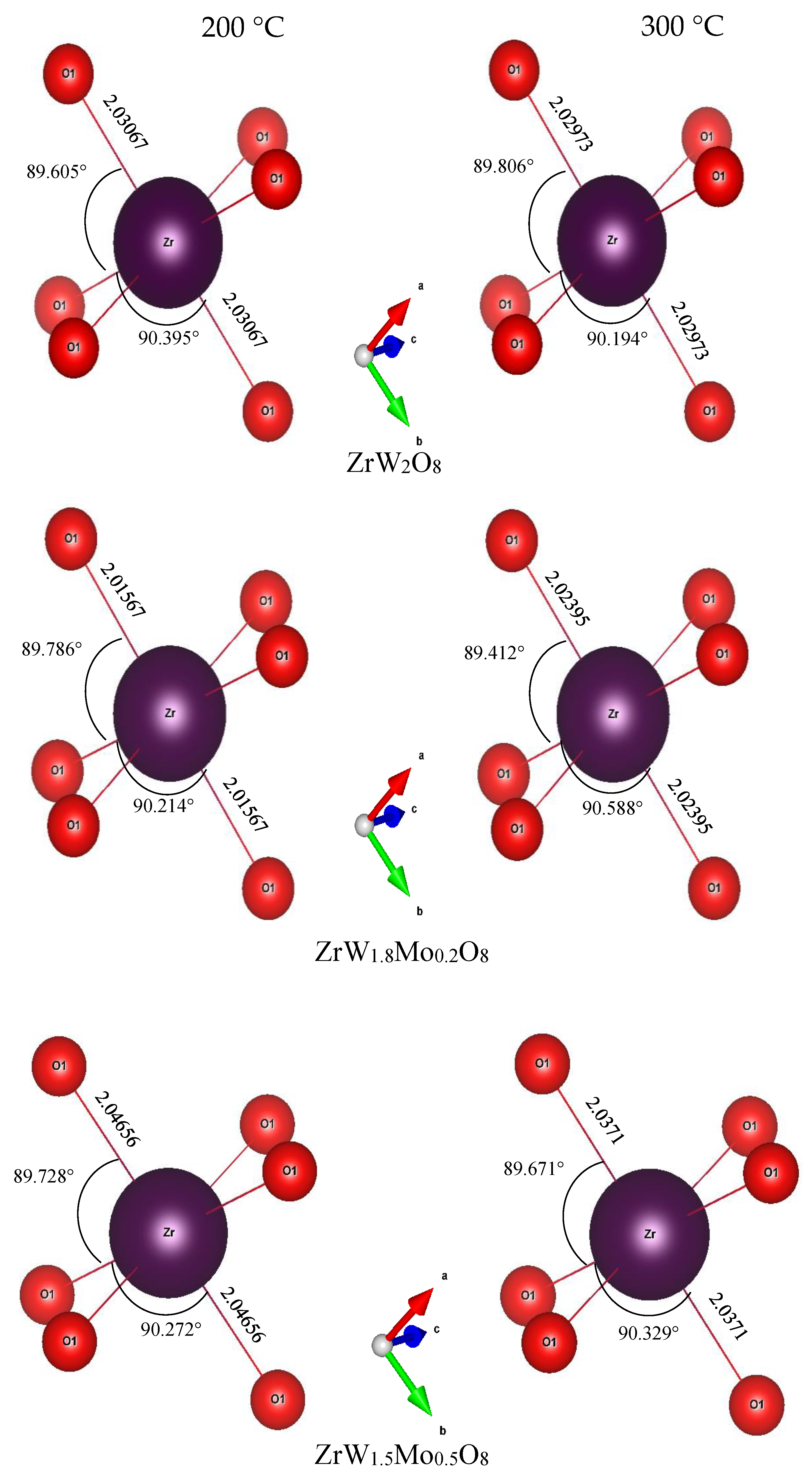
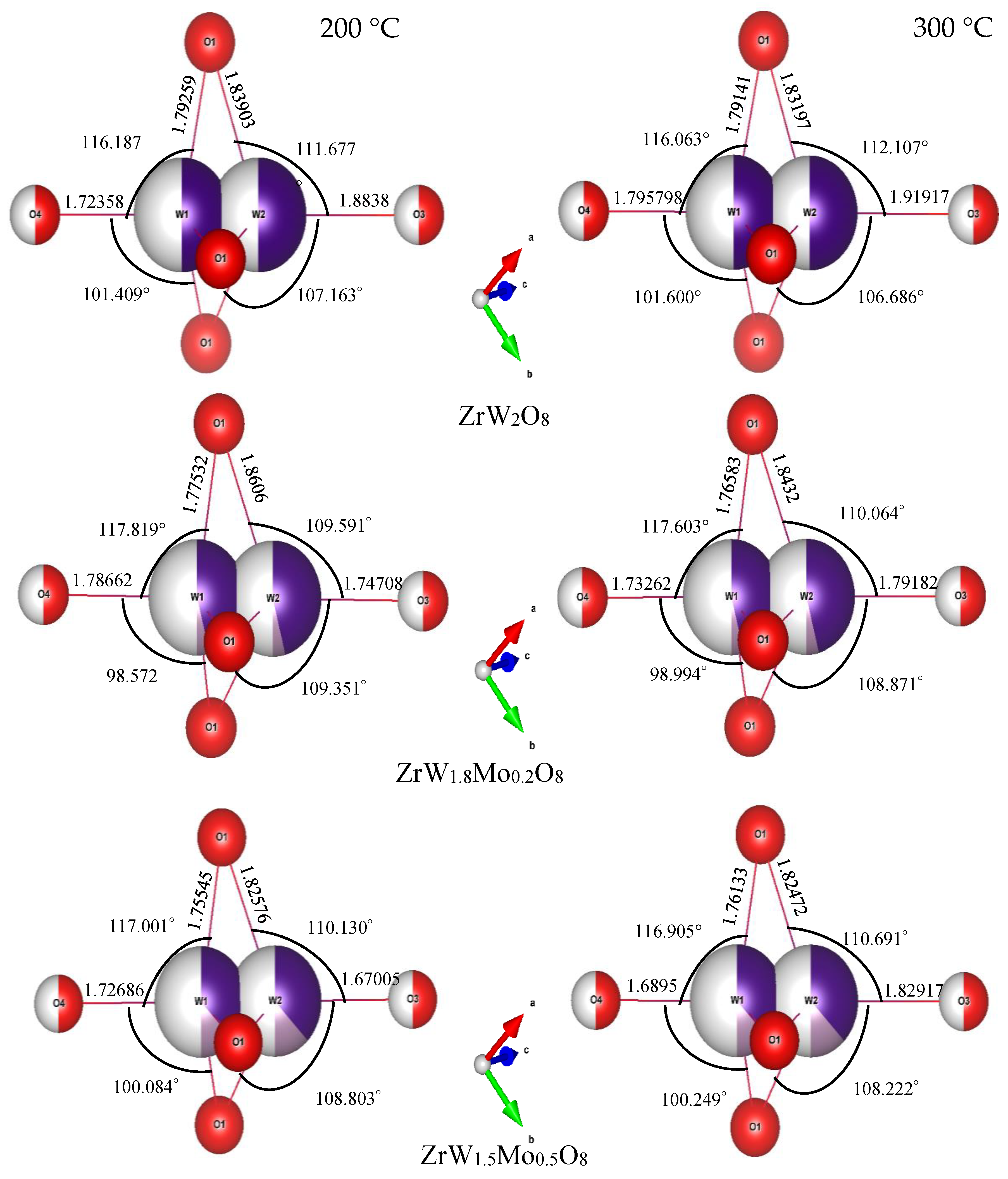
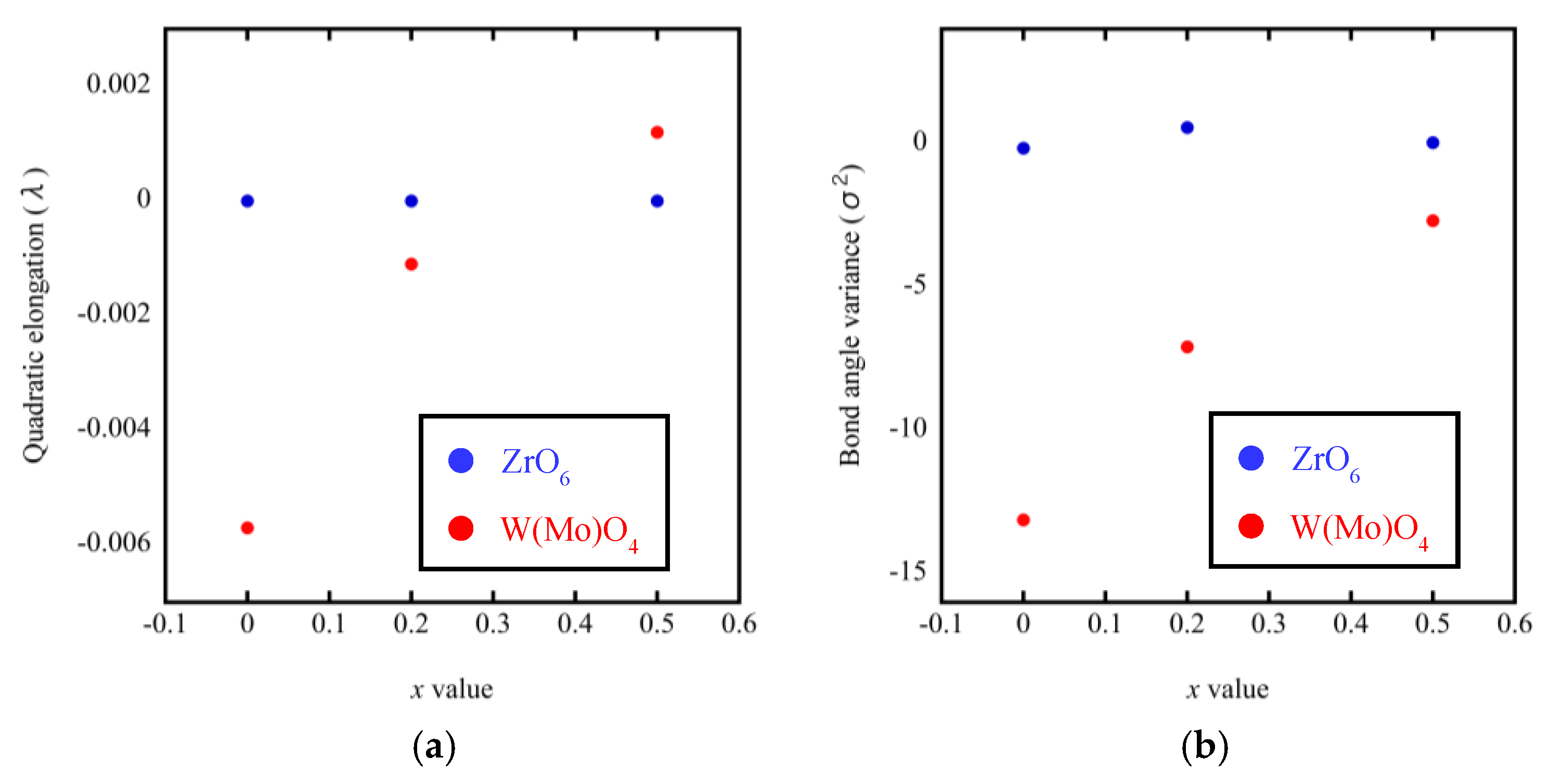
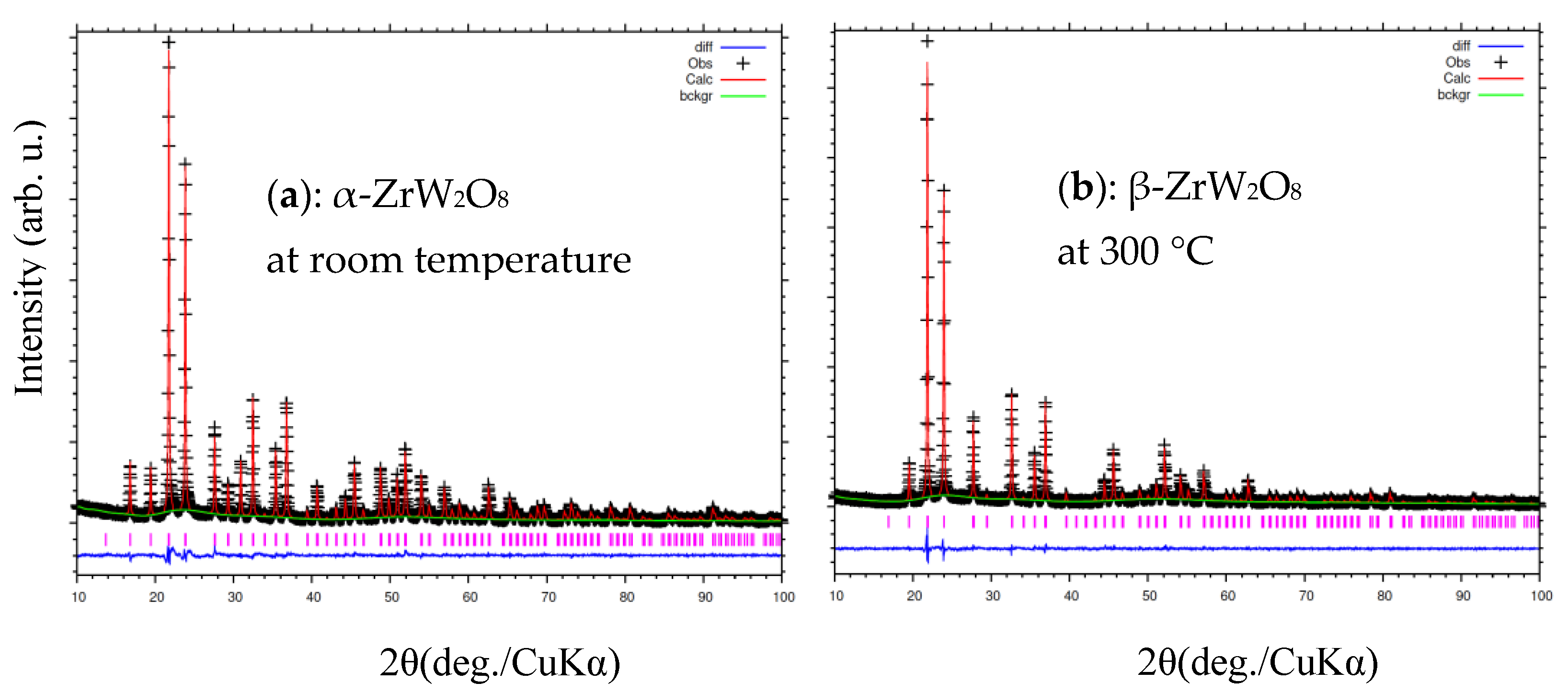
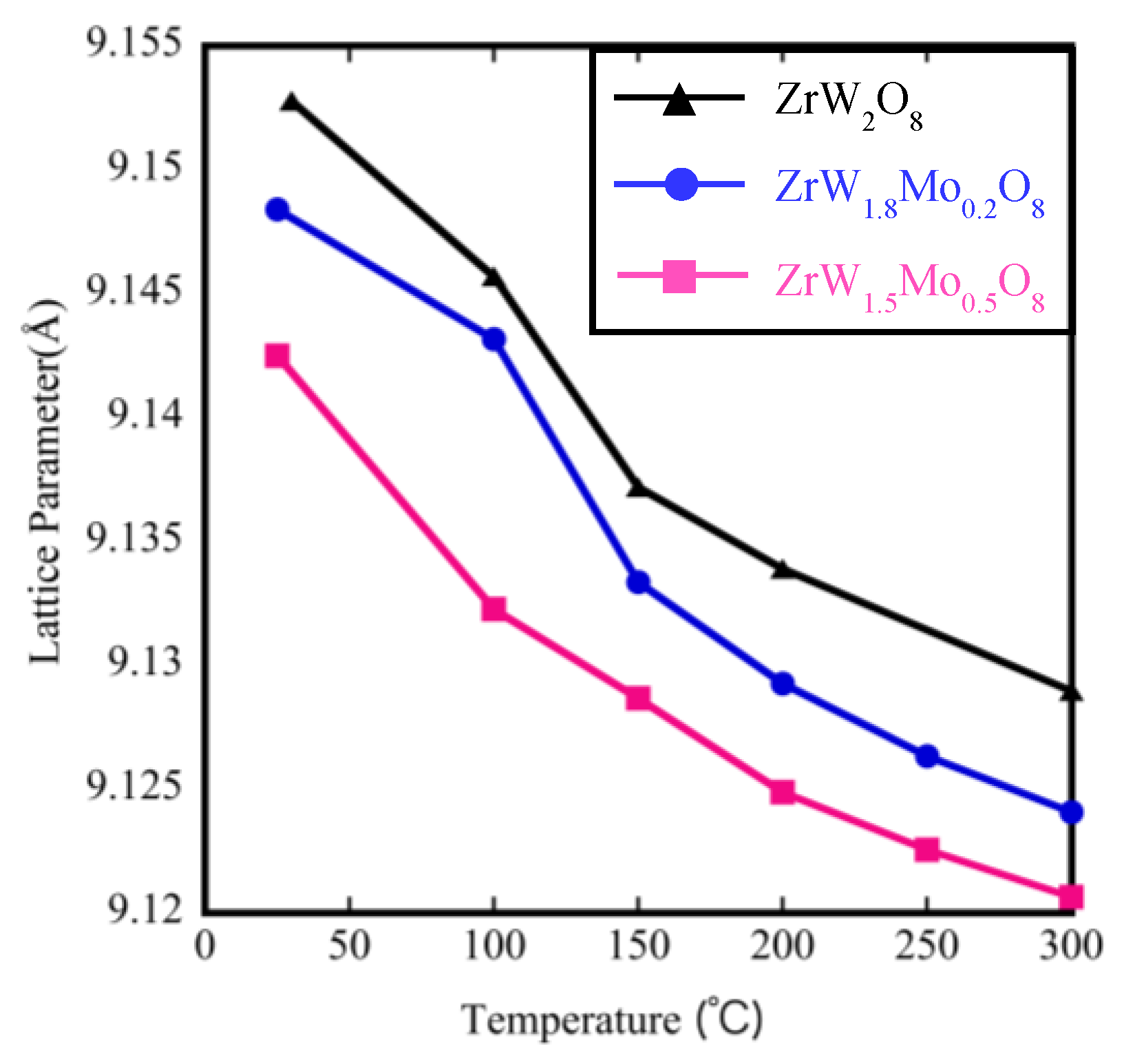
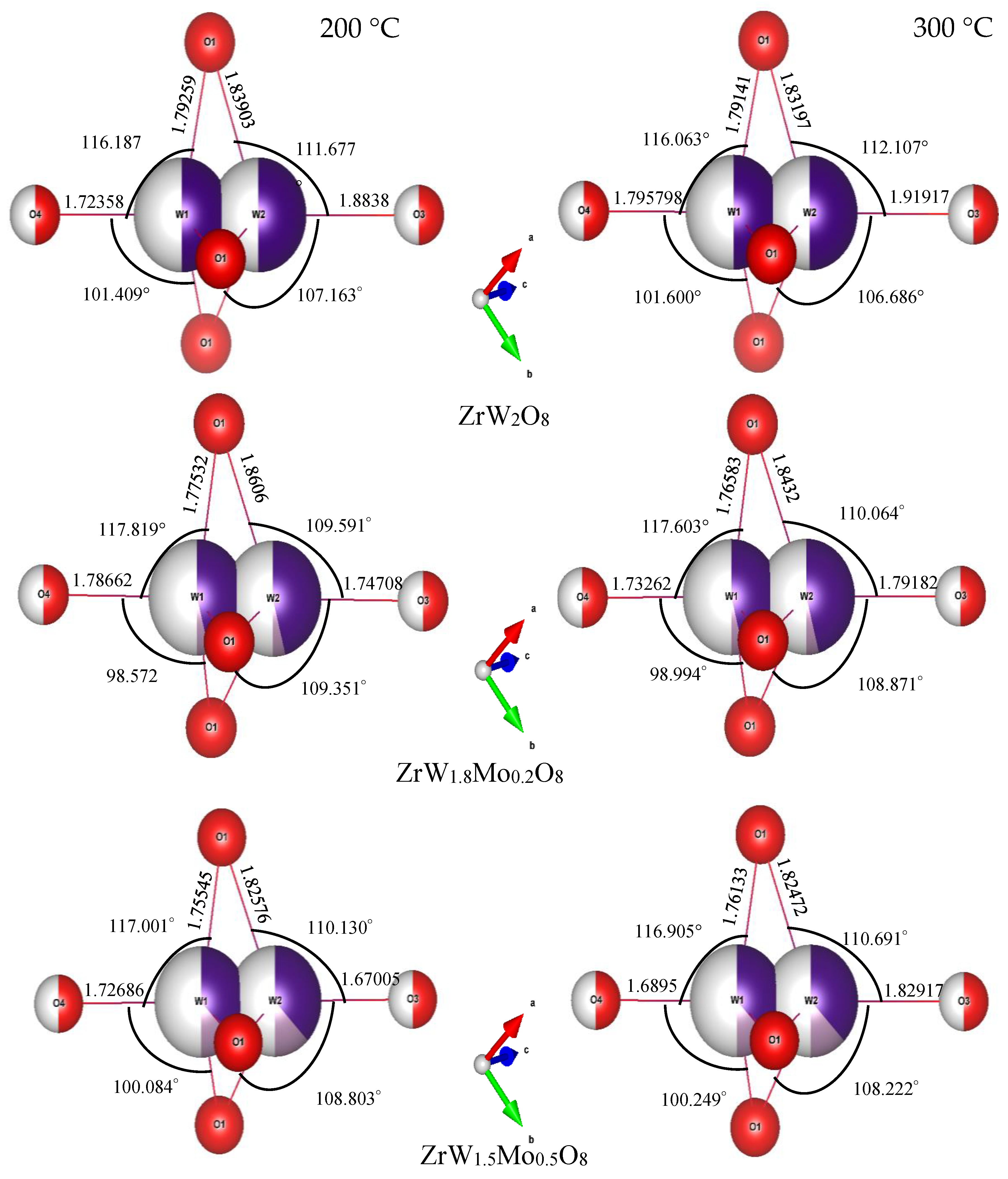
3.3. Rietveld Structure Refinement of ZrW2−xMoxO8 Compounds
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Ouyang, J.S.; Li, Y.B.; Chen, B.; Huang, D.H. Macro-Scale Strength and Microstructure of ZrW2O8 Cementitious Composites with Tunable Low Thermal Expansion. Materials 2018, 11, 748. [Google Scholar] [CrossRef] [PubMed]
- Holzer, H.; Dunand, D.C. Phase Transformation and Thermal Expansion of Cu/ZrW2O8 Metal Matrix Composites. J. Mater. Res. 1999, 14, 780–789. [Google Scholar] [CrossRef]
- Wu, Y.; Wang, M.L.; Chen, Z.; Ma, N.H.; Wang, H.W. The effect of phase transformation on the thermal expansion property in Al/ZrW2O8 composites. J. Mater. Sci. 2013, 48, 2928–2933. [Google Scholar] [CrossRef]
- Kanamori, K.; Kineri, T.; Fukuda, R.; Kawano, T.; Nishio, K. Low-temperature sintering of ZrW2O8–SiO2 by spark plasma sintering. J. Mater. Sci. 2009, 44, 855–860. [Google Scholar] [CrossRef]
