Deduction of Novel Genes Potentially Involved in Keratinocytes of Type 2 Diabetes Using Next-Generation Sequencing and Bioinformatics Approaches

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
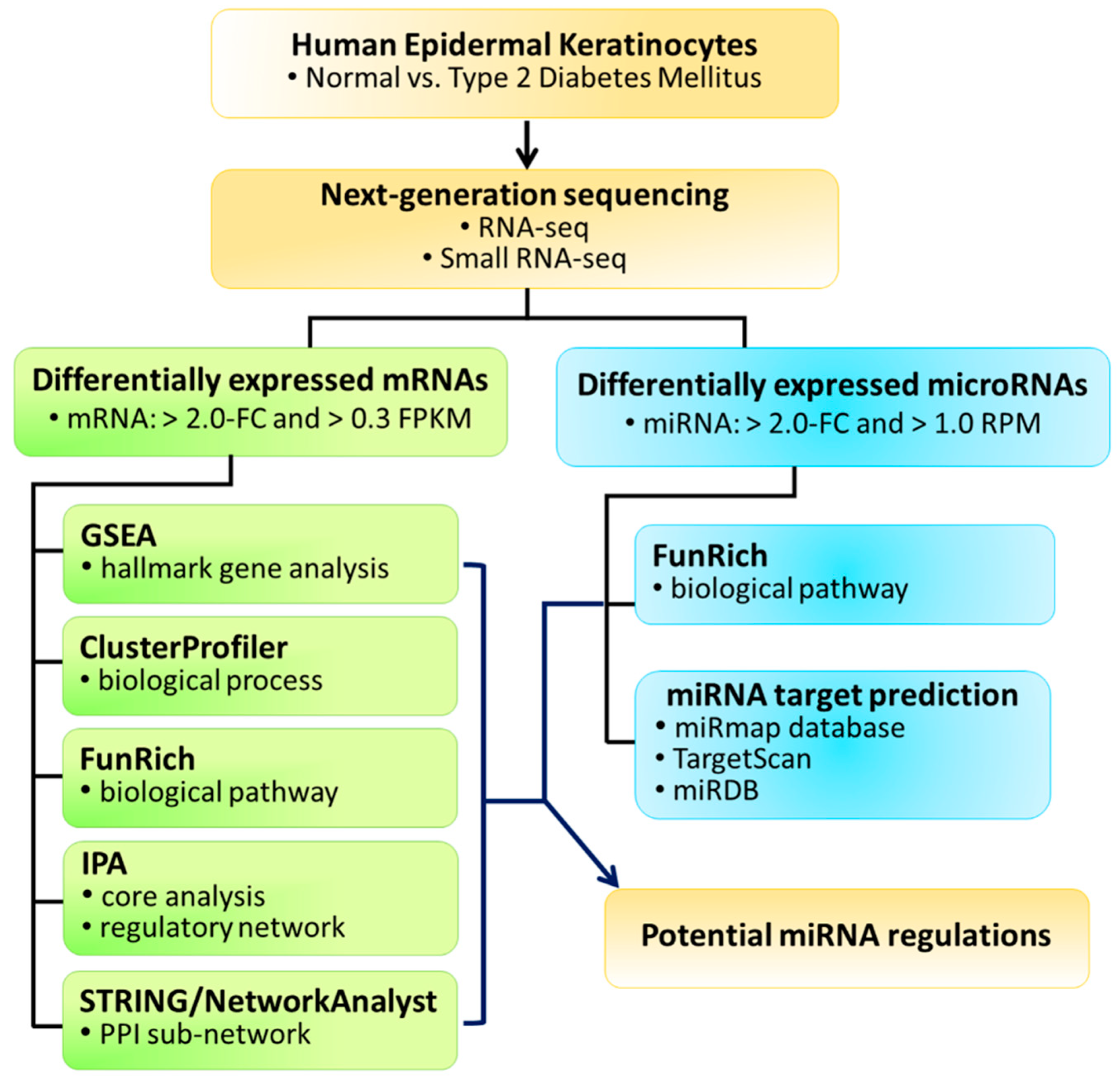
2.1. Experimental Design
2.2. Cell Culture of Primary Human Epidermal Keratinocytes
2.3. Next-Generation Sequencing
2.4. clusterProfiler
2.5. Gene Set Enrichment Analysis (GSEA)
2.6. Functional Enrichment Analysis Tool (FunRich)
2.7. STRING and NetworkAnalyst Databases for Protein–Protein Interaction Network Analysis Database
2.8. Ingenuity Pathway Analysis (IPA)
2.9. miRmap Target Prediction Database
3. Results
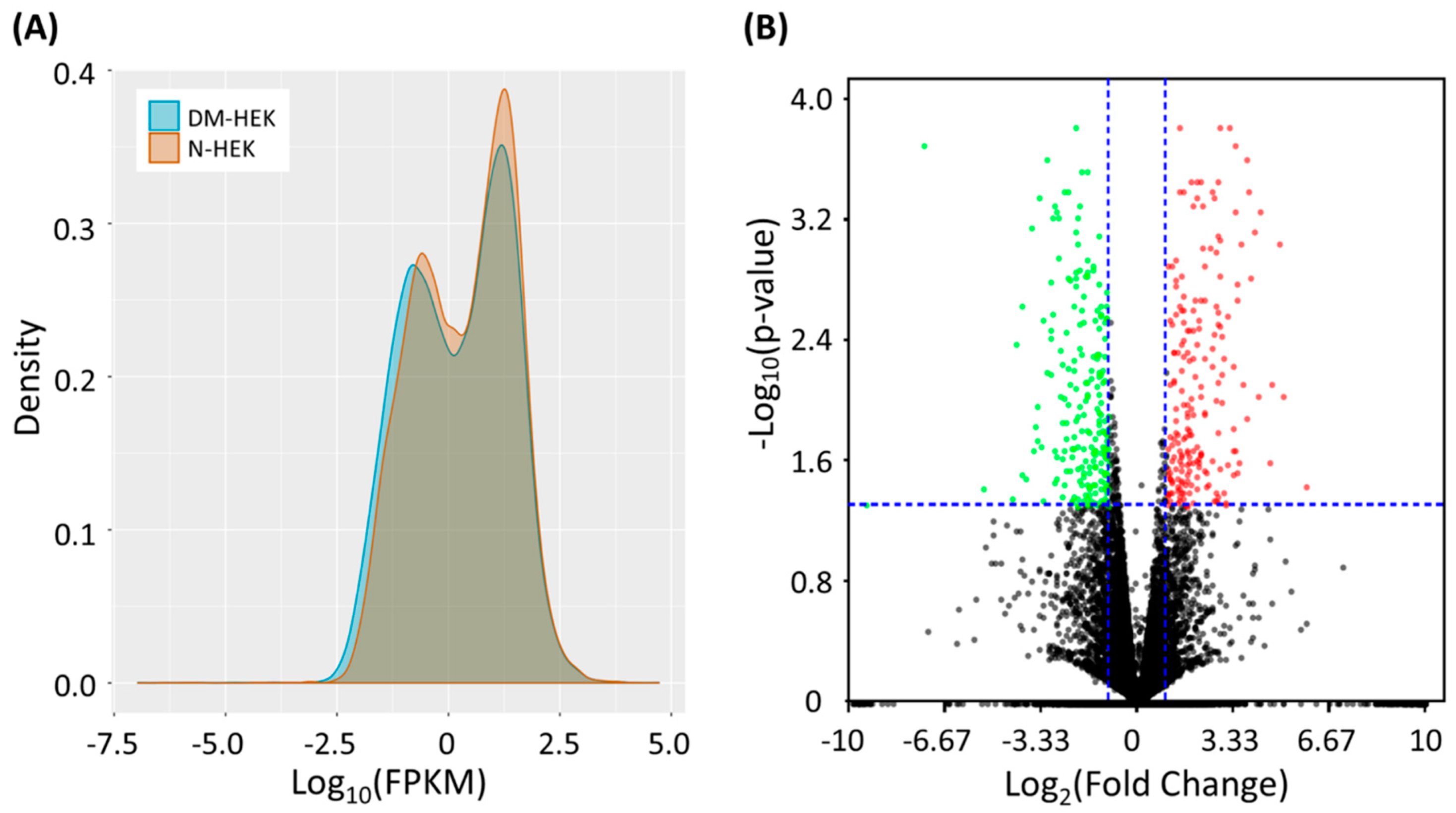
3.1. Identification of Differentially Expressed Genes in Human Type 2 Diabetic Epidermal Keratinocytes (DM-HEK)
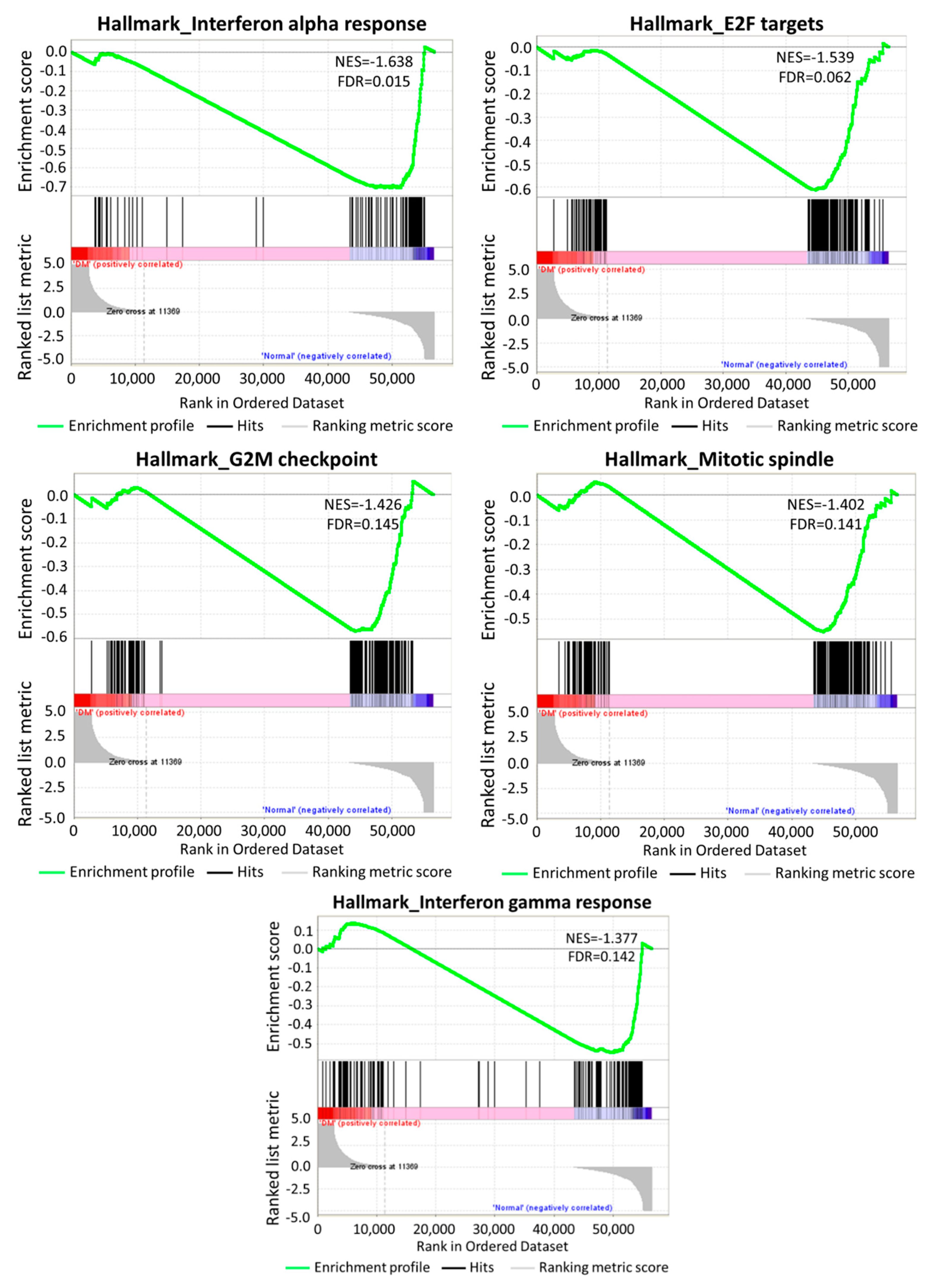
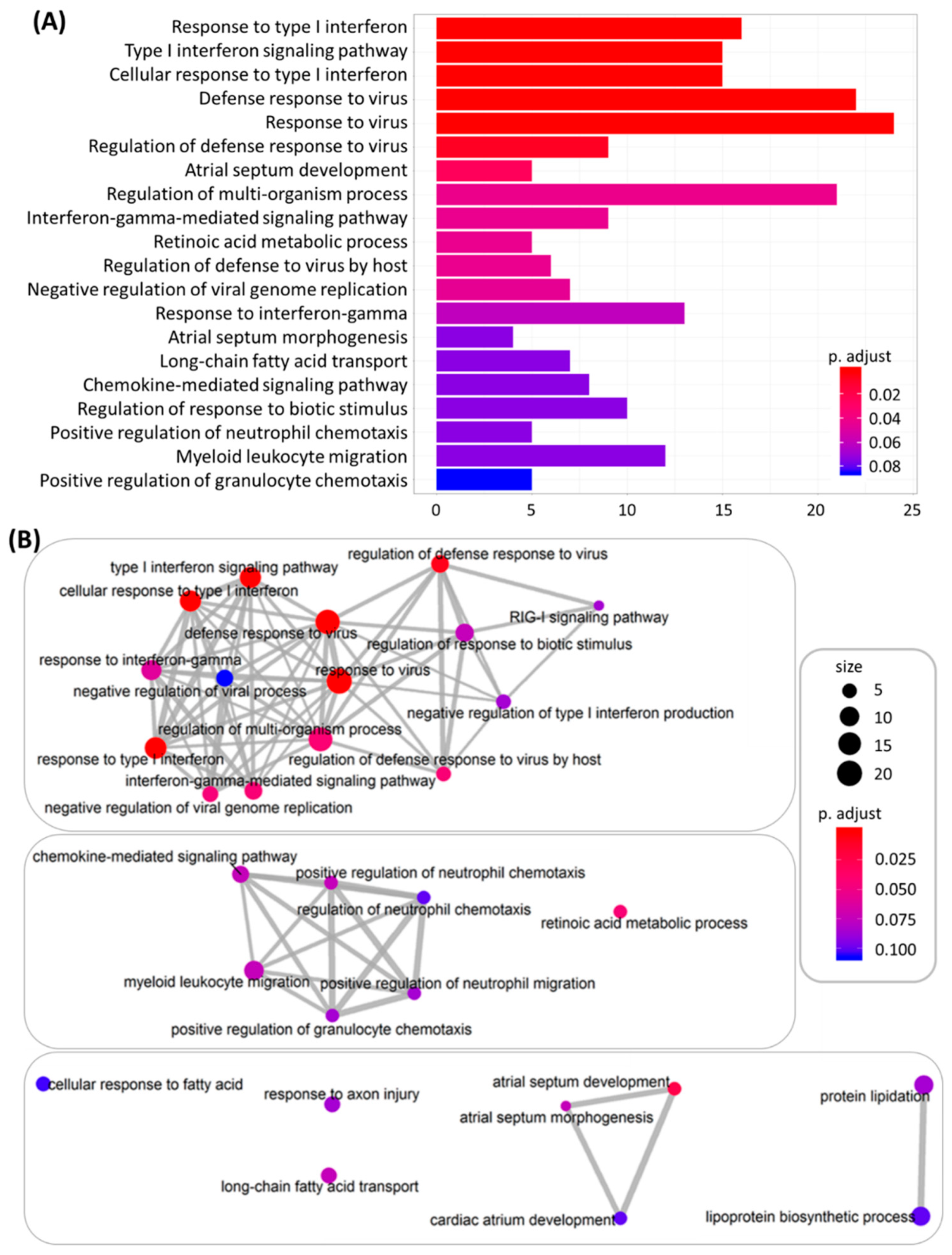
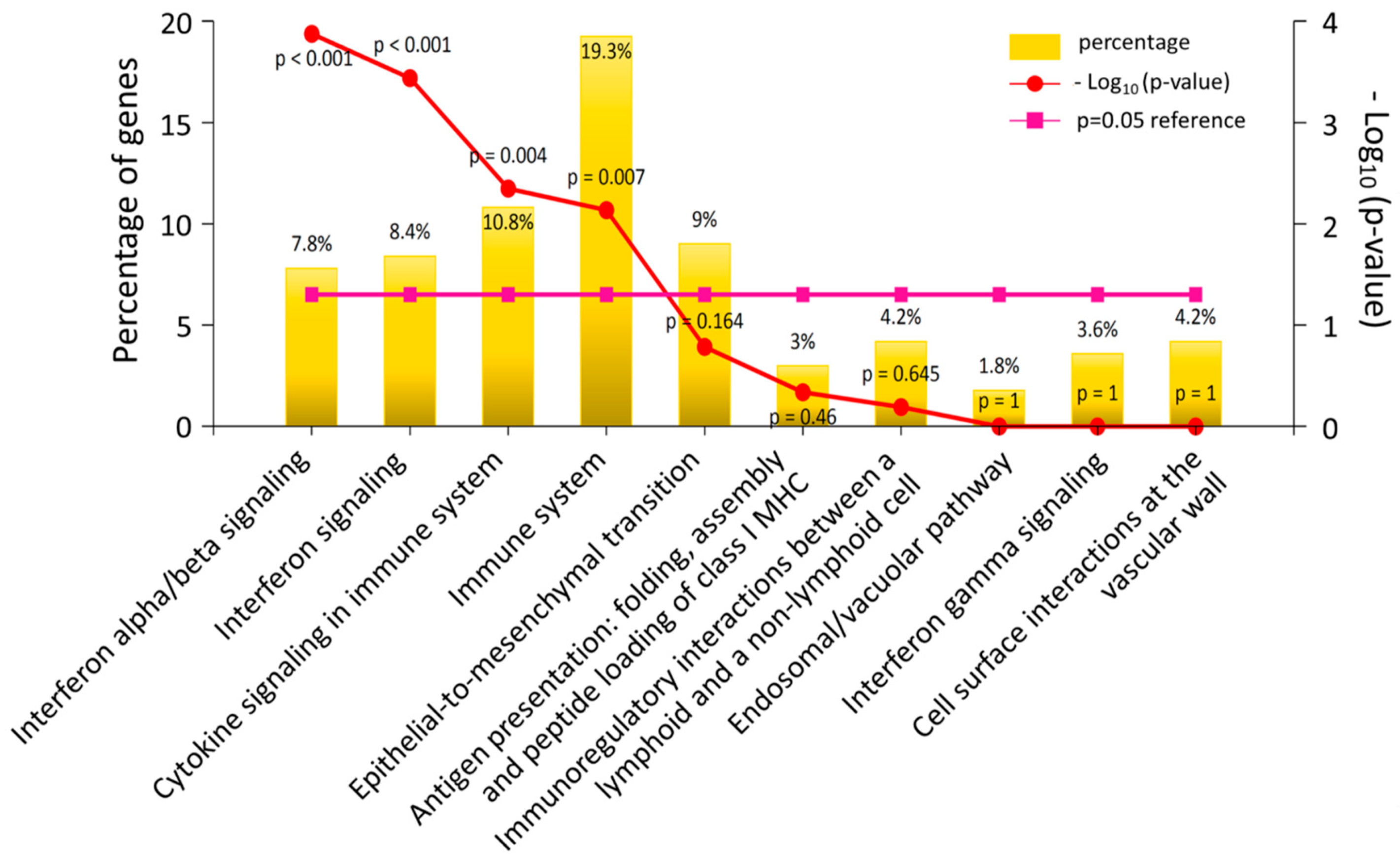
3.2. The Differentially Expressed Genes in DM-HEK Were Enriched in Interferon (IFN) Signaling and Viral Defense Response
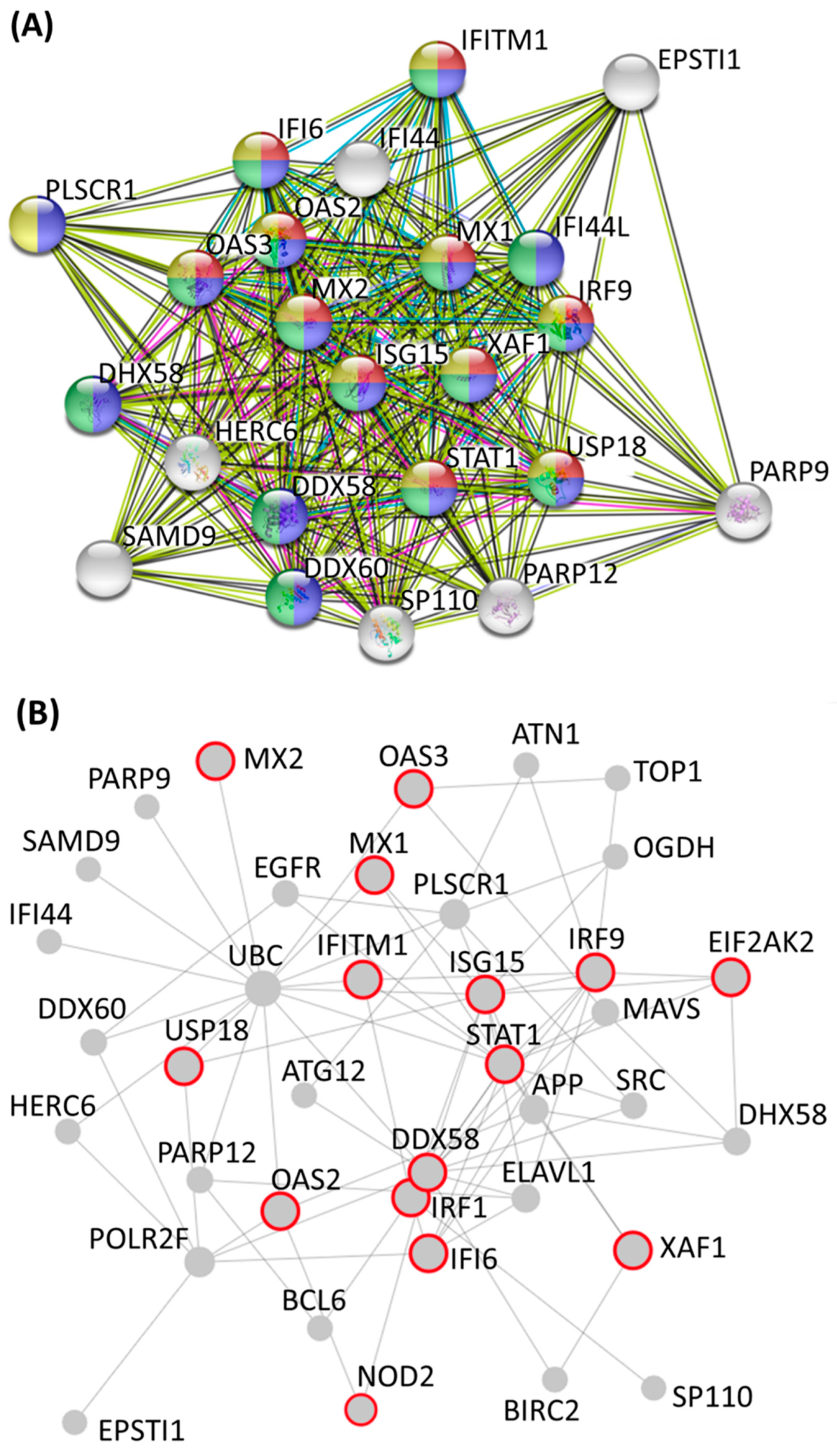
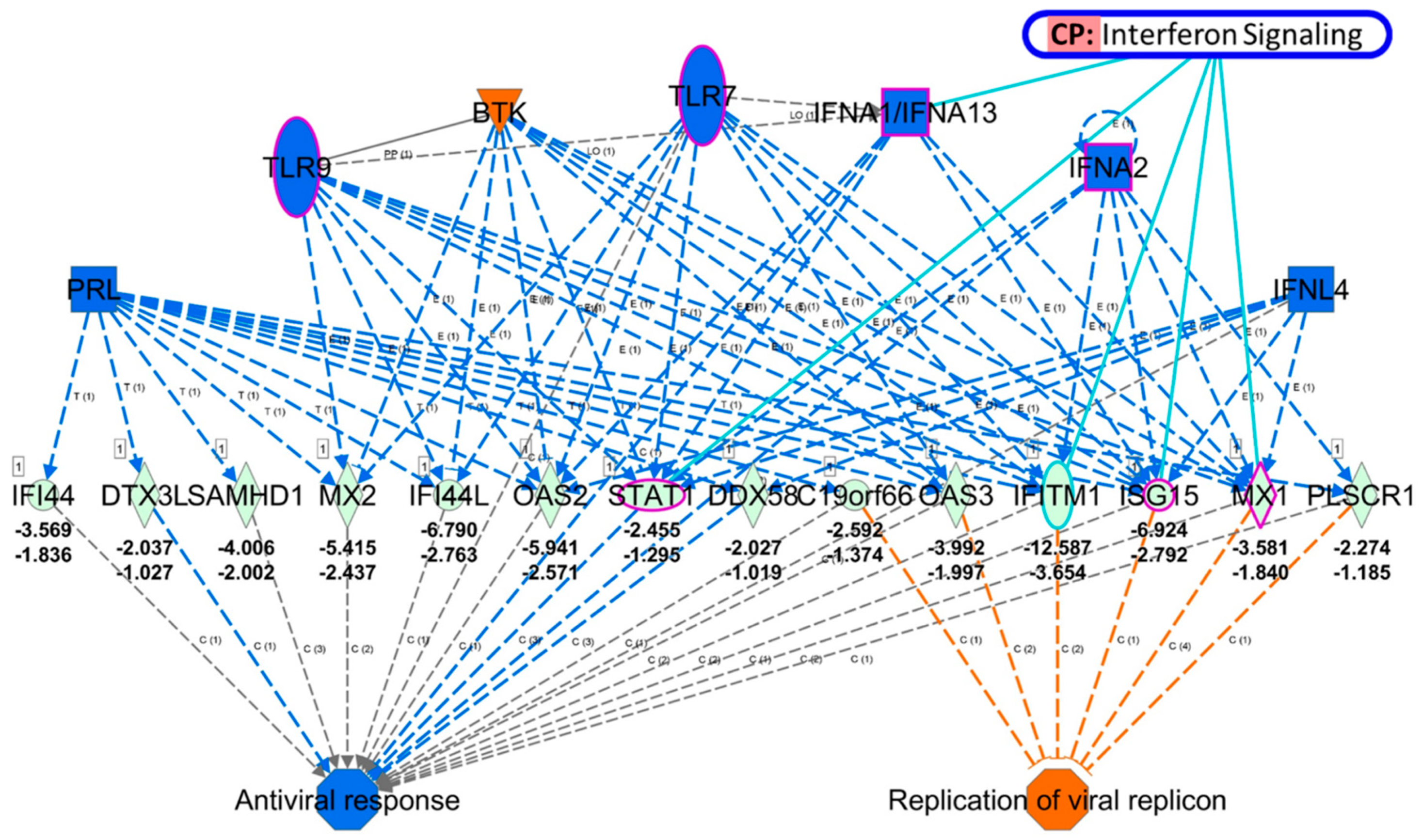
3.3. Identification of Potential Mechanistic Regulatory Network and Gene Clusters Involved in IFN Signaling and Defense Response of HEK Cells
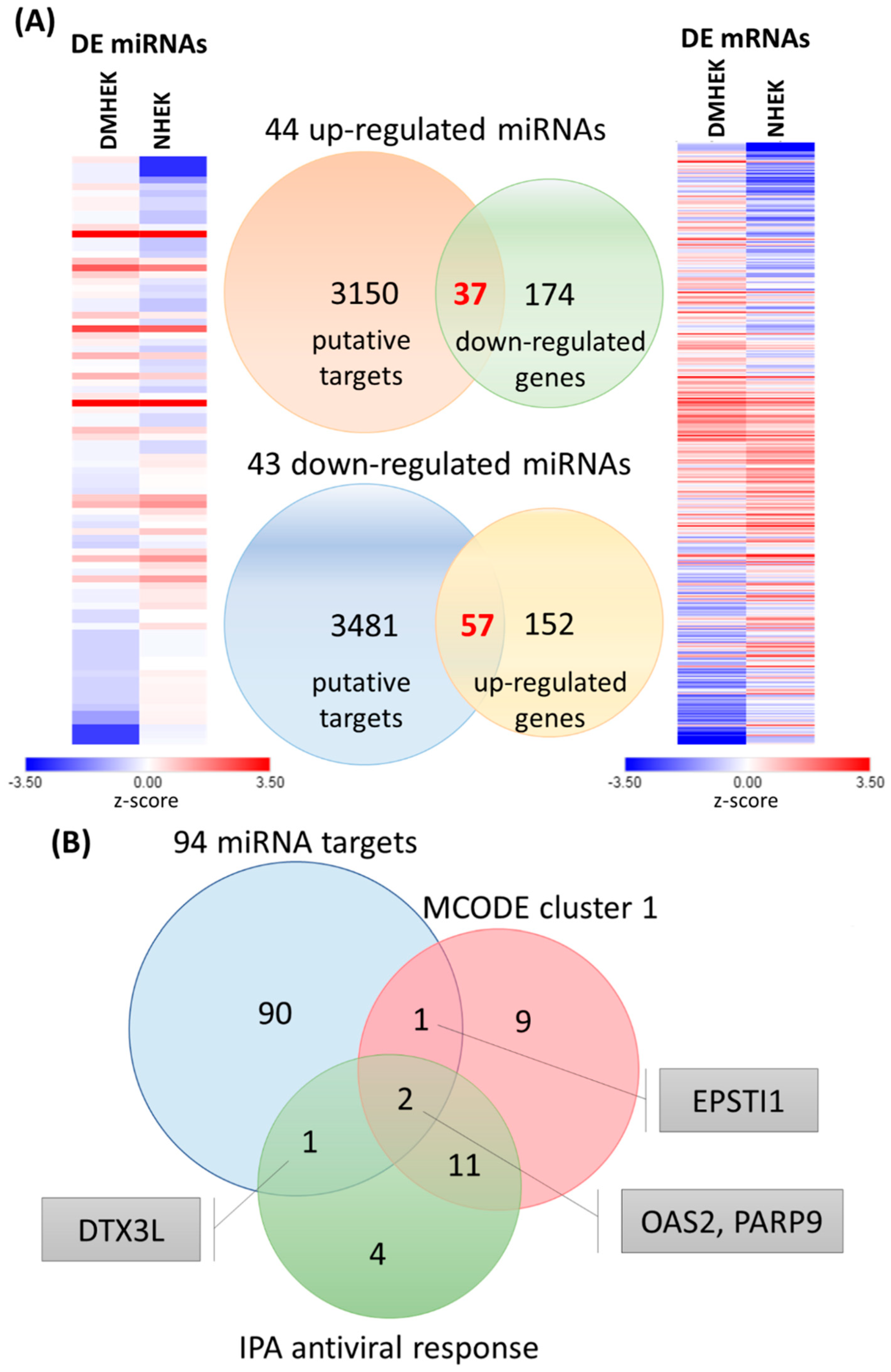
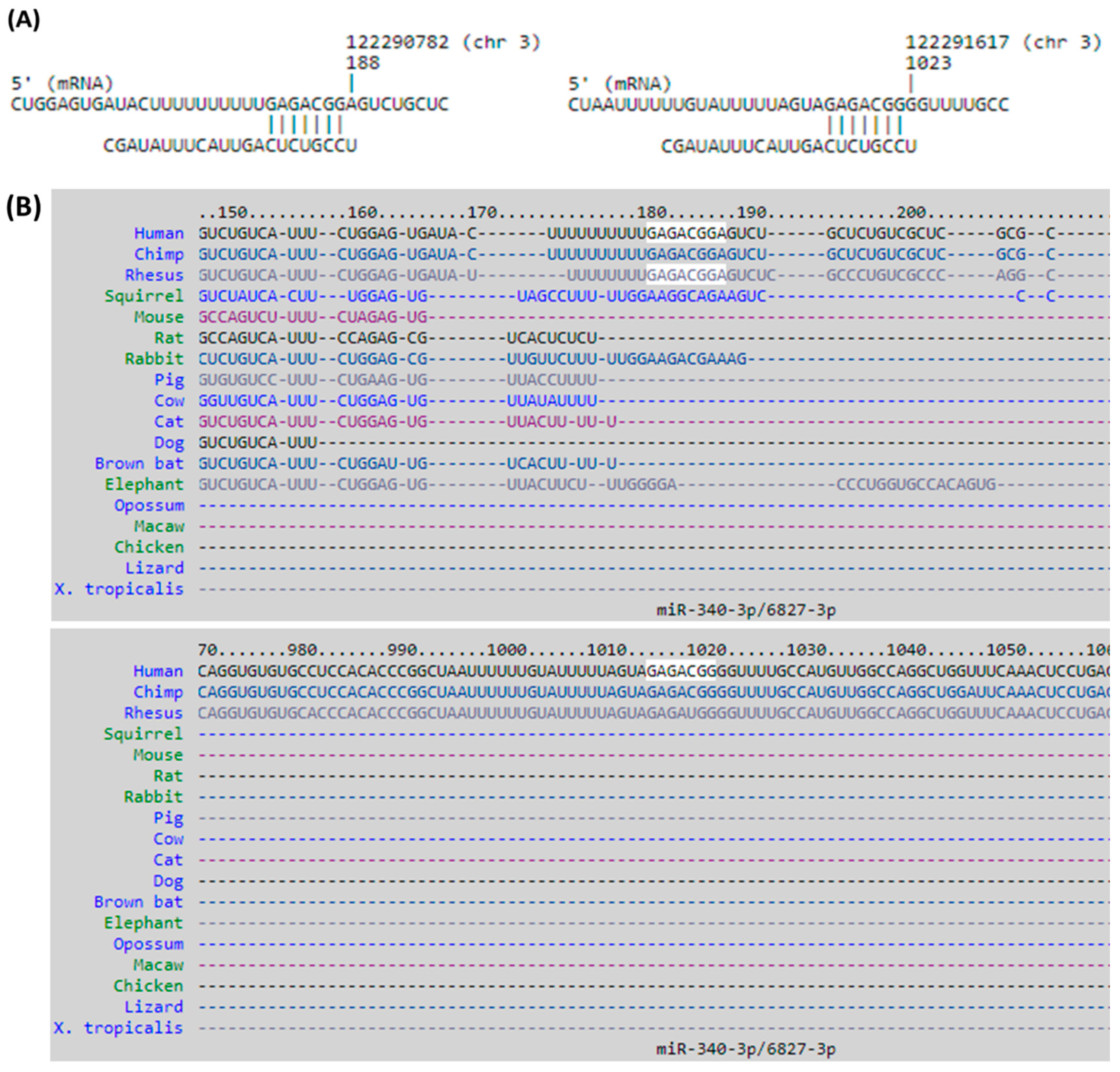
3.4. Identification of Potential miRNA–mRNA Interactions in DM-HEK Cells
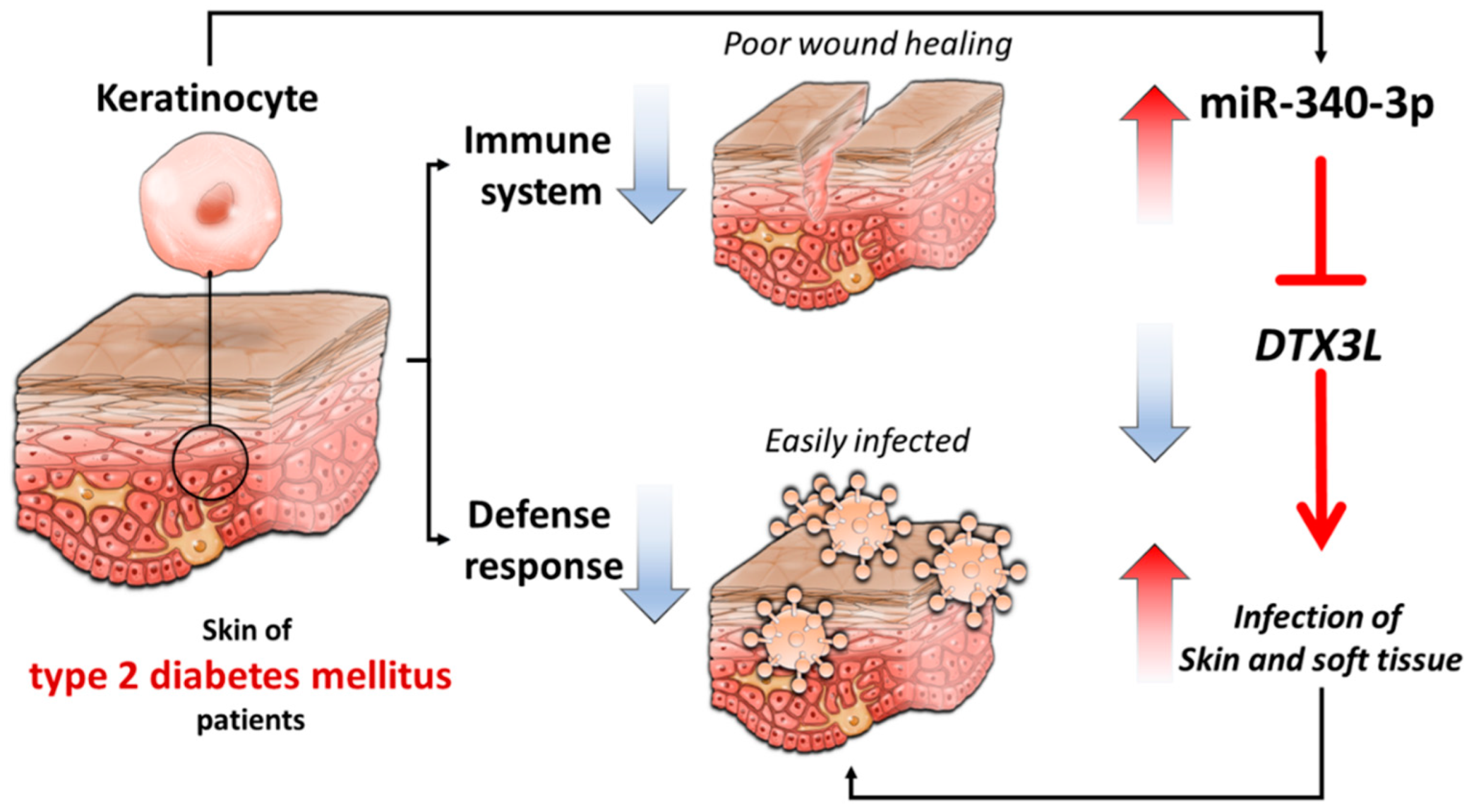
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Weisman, A.; Fazli, G.S.; Johns, A.; Booth, G.L. Evolving Trends in the Epidemiology, Risk Factors, and Prevention of Type 2 Diabetes: A Review. Can. J. Cardiol. 2018, 34, 552–564. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Ley, S.H.; Hu, F.B. Global aetiology and epidemiology of type 2 diabetes mellitus and its complications. Nat. Rev. Endocrinol. 2018, 14, 88–98. [Google Scholar] [CrossRef] [PubMed]
- De Macedo, G.M.; Nunes, S.; Barreto, T. Skin disorders in diabetes mellitus: An epidemiology and physiopathology review. Diabetol. Metab. Syndr. 2016, 8, 63. [Google Scholar] [CrossRef] [PubMed]
- Makrantonaki, E.; Jiang, D.; Hossini, A.M.; Nikolakis, G.; Wlaschek, M.; Scharffetter-Kochanek, K.; Zouboulis, C.C. Diabetes mellitus and the skin. Rev. Endocr. Metab. Disord. 2016, 17, 269–282. [Google Scholar] [CrossRef]
