Very Low-Density Lipoproteins of Metabolic Syndrome Modulates STIM1, Suppresses Store-Operated Calcium Entry, and Deranges Myofilament Proteins in Atrial Myocytes

 , and
, and 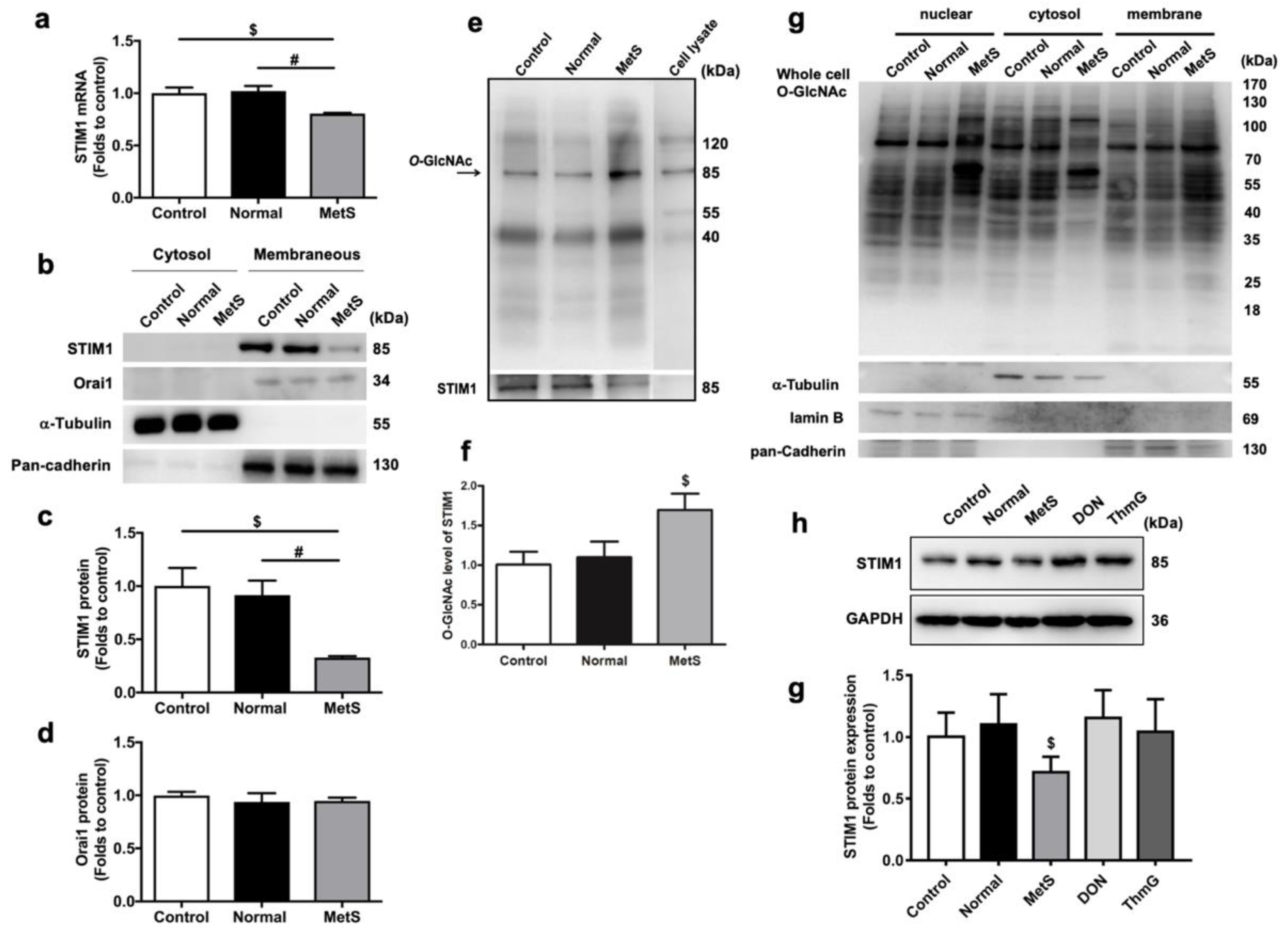{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. VLDL Isolation
2.2. HL-1 Atrial Myocyte Culture and Incubation with Isolated VLDLs
2.3. Quantitative Real-Time Reverse Transcriptase PCR
2.4. Isolation of Nuclear, Cytoplasmic, and Membrane Proteins from HL-1 Cells
2.5. Western Blot
2.6. Detection of O-GlcNAcylation STIM1 by Immunoprecipitation
2.7. Measurement of Calcineurin Activity
2.8. Measurement of SR Calcium Load and SOCE
2.9. Tissue Protein Isolation from Mice Atrial Tissue
2.10. Myofilament and Contractile Protein Expression and Phosphorylation Analysis in VLDL-Treated HL-1 Cells and Atrial Tissues of VLDL-Injected Mice
2.11. Transmission Electron Microscopy (TEM)
2.12. Data Analysis and Statistics
3. Results
3.1. MetS-VLDLs, but Not Normal-VLDLs, Induced the Downregulation of STIM1 at the Transcriptional and Translational Levels in HL-1 Cells
3.2. MetS-VLDLs Enhanced the O-GlcNAcylation of STIM1 Proteins
3.3. MetS-VLDLs Suppressed SOCE in HL-1 Cells
3.4. MetS-VLDLs Inhibited the Calcineurin–NFAT Pathway
3.5. MetS-VLDLs Affected Myofilament Protein Expression and Caused Sarcomere Derangement
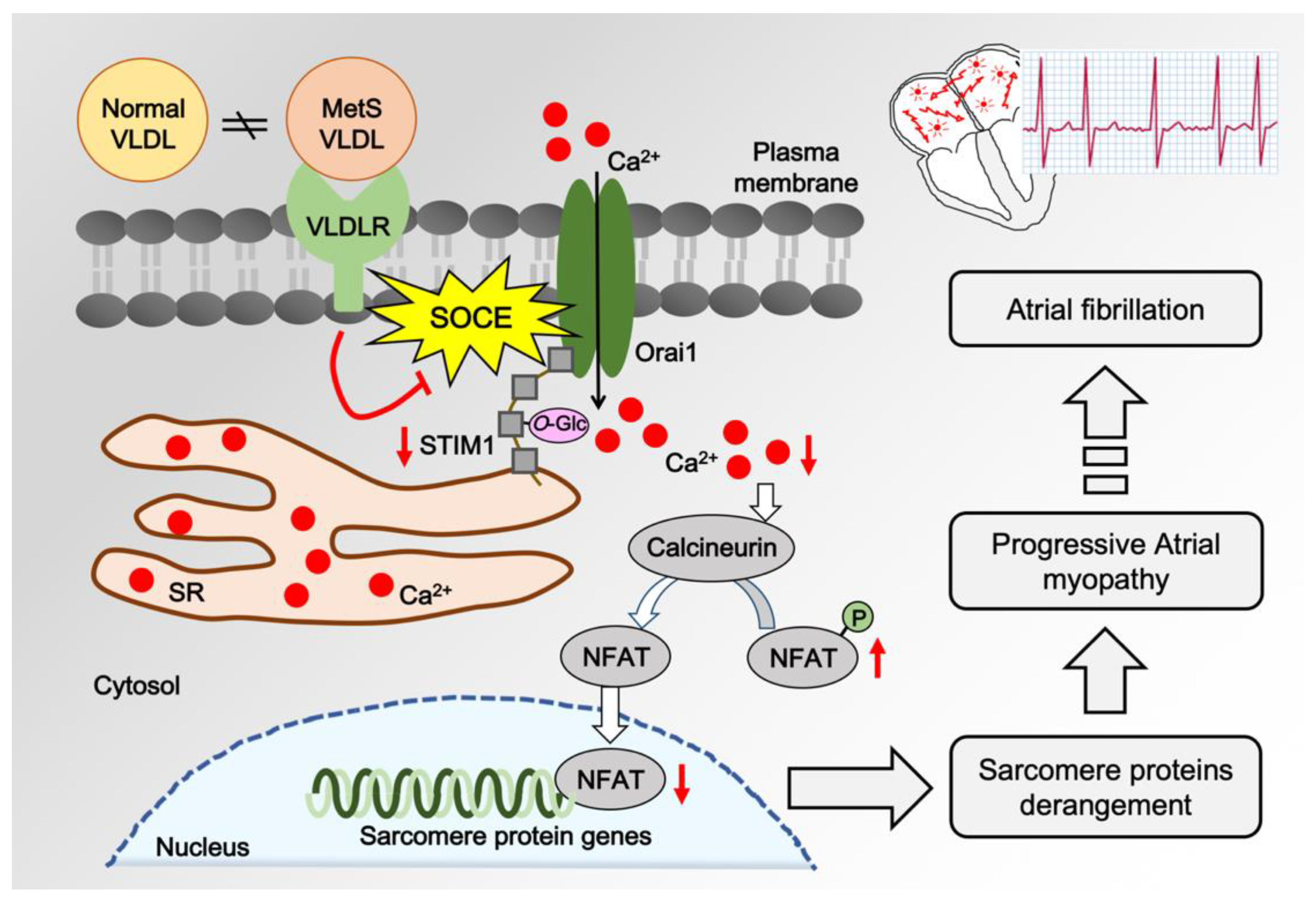
4. Discussions
Clinical Implications
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Nattel, S.; Dobrev, D. The multidimensional role of calcium in atrial fibrillation pathophysiology: Mechanistic insights and therapeutic opportunities. Eur. Heart J. 2012, 33, 1870–1877. [Google Scholar] [CrossRef] [PubMed]
- Hunton, D.L.; Lucchesi, P.A.; Pang, Y.; Cheng, X.; Dell’Italia, L.J.; Marchase, R.B. Capacitative calcium entry contributes to nuclear factor of activated T-cells nuclear translocation and hypertrophy in cardiomyocytes. J. Biol. Chem. 2002, 277, 14266–14273. [Google Scholar] [CrossRef] [PubMed]