- Wu, G.; Zhou, C.; Zhang, Q.; Pei, R. Decomposition of ZrW2O8 in Al matrix and the influence of heat treatment on ZrW2O8/Al–Si thermal expansion. Scr. Mater. 2015, 96, 29–32. [Google Scholar] [CrossRef]
- Yilmaz, S. Thermal mismatch stress development in Cu-ZrW2O8 composite investigated by synchrotron X-ray diffraction. Compos. Sci. Technol. 2002, 62, 1835–1839. [Google Scholar] [CrossRef]
- Mary, T.A.; Evans, J.S.O.; Vogt, T.; Sleight, A.W. Negative thermal expansion from 0.3 to 1050 Kelvin in ZrW2O8. Science 1996, 272, 90–92. [Google Scholar] [CrossRef]
- Evans, J.S.O.; Mary, T.A.; Vogt, T.; Subramanian, M.A.; Sleight, A.W. Negative Thermal Expansion in ZrW2O8 and HfW2O8. Chem. Mater. 1996, 8, 2809–2823. [Google Scholar] [CrossRef]
- Evans, J.S.O.; David, W.I.F.; Sleight, A.W. Structural investigation of the negative-thermal-expansion material ZrW2O8. Acta Crystallogr. B 1999, 55, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Meyer, C.D.; Bouree, F.; Evans, J.S.O.; Buysser, K.D.; Bruneel, E.; Driessche, I.V.; Hoste, S. Structure and phase transition of Sn-substituted Zr(1−x)SnxW2O8. J. Mater. Chem. 2004, 14, 2988–2994. [Google Scholar] [CrossRef]