- Hu, H.; Jiang, H.; Ren, H.; Hu, X.; Wang, X.; Han, C. AGEs and chronic subclinical inflammation in diabetes: Disorders of immune system. Diabetes. Metab. Res. Rev. 2015, 31, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Nestle, F.O.; Di Meglio, P.; Qin, J.Z.; Nickoloff, B.J. Skin immune sentinels in health and disease. Nat. Rev. Immunol. 2009, 9, 679–691. [Google Scholar] [CrossRef] [PubMed]
- Werner, S.; Krieg, T.; Smola, H. Keratinocyte-fibroblast interactions in wound healing. J. Investig. Dermatol. 2007, 127, 998–1008. [Google Scholar] [CrossRef]
- Spravchikov, N.; Sizyakov, G.; Gartsbein, M.; Accili, D.; Tennenbaum, T.; Wertheimer, E. Glucose effects on skin keratinocytes: Implications for diabetes skin complications. Diabetes 2001, 50, 1627–1635. [Google Scholar] [CrossRef]
- Zhu, P.; Yang, C.; Chen, L.H.; Ren, M.; Lao, G.J.; Yan, L. Impairment of human keratinocyte mobility and proliferation by advanced glycation end products-modified BSA. Arch. Dermatol. Res. 2011, 303, 339–350. [Google Scholar] [CrossRef]
- Huang, S.M.; Wu, C.S.; Chiu, M.H.; Yang, H.J.; Chen, G.S.; Lan, C.E. High-glucose environment induced intracellular O-GlcNAc glycosylation and reduced galectin-7 expression in keratinocytes: Implications on impaired diabetic wound healing. J. Dermatol. Sci. 2017, 87, 168–175. [Google Scholar] [CrossRef]
- Arya, A.K.; Tripathi, K.; Das, P. Promising role of ANGPTL4 gene in diabetic wound healing. Int. J. Low. Extremity Wounds 2014, 13, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Jhamb, S.; Vangaveti, V.N.; Malabu, U.H. Genetic and molecular basis of diabetic foot ulcers: Clinical review. J. Tissue Viability. 2016, 25, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liang, Y.; Lu, Q. MicroRNA epigenetic alterations: Predicting biomarkers and therapeutic targets in human diseases. Clin. Genet. 2008, 74, 307–315. [Google Scholar] [CrossRef]
- Zgheib, C.; Hodges, M.; Hu, J.; Beason, D.P.; Soslowsky, L.J.; Liechty, K.W.; Xu, J. Mechanisms of mesenchymal stem cell correction of the impaired biomechanical properties of diabetic skin: The role of miR-29a. Wound Repair Regen. 2016, 24, 237–246. [Google Scholar] [CrossRef]
- Wang, W.; Yang, C.; Wang, X.Y.; Zhou, L.Y.; Lao, G.J.; Liu, D.; Wang, C.; Hu, M.D.; Zeng, T.T.; Yan, L.; et al. MicroRNA-129 and -335 Promote Diabetic Wound Healing by Inhibiting Sp1-Mediated MMP-9 Expression. Diabetes 2018, 67, 1627–1638. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Li, D.; Wang, A.; Chu, T.; Lohcharoenkal, W.; Zheng, X.; Grunler, J.; Narayanan, S.; Eliasson, S.; Herter, E.K.; et al. MicroRNA-132 with Therapeutic Potential in Chronic Wounds. J. Investig. Dermatol. 2017, 137, 2630–2638. [Google Scholar] [CrossRef]