- Palty, R.; Raveh, A.; Kaminsky, I.; Meller, R.; Reuveny, E. SARAF inactivates the store operated calcium entry machinery to prevent excess calcium refilling. Cell 2012, 149, 425–438. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Hojayev, B.; Jiang, N.; Wang, Z.V.; Tandan, S.; Rakalin, A.; Rothermel, B.A.; Gillette, T.G.; Hill, J.A. STIM1-dependent store-operated Ca(2)(+) entry is required for pathological cardiac hypertrophy. J. Mol. Cell Cardiol. 2012, 52, 136–147. [Google Scholar] [CrossRef] [PubMed]
- Calloway, N.; Owens, T.; Corwith, K.; Rodgers, W.; Holowka, D.; Baird, B. Stimulated association of STIM1 and Orai1 is regulated by the balance of PtdIns(4,5)P(2) between distinct membrane pools. J. Cell Sci. 2011, 124, 2602–2610. [Google Scholar] [CrossRef] [PubMed]
- Onohara, N.; Nishida, M.; Inoue, R.; Kobayashi, H.; Sumimoto, H.; Sato, Y.; Mori, Y.; Nagao, T.; Kurose, H. TRPC3 and TRPC6 are essential for angiotensin II-induced cardiac hypertrophy. EMBO J. 2006, 25, 5305–5316. [Google Scholar] [CrossRef] [PubMed]
- Collins, H.E.; Zhu-Mauldin, X.; Marchase, R.B.; Chatham, J.C. STIM1/Orai1-mediated SOCE: Current perspectives and potential roles in cardiac function and pathology. Am. J. Physiol. Heart Circ. Physiol. 2013, 305, H446–H458. [Google Scholar] [CrossRef] [PubMed]
- Cahalan, M.D. STIMulating store-operated Ca(2+) entry. Nat. Cell Biol. 2009, 11, 669–677. [Google Scholar] [CrossRef]
- Soboloff, J.; Rothberg, B.S.; Madesh, M.; Gill, D.L. STIM proteins: Dynamic calcium signal transducers. Nat. Rev. Mol. Cell Biol. 2012, 13, 549–565. [Google Scholar] [CrossRef]
- Feske, S.; Prakriya, M. Conformational dynamics of STIM1 activation. Nat. Struct. Mol. Biol. 2013, 20, 918–919. [Google Scholar] [CrossRef]
- Vaidyanathan, K.; Wells, L. Multiple tissue-specific roles for the O-GlcNAc post-translational modification in the induction of and complications arising from type II diabetes. J. Biol. Chem. 2014, 289, 34466–34471. [Google Scholar] [CrossRef] [PubMed]