- Evans, J.S.O.; Hanson, P.A.; Ibberson, R.M.; Kameswari, U.; Duan, N.; Sleight, A.W. Low-Temperature Oxygen Migration and Negative Thermal Expansion in ZrW2−xMoxO8. J. Am. Chem. Soc. 2000, 122, 8694–8699. [Google Scholar] [CrossRef]
- Korthuis, V.; Khosrovani, N.; Sleight, A.W.; Roberts, N.; Dupree, R.; Warren, W.W. Negative Thermal Expansion and Phase Transitions in the ZrV2−xPxO7 Series. Chem. Mater. 1995, 7, 412–417. [Google Scholar] [CrossRef]
- Khosrovani, N.; Korthuis, V.; Sleight, A.W.; Vogt, T. Unusual 180° P−O−P Bond Angles in ZrP2O7. Inorg. Chem. 1996, 35, 485–489. [Google Scholar] [CrossRef] [PubMed]
- Evans, J.S.O.; Hanson, J.C.; Sleight, A.W. Room-Temperature Superstructure of ZrV2O7. Acta. Cryst. 1998, B54, 705–713. [Google Scholar] [CrossRef]
- Turquat, C.; Muller, C.; Nigrelli, E.; Leroux, C.; Soubeyroux, J.L.; Nihoul, G. Structural investigation of temperature-induced phase transitions in HfV2O7. Eur. Phys. J. Appl. Phys. 2000, 10, 15–27. [Google Scholar] [CrossRef]
- Huang, C.H.; Knop, O.; Othen, D.A.; Woodhams, F.W.D.; Howie, R.A. Pyrophosphates of Tetravalent Elements and a Mössbauer Study of SnP2O7. Can. J. Chem. 1975, 53, 79–91. [Google Scholar] [CrossRef]