- Liang, L.; Stone, R.C.; Stojadinovic, O.; Ramirez, H.; Pastar, I.; Maione, A.G.; Smith, A.; Yanez, V.; Veves, A.; Kirsner, R.S.; et al. Integrative analysis of miRNA and mRNA paired expression profiling of primary fibroblast derived from diabetic foot ulcers reveals multiple impaired cellular functions. Wound Repair Regen. 2016, 24, 943–953. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Wu, W.; Zhang, L.; Dorset-Martin, W.; Morris, M.W.; Mitchell, M.E.; Liechty, K.W. The role of microRNA-146a in the pathogenesis of the diabetic wound-healing impairment: Correction with mesenchymal stem cell treatment. Diabetes 2012, 61, 2906–2912. [Google Scholar] [CrossRef]
- Caporali, A.; Emanueli, C. MicroRNA-503 and the extended microRNA-16 family in angiogenesis. Trends Cardiovasc. Med. 2011, 21, 162–166. [Google Scholar] [CrossRef]
- Ross, K. Towards topical microRNA-directed therapy for epidermal disorders. J. Control. Release 2018, 269, 136–147. [Google Scholar] [CrossRef]
- Desmet, E.; Bracke, S.; Forier, K.; Taevernier, L.; Stuart, M.C.; De Spiegeleer, B.; Raemdonck, K.; Van Gele, M.; Lambert, J. An elastic liposomal formulation for RNAi-based topical treatment of skin disorders: Proof-of-concept in the treatment of psoriasis. Int. J. Pharm. 2016, 500, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Zewde, M.; Kiyotani, K.; Park, J.H.; Fang, H.; Yap, K.L.; Yew, P.Y.; Alachkar, H.; Kato, T.; Mai, T.H.; Ikeda, Y.; et al. The era of immunogenomics/immunopharmacogenomics. J. Hum. Genet. 2018, 63, 865–875. [Google Scholar] [CrossRef] [PubMed]
- Titeux, M.; Izmiryan, A.; Hovnanian, A. The Molecular Revolution in Cutaneous Biology: Emerging Landscape in Genomic Dermatology: New Mechanistic Ideas, Gene Editing, and Therapeutic Breakthroughs. J. Investig. Dermatol. 2017, 137, e123–e129. [Google Scholar] [CrossRef] [PubMed]
- Shukla, V.; Varghese, V.K.; Kabekkodu, S.P.; Mallya, S.; Satyamoorthy, K. A compilation of Web-based research tools for miRNA analysis. Brief. Funct. Genom. 2017, 16, 249–273. [Google Scholar] [CrossRef] [PubMed]