- Marsh, S.A.; Collins, H.E.; Chatham, J.C. Protein O-GlcNAcylation and Cardiovascular (Patho)physiology. J. Biol. Chem. 2014, 289, 34449–34456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu-Mauldin, X.; Marsh, S.A.; Zou, L.; Marchase, R.B.; Chatham, J.C. Modification of STIM1 by O-linked N-acetylglucosamine (O-GlcNAc) attenuates store-operated calcium entry in neonatal cardiomyocytes. J. Biol. Chem. 2012, 287, 39094–39106. [Google Scholar] [CrossRef] [PubMed]
- Nagy, T.; Champattanachai, V.; Marchase, R.B.; Chatham, J.C. Glucosamine inhibits angiotensin II-induced cytoplasmic Ca2+ elevation in neonatal cardiomyocytes via protein-associated O-linked Nacetylglucosamine. Am. J. Physiol. Cell Physiol. 2006, 290, C57–C65. [Google Scholar] [CrossRef]
- Clark, R.J.; McDonough, P.M.; Swanson, E.; Trost, S.U.; Suzuki, M.; Fukuda, M.; Dillmann, W.H. Diabetes and the Accompanying Hyperglycemia Impairs Cardiomyocyte Calcium Cycling through Increased Nuclear O-GlcNAcylation. J. Biol. Chem. 2003, 278, 44230–44237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erickson, J.R.; Pereira, L.; Wang, L.; Han, G.; Ferguson, A.; Dao, K.; Copeland, R.J.; Despa, F.; Hart, G.W.; Ripplinger, C.M.; et al. Diabetic hyperglycaemia activates CaMKII and arrhythmias by O-linked glycosylation. Nature 2013, 502, 372–376. [Google Scholar] [CrossRef] [PubMed]
- Bond, M.R.; Hanover, J.A. A little sugar goes a long way: The cell biology of O-GlcNAc. J. Cell Biol. 2015, 208, 869–880. [Google Scholar] [CrossRef] [Green Version]
- Yokoe, S.; Asahi, M.; Takeda, T.; Otsu, K.; Taniguchi, N.; Miyoshi, E.; Suzuki, K. Inhibition of phospholamban phosphorylation by O-GlcNAcylation: Implications for diabetic cardiomyopathy. Glycobiology 2010, 20, 1217–1226. [Google Scholar] [CrossRef]
- Lee, H.C.; Lin, H.T.; Ke, L.Y.; Wei, C.; Hsiao, Y.L.; Chu, C.S.; Lai, W.T.; Shin, S.J.; Chen, C.H.; Sheu, S.H.; et al. VLDL from Metabolic Syndrome Individuals Enhanced Lipid Accumulation in Atria with Association of Susceptibility to Atrial Fibrillation. Int. J. Mol. Sci. 2016, 17, 134. [Google Scholar] [CrossRef]
- Lee, H.C.; Chen, C.C.; Tsai, W.C.; Lin, H.T.; Shiao, Y.L.; Sheu, S.H.; Wu, B.N.; Chen, C.H.; Lai, W.T. Very-Low-Density Lipoprotein of Metabolic Syndrome Modulates Gap Junctions and Slows Cardiac Conduction. Sci. Rep. 2017, 7, 12050. [Google Scholar] [CrossRef] [Green Version]