- Evans, J.S.O.; Mary, T.A. Structural phase transitions and negative thermal expansion in Sc2(MoO4)3. Int. J. Inorg. Mater. 2000, 2, 143–151. [Google Scholar] [CrossRef]
- Abrahams, S.C.; Bernstein, J.L. Crystal Structure of the Transition-Metal Molybdates and Tungstates. II. Diamagnetic SC2(WO4)3. J. Chem. Phys. 1966, 45, 2745–2752. [Google Scholar] [CrossRef]
- Gates, S.D.; Colin, J.A.; Lind, C. Non-hydrolytic sol-gel synthesis, properties, and high-pressure behavior of gallium molybdate. J. Mater. Chem. 2006, 16, 4214–4219. [Google Scholar] [CrossRef]
- Allen, S.; Evans, J.S.O. Negative thermal expansion and oxygen disorder in cubic ZrMo2O8. Phys. Rev. B 2003, 68, 134101–134103. [Google Scholar] [CrossRef]
- Ahmad, M.I.; Lindley, K.; Akinc, M. Hydrothermal Synthesis of ZrW2−δMoδO8 (δ = 0–0.91) and its α→β Transformation. J. Am. Ceram. Soc. 2011, 94, 2619–2624. [Google Scholar] [CrossRef]
- Zhao, R.Q.; Yang, X.J.; Wang, H.L.; Han, J.S.; Ma, H.; Zhao, X. A novel route to synthesize cubic ZrW2−xMoxO8 (x = 0–1.3) solid solutions and their negative thermal expansion properties. J. Solid State Chem. 2007, 180, 3160–3165. [Google Scholar] [CrossRef]