- Gligorijevic, V.; Malod-Dognin, N.; Przulj, N. Integrative methods for analyzing big data in precision medicine. Proteomics 2016, 16, 741–758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Campos, M.A.; Espinal-Enriquez, J.; Hernandez-Lemus, E. Pathway Analysis: State of the Art. Front. Physiol. 2015, 6, 383. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. Omics 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- Pathan, M.; Keerthikumar, S.; Ang, C.S.; Gangoda, L.; Quek, C.Y.; Williamson, N.A.; Mouradov, D.; Sieber, O.M.; Simpson, R.J.; Salim, A.; et al. FunRich: An open access standalone functional enrichment and interaction network analysis tool. Proteomics 2015, 15, 2597–2601. [Google Scholar] [CrossRef]
- Pathan, M.; Keerthikumar, S.; Chisanga, D.; Alessandro, R.; Ang, C.S.; Askenase, P.; Batagov, A.O.; Benito-Martin, A.; Camussi, G.; Clayton, A.; et al. A novel community driven software for functional enrichment analysis of extracellular vesicles data. J. Extracell. Vesicles 2017, 6, 1321455. [Google Scholar] [CrossRef] [Green Version]
- Szklarczyk, D.; Morris, J.H.; Cook, H.; Kuhn, M.; Wyder, S.; Simonovic, M.; Santos, A.; Doncheva, N.T.; Roth, A.; Bork, P.; et al. The STRING database in 2017: Quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. 2017, 45, D362–D368. [Google Scholar] [CrossRef] [PubMed]
- Bader, G.D.; Hogue, C.W. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinform. 2003, 4, 2. [Google Scholar] [CrossRef]
- Xia, J.; Benner, M.J.; Hancock, R.E. NetworkAnalyst--integrative approaches for protein-protein interaction network analysis and visual exploration. Nucleic Acids Res. 2014, 42, W167–W174. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.; Gill, E.E.; Hancock, R.E. NetworkAnalyst for statistical, visual and network-based meta-analysis of gene expression data. Nat. Protoc. 2015, 10, 823–844. [Google Scholar] [CrossRef]
- Thomas, S.; Bonchev, D. A survey of current software for network analysis in molecular biology. Hum. Genom. 2010, 4, 353–360. [Google Scholar] [CrossRef]
- Kramer, A.; Green, J.; Pollard, J., Jr.; Tugendreich, S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 2014, 30, 523–530. [Google Scholar] [CrossRef] [PubMed]
- Vejnar, C.E.; Zdobnov, E.M. MiRmap: Comprehensive prediction of microRNA target repression strength. Nucleic Acids Res. 2012, 40, 11673–11683. [Google Scholar] [CrossRef]
- Hu, S.C.; Lan, C.E. High-glucose environment disturbs the physiologic functions of keratinocytes: Focusing on diabetic wound healing. J. Dermatol. Sci. 2016, 84, 121–127. [Google Scholar] [CrossRef]