- Touchberry, C.D.; Elmore, C.J.; Nguyen, T.M.; Andresen, J.J.; Zhao, X.; Orange, M.; Weisleder, N.; Brotto, M.; Claycomb, W.C.; Wacker, M.J. Store-operated calcium entry is present in HL-1 cardiomyocytes and contributes to resting calcium. Biochem. Biophys. Res. Commun. 2011, 416, 45–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Executive Summary of the Third Report of The National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, And Treatment of High Blood Cholesterol In Adults (Adult Treatment Panel III). JAMA 2001, 285, 2486–2497. [CrossRef]
- Chen, C.H.; Lu, J.; Chen, S.H.; Huang, R.Y.; Yilmaz, H.R.; Dong, J.; Elayda, M.A.; Dixon, R.A.; Yang, C.Y. Effects of electronegative VLDL on endothelium damage in metabolic syndrome. Diabetes Care 2012, 35, 648–653. [Google Scholar] [CrossRef] [PubMed]
- White, S.M.; Constantin, P.E.; Claycomb, W.C. Cardiac physiology at the cellular level: Use of cultured HL-1 cardiomyocytes for studies of cardiac muscle cell structure and function. Am. J. Physiol. Heart Circ. Physiol. 2004, 286, H823–H829. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Wang, X.; Wang, X.; Loktionova, N.A.; Cai, X.; Nwokonko, R.M.; Vrana, E.; Wang, Y.; Rothberg, B.S.; Gill, D.L. STIM1 dimers undergo unimolecular coupling to activate Orai1 channels. Nat. Commun. 2015, 6, 8395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaremba, R.; Merkus, D.; Hamdani, N.; Lamers, J.M.; Paulus, W.J.; Dos Remedios, C.; Duncker, D.J.; Stienen, G.J.; van der Velden, J. Quantitative analysis of myofilament protein phosphorylation in small cardiac biopsies. Proteom. Clin. Appl. 2007, 1, 1285–1290. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Hildebrand, M.E.; Garcia, E.; Snutch, T.P. The transient receptor potential channel antagonist SKF96365 is a potent blocker of low-voltage-activated T-type calcium channels. Br. J. Pharm. 