- Huang, L.; Xiao, Q.G.; Ma, H.; Li, G.B.; Liao, F.H.; Qi, C.M.; Zhao, X.H. Phase Behaviors of the ZrMo2–xWxO8 (x = 0.2–2.0) System and the Preparation of an Mo-Rich Cubic Phase. Eur. J. Inorg. Chem. 2005, 2005, 4521–4526. [Google Scholar] [CrossRef]
- Kanamori, K.; Kineri, T.; Fukuda, R.; Nishio, K.; Hashimoto, M.; Mae, H. Spark Plasma Sintering of Sol-Gel Derived Amorphous ZrW2O8 Nanopowder. J. Am. Ceram. Soc. 2009, 92, 32–35. [Google Scholar] [CrossRef]
- Larson, A.C.; Von Dreele, R.B.; Alamos, L. General Structural Analysis System (GSAS); Los Alamos National Laboratory Report LAUR; Los Alamos National Laboratory: Los Alamos, NM, USA, 2000. [Google Scholar]
- Momma, K.; Izumi, F. VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data. J. Appl. Crystallogr. 2011, 44, 1272–1276. [Google Scholar] [CrossRef]
- Robinson, K.; Gibbs, G.V.; Ribbe, P.H. Quadratic elongation: A quantitative measure of distortion in coordination polyhedra. Science 1971, 172, 567–570. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | x = 0.0 | x = 0.2 | x = 0.5 | x = 0.6 | x = 0.7 | x = 1.0 |
|---|---|---|---|---|---|---|
| Sintering temperature (°C) | 600 | 600 | 550 | 550 | 550 | 500 |