- Thimmappaiah Jagadeesh, A.; Prakash, P.Y.; Karthik Rao, N.; Ramya, V. Culture characterization of the skin microbiome in Type 2 diabetes mellitus: A focus on the role of innate immunity. Diabetes Res. Clin. Pract. 2017, 134, 1–7. [Google Scholar] [CrossRef]
- Chehoud, C.; Rafail, S.; Tyldsley, A.S.; Seykora, J.T.; Lambris, J.D.; Grice, E.A. Complement modulates the cutaneous microbiome and inflammatory milieu. Proc. Natl. Acad. Sci. USA 2013, 110, 15061–15066. [Google Scholar] [CrossRef] [Green Version]
- Touch, S.; Clement, K.; Andre, S. T Cell Populations and Functions Are Altered in Human Obesity and Type 2 Diabetes. Curr. Diabetes Rep. 2017, 17, 81. [Google Scholar] [CrossRef] [PubMed]
- Huang da, W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Muller, L.M.; Gorter, K.J.; Hak, E.; Goudzwaard, W.L.; Schellevis, F.G.; Hoepelman, A.I.; Rutten, G.E. Increased risk of common infections in patients with type 1 and type 2 diabetes mellitus. Clin. Infect. Dis. 2005, 41, 281–288. [Google Scholar] [CrossRef] [PubMed]
- Hine, J.L.; de Lusignan, S.; Burleigh, D.; Pathirannehelage, S.; McGovern, A.; Gatenby, P.; Jones, S.; Jiang, D.; Williams, J.; Elliot, A.J.; et al. Association between glycaemic control and common infections in people with Type 2 diabetes: A cohort study. Diabet. Med. 2017, 34, 551–557. [Google Scholar] [CrossRef] [PubMed]
- Lima, A.L.; Illing, T.; Schliemann, S.; Elsner, P. Cutaneous Manifestations of Diabetes Mellitus: A Review. Am. J. Clin. Dermatol. 2017, 18, 541–553. [Google Scholar] [CrossRef] [PubMed]
- Schommer, N.N.; Gallo, R.L. Structure and function of the human skin microbiome. Trends Microbiol. 2013, 21, 660–668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byrd, A.L.; Belkaid, Y.; Segre, J.A. The human skin microbiome. Nat. Rev. Microbiol. 2018, 16, 143–155. [Google Scholar] [CrossRef]
- Grice, E.A.; Snitkin, E.S.; Yockey, L.J.; Bermudez, D.M.; Liechty, K.W.; Segre, J.A. Longitudinal shift in diabetic wound microbiota correlates with prolonged skin defense response. Proc. Natl. Acad. Sci. USA 2010, 107, 14799–14804. [Google Scholar] [CrossRef] [Green Version]
- Redel, H.; Gao, Z.; Li, H.; Alekseyenko, A.V.; Zhou, Y.; Perez-Perez, G.I.; Weinstock, G.; Sodergren, E.; Blaser, M.J. Quantitation and composition of cutaneous microbiota in diabetic and nondiabetic men. J. Infect. Dis. 2013, 207, 1105–1114. [Google Scholar] [CrossRef]