2010, 160, 1464–1475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kappel, S.; Borgstrom, A.; Stoklosa, P.; Dorr, K.; Peinelt, C. Store-operated calcium entry in disease: Beyond STIM/Orai expression levels. Semin. Cell Dev. Biol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Go, C.K.; Gross, S.; Hooper, R.; Soboloff, J. EGR-mediated control of STIM expression and function. Cell Calcium 2019, 77, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Fang, M.; Li, Y.; Wu, Y.; Ning, Z.; Wang, X.; Li, X. miR-185 silencing promotes the progression of atherosclerosis via targeting stromal interaction molecule 1. Cell Cycle 2019, 18, 682–695. [Google Scholar] [CrossRef] [Green Version]
- Rosenberg, P.; Katz, D.; Bryson, V. SOCE and STIM1 signaling in the heart: Timing and location matter. Cell Calcium 2019, 77, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Marsh, S.A.; Dell’Italia, L.J.; Chatham, J.C. Activation of the hexosamine biosynthesis pathway and protein O-GlcNAcylation modulate hypertrophic and cell signaling pathways in cardiomyocytes from diabetic mice. Amino Acids 2011, 40, 819–828. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Belke, D.; Suarez, J.; Swanson, E.; Clark, R.; Hoshijima, M.; Dillmann, W.H. Adenovirus-mediated overexpression of O-GlcNAcase improves contractile function in the diabetic heart. Circ. Res. 2005, 96, 1006–1013. [Google Scholar] [CrossRef] [PubMed]
- Fulop, N.; Marchase, R.B.; Chatham, J.C. Role of protein O-linked N-acetyl-glucosamine in mediating cell function and survival in the cardiovascular system. Cardiovasc. Res. 2007, 73, 288–297. [Google Scholar] [CrossRef] [PubMed]
- Schott, P.; Asif, A.; Gräf, C.; Toischer, K.; Hasenfuss, G.; Kögler, H. Myocardial adaptation of energy metabolism to elevated preload depends on calcineurin activity. Basic Res. Cardiol. 2008, 103, 232–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Cui, G.M.; Zhou, N.N.; Li, C.; Zhang, Q.; Sun, H.; Han, B.; Zou, C.W.; Wang, L.J.; Li, X.D.; et al. Calpain-Calcineurin-Nuclear Factor Signaling and the Development of Atrial Fibrillation in Patients with Valvular Heart Disease and Diabetes. J. Diabetes Res. 2016, 2016, 4639654. [Google Scholar] [CrossRef] [PubMed]