| Density (g/cm3) | 4.6271 | 4.6832 | 4.4217 | 4.4855 | 4.2760 | 4.1689 |
| Theoretical density (g/cm3) | 5.08 | 4.93 | 4.71 | 4.63 | 4.56 | 4.33 |
| Relative density (%) | 91.0846 | 94.9939 | 93.8790 | 96.8790 | 93.7719 | 96.2794 |
| Coefficient of Thermal Expansion (×10–6 °C−1) | Phase Transition Temperature (°C) | ||
|---|---|---|---|
| Compound | α Phase | β Phase | |
| x = 0.0 | −7.85 (50–130 °C) | −3.22 (200–400 °C) | 170 |
| x = 0.2 | −11.79 (50–130 °C) | −4.17 (200–400 °C) | 145 |
| x = 0.5 | −15.34 (50–70 °C) | −3.16 (200–400 °C) | 78 |
| x = 0.6 | −9.01 (50–70 °C) | −3.21 (200–400 °C) | 60 |
| x = 0.7 | - | −2.91 (200–400 °C) | - |
| x = 1.0 | - | −2.50 (200–400 °C) | - |
| Temperature | Room Temperature | 300 °C | ||||
|---|---|---|---|---|---|---|
| Compound | ZrW2O8 | ZrW1.8Mo0.2O8 | ZrW1.5Mo0.5O8 | ZrW2O8 | ZrW1.8Mo0.2O8 | ZrW1.5Mo0.5O8 |
| Goodness-of-fit (x2) | 1.776 | 6.993 | 3.983 | 1.375 | 2.928 | 3.194 |
| Space Group | P 21 3 | P 21 3 | P 21 3 | P a | P a | P a |
| Atom | Zr | Zr | Zr | Zr | Zr | Zr |
| x = y = z | 0.000319 | 0.004534 | 0.002891 | 0.000000 | 0.000000 | 0.000000 |
| Occupancy | 1 | 1 | 1 | 1 | 1 | 1 |