- Strom, A.; Bruggemann, J.; Ziegler, I.; Jeruschke, K.; Weiss, J.; Al-Hasani, H.; Roden, M.; Ziegler, D. Pronounced reduction of cutaneous Langerhans cell density in recently diagnosed type 2 diabetes. Diabetes 2014, 63, 1148–1153. [Google Scholar] [CrossRef]
- Miller, L.S.; Modlin, R.L. Human keratinocyte Toll-like receptors promote distinct immune responses. J. Investig. Dermatol. 2007, 127, 262–263. [Google Scholar] [CrossRef] [PubMed]
- Takaoka, A.; Yanai, H. Interferon signalling network in innate defence. Cell. Microbiol. 2006, 8, 907–922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, P.L.; Lombardi, G.; Foster, G.R. Type I interferons and the innate immune response—more than just antiviral cytokines. Mol. Immunol. 2005, 42, 869–877. [Google Scholar] [CrossRef] [PubMed]
- Lan, C.C.; Wu, C.S.; Huang, S.M.; Kuo, H.Y.; Wu, I.H.; Liang, C.W.; Chen, G.S. High-glucose environment reduces human beta-defensin-2 expression in human keratinocytes: Implications for poor diabetic wound healing. Br. J. Dermatol. 2012, 166, 1221–1229. [Google Scholar] [CrossRef] [PubMed]
- Rebane, A.; Zimmermann, M.; Aab, A.; Baurecht, H.; Koreck, A.; Karelson, M.; Abram, K.; Metsalu, T.; Pihlap, M.; Meyer, N.; et al. Mechanisms of IFN-gamma-induced apoptosis of human skin keratinocytes in patients with atopic dermatitis. J. Allergy Clin Immunol. 2012, 129, 1297–1306. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.Y.; Liu, Q.; Yang, H.; Tan, Q.; Gan, L.Q.; Ren, F.L.; Wang, H. OSMR gene effect on the pathogenesis of chronic autoimmune Urticaria via the JAK/STAT3 pathway. Mol. Med. 2018, 24, 28. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.S.; Jividen, K.; Spencer, A.; Dworak, N.; Ni, L.; Oostdyk, L.T.; Chatterjee, M.; Kusmider, B.; Reon, B.; Parlak, M.; et al. Ubiquitin Modification by the E3 Ligase/ADP-Ribosyltransferase Dtx3L/Parp9. Mol. Cell 2017, 66, 503–516. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Sun, Y.; Zhang, L.; Liu, H. Effects of DTX3L on the cell proliferation, adhesion, and drug resistance of multiple myeloma cells. Tumour Biol. 2017, 39, 39. [Google Scholar] [CrossRef] [PubMed]
- Bachmann, S.B.; Frommel, S.C.; Camicia, R.; Winkler, H.C.; Santoro, R.; Hassa, P.O. DTX3L and ARTD9 inhibit IRF1 expression and mediate in cooperation with ARTD8 survival and proliferation of metastatic prostate cancer cells. Mol. Cancer 2014, 13, 125. [Google Scholar] [CrossRef] [Green Version]