- Heijman, J.; Ghezelbash, S.; Wehrens, X.H.; Dobrev, D. Serine/Threonine Phosphatases in Atrial Fibrillation. J. Mol. Cell. Cardiol. 2017, 103, 110–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molkentin, J.D. Calcineurin–NFAT signaling regulates the cardiac hypertrophic response in coordination with the MAPKs. Cardiovasc. Res. 2004, 63, 467–475. [Google Scholar] [CrossRef] [PubMed]
- Machackova, J.; Barta, J.; Dhalla, N.S. Myofibrillar remodelling in cardiac hypertrophy, heart failure and cardiomyopathies. Can. J. Cardiol. 2006, 22, 953–968. [Google Scholar] [CrossRef] [Green Version]
- Willis, M.S.; Schisler, J.C.; Portbury, A.L.; Patterson, C. Build it up–Tear it down: Protein quality control in the cardiac sarcomere. Cardiovasc. Res. 2009, 81, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Wen, H.; Zhao, Z.; Fefelova, N.; Xie, L.H. Potential Arrhythmogenic Role of TRPC Channels and Store-Operated Calcium Entry Mechanism in Mouse Ventricular Myocytes. Front. Physiol. 2018, 9, 1785. [Google Scholar] [CrossRef] [PubMed]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shiou, Y.-L.; Lin, H.-T.; Ke, L.-Y.; Wu, B.-N.; Shin, S.-J.; Chen, C.-H.; Tsai, W.-C.; Chu, C.-S.; Lee, H.-C. Very Low-Density Lipoproteins of Metabolic Syndrome Modulates STIM1, Suppresses Store-Operated Calcium Entry, and Deranges Myofilament Proteins in Atrial Myocytes. J. Clin. Med. 2019, 8, 881. https://doi.org/10.3390/jcm8060881
Shiou Y-L, Lin H-T, Ke L-Y, Wu B-N, Shin S-J, Chen C-H, Tsai W-C, Chu C-S, Lee H-C. Very Low-Density Lipoproteins of Metabolic Syndrome Modulates STIM1, Suppresses Store-Operated Calcium Entry, and Deranges Myofilament Proteins in Atrial Myocytes. Journal of Clinical Medicine. 2019; 8(6):881. https://doi.org/10.3390/jcm8060881
Chicago/Turabian StyleShiou, Yi-Lin, Hsin-Ting Lin, Liang-Yin Ke, Bin-Nan Wu, Shyi-Jang Shin, Chu-Huang Chen, Wei-Chung Tsai, Chih-Sheng Chu, and Hsiang-Chun Lee. 2019. "Very Low-Density Lipoproteins of Metabolic Syndrome Modulates STIM1, Suppresses Store-Operated Calcium Entry, and Deranges Myofilament Proteins in Atrial Myocytes" Journal of Clinical Medicine 8, no. 6: 881. https://doi.org/10.3390/jcm8060881