| Atom | W1 | W(Mo)1 | W(Mo)1 | W1 | W(Mo)1 | W(Mo)1 |
| x = y = z | 0.342614 | 0.343947 | 0.351651 | 0.340273 | 0.339711 | 0.338886 |
| Occupancy | 1 | W1:0.9, Mo1:0.1 | W1:0.75, Mo1:0.25 | 0.5 | W1:0.45, Mo1:0.05 | W1:0.375, Mo1:0.125 |
| Atom | W2 | W(Mo)2 | W(Mo)2 | W2 | W(Mo)2 | W(Mo)2 |
| x = y = z | 0.601688 | 0.603332 | 0.608726 | 0.603304 | 0.605205 | 0.606212 |
| Occupancy | 1 | W2:0.9, Mo2:0.1 | W2:0.75, Mo2:0.25 | 0.5 | W2:0.45, Mo2:0.05 | W2:0.375, Mo2:0.125 |
| Atom | O1 | O1 | O1 | O1 | O1 | O1 |
| x | 0.042345 | 0.021103 | 0.008577 | 0.050445 | 0.055887 | 0.05519 |
| y | −0.19759 | −0.179137 | −0.183323 | −0.205553 | −0.201601 | −0.202851 |
| z | −0.058376 | −0.066893 | −0.057469 | −0.069239 | −0.073105 | −0.0735229 |
| Occupancy | 1 | 1 | 1 | 1 | 1 | 1 |
| Atom | O2 | O2 | O2 | - | - | - |
| x | 0.078837 | 0.094246 | 0.121337 | - | - | - |
| y | −0.057928 | −0.071485 | −0.094012 | - | - | - |
| z | 0.211437 | 0.221976 | 0.205825 | - | - | - |
| Occupancy | 1 | 1 | 1 | - | - | - |
| Atom | O3 | O3 | O3 | O3 | O3 | O3 |
| x = y = z | 0.486051 | 0.479937 | 0.469176 | 0.509573 | 0.510211 | 0.512884 |
| Occupancy | 1 | 1 | 1 | 0.5 | 0.5 | 0.5 |
| Atom | O4 | O4 | O4 | O4 | O4 | O4 |