- Thang, N.D.; Yajima, I.; Kumasaka, M.Y.; Iida, M.; Suzuki, T.; Kato, M. Deltex-3-like (DTX3L) stimulates metastasis of melanoma through FAK/PI3K/AKT but not MEK/ERK pathway. Oncotarget 2015, 6, 14290–14299. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Mao, D.; Roswit, W.T.; Jin, X.; Patel, A.C.; Patel, D.A.; Agapov, E.; Wang, Z.; Tidwell, R.M.; Atkinson, J.J.; et al. PARP9-DTX3L ubiquitin ligase targets host histone H2BJ and viral 3C protease to enhance interferon signaling and control viral infection. Nat. Immunol. 2015, 16, 1215–1227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richmond, J.M.; Harris, J.E. Immunology and skin in health and disease. Cold Spring Harb. Perspect Med. 2014, 4, a015339. [Google Scholar] [CrossRef] [PubMed]
- Choi, D.; Park, E.; Jung, E.; Seong, Y.J.; Yoo, J.; Lee, E.; Hong, M.; Lee, S.; Ishida, H.; Burford, J.; et al. Laminar flow downregulates Notch activity to promote lymphatic sprouting. J. Clin. InvestIG. 2017, 127, 1225–1240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jian, Q.; An, Q.; Zhu, D.; Hui, K.; Liu, Y.; Chi, S.; Li, C. MicroRNA 340 is involved in UVB-induced dendrite formation through the regulation of RhoA expression in melanocytes. Mol. Cell. Biol. 2014, 34, 3407–3420. [Google Scholar] [CrossRef]
- Yan, J.; Qin, Y.; Yu, J.; Peng, Q.; Chen, X. MiR-340/iASPP axis affects UVB-mediated retinal pigment epithelium (RPE) cell damage. J. Photochem. Photobiol. B 2018, 186, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Hou, T.; Liao, J.; Zhang, C.; Sun, C.; Li, X.; Wang, G. Elevated expression of miR-146, miR-139 and miR-340 involved in regulating Th1/Th2 balance with acute exposure of fine particulate matter in mice. Int. Immunopharmacol. 2018, 54, 68–77. [Google Scholar] [CrossRef]
- Bian, J.; Liu, R.; Fan, T.; Liao, L.; Wang, S.; Geng, W.; Wang, T.; Shi, W.; Ruan, Q. miR-340 Alleviates Psoriasis in Mice through Direct Targeting of IL-17A. J. Immunol. 2018, 201, 1412–1420. [Google Scholar] [CrossRef]
- Carreras-Badosa, G.; Bonmati, A.; Ortega, F.J.; Mercader, J.M.; Guindo-Martinez, M.; Torrents, D.; Prats-Puig, A.; Martinez-Calcerrada, J.M.; Platero-Gutierrez, E.; De Zegher, F.; et al. Altered Circulating miRNA Expression Profile in Pregestational and Gestational Obesity. J. Clin. Endocrinol. Metab. 2015, 100, E1446–E1456. [Google Scholar] [CrossRef] [Green Version]
- Giardina, S.; Hernandez-Alonso, P.; Diaz-Lopez, A.; Salas-Huetos, A.; Salas-Salvado, J.; Bullo, M. Changes in circulating miRNAs in healthy overweight and obese subjects: Effect of diet composition and weight loss. Clin. Nutr. 2017. [Google Scholar] [CrossRef]