| x = y = z | 0.250373 | 0.262464 | 0.262221 | 0.231004 | 0.228680 | 0.231430 |
| Occupancy | 1 | 1 | 1 | 0.5 | 0.5 | 0.5 |
| Compound | Calculated Coefficient of Thermal Expansion by Lattice Parameter (×10−6 °C−1) | |
|---|---|---|
| α Phase | β Phase | |
| ZrW2O8 | −9.88 | −5.35 |
| ZrW1.8Mo0.2O8 | −7.4 | −4.72 |
| ZrW1.5Mo0.5O8 | - | −3.95 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wei, H.; Hasegawa, M.; Mizutani, S.; Aimi, A.; Fujimoto, K.; Nishio, K. Low-Temperature Spark Plasma Sintering of ZrW2−xMoxO8 Exhibiting Controllable Negative Thermal Expansion. Materials 2018, 11, 1582. https://doi.org/10.3390/ma11091582
Wei H, Hasegawa M, Mizutani S, Aimi A, Fujimoto K, Nishio K. Low-Temperature Spark Plasma Sintering of ZrW2−xMoxO8 Exhibiting Controllable Negative Thermal Expansion. Materials. 2018; 11(9):1582. https://doi.org/10.3390/ma11091582
Chicago/Turabian StyleWei, Hui, Marin Hasegawa, Shunsuke Mizutani, Akihisa Aimi, Kenjiro Fujimoto, and Keishi Nishio. 2018. "Low-Temperature Spark Plasma Sintering of ZrW2−xMoxO8 Exhibiting Controllable Negative Thermal Expansion" Materials 11, no. 9: 1582. https://doi.org/10.3390/ma11091582