- Li, S.; Zhang, R.; Yuan, Y.; Yi, S.; Chen, Q.; Gong, L.; Liu, J.; Ding, F.; Cao, Z.; Gu, X. MiR-340 Regulates Fibrinolysis and Axon Regrowth Following Sciatic Nerve Injury. Mol. Neurobiol. 2017, 54, 4379–4389. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cluster | Score (Density *#Nodes) | Nodes | Edges | Node IDs |
|---|---|---|---|---|
| 1 | 20.273 | 23 | 223 | PLSCR1, IFITM1, PARP12, STAT1, IRF9, XAF1, DDX58, DHX58, OAS2, IFI44L, MX1, SP110, HERC6, PARP9, MX2, USP18, IFI44, ISG15, DDX60, EPSTI1, OAS3, IFI6, SAMD9 |
| 2 | 4.4 | 11 | 22 | TOR4A, CXCL5, FGF2, CXCR2, LGALS3BP, CXCL1, MMP7, PPBP, MMP1, CXCL3, CXCR1 |
| 3 | 4 | 17 | 32 | MITF, COL8A2, TEK, ITGA4, COL5A3, COL6A2, ITGB2, EDNRA, ITGAL, EGF, NTSR1, TIMP3, LYN, COL9A2, VAV1, HRH1, PLCB1 |
| 4 | 3.333 | 4 | 5 | KDM6A, SMARCA2, NAP1L4, HIST2H2AC |
| 5 | 3.333 | 4 | 5 | RRAD, PRKX, RTN4RL1, LGR6 |
| 6 | 3 | 3 | 3 | SAMHD1, HLA-A, HLA-C |
| 7 | 3 | 3 | 3 | GBP6, B2M, TRIM2 |
| 8 | 3 | 3 | 3 | ALDH1A3, DIP2C, ACOXL |
| 9 | 3 | 5 | 6 | ACACA, CARS, DPYSL4, SHMT1, CYB5R2 |
| Biological Process | Gene Count | False Discovery Rate |
|---|---|---|
| Response to virus | 14 | 7.09 × 10−19 |
| Defense response to virus | 13 | 7.09 × 10−19 |
| Type I interferon signaling pathway | 11 | 7.09 × 10−19 |
| Cellular response to type I interferon | 11 | 7.09 × 10−19 |
| Defense response | 16 | 1.10 × 10−11 |
| Innate immune response | 14 | 3.42 × 10−11 |
| Immune system process | 17 | 6.53 × 10−11 |
| Immune response | 15 | 9.23 × 10−11 |
| Response to cytokine | 12 | 2.49 × 10−10 |
| Negative regulation of multi-organism process | 7 | 9.85 × 10−8 |
| miRNA | Fold Change (DM-HEK/N-HEK) | Putative Target | miRmap Score | TargetScan | miRDB |
|---|---|---|---|---|---|
| hsa-miR-2116-3p | 2.41 | DTX3L | 98.87 | + | − |
| hsa-miR-296-3p | 2.30 | OAS2 | 97.82 | − | − |
| hsa-miR-340-3p | 2.00 | DTX3L | 97.39 | + | + |
| hsa-miR-4642 | 9.17 | OAS2 | 97.20 | − | − |
| hsa-miR-5010-3p | 2.97 | EPSTI1 | 97.49 | + | − |
| hsa-miR-548b-5p | 2.24 | PARP9 | 97.23 | − | + |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, E.-S.; Chang, W.-A.; Chen, Y.-Y.; Wu, L.-Y.; Chen, Y.-J.; Kuo, P.-L. Deduction of Novel Genes Potentially Involved in Keratinocytes of Type 2 Diabetes Using Next-Generation Sequencing and Bioinformatics Approaches. J. Clin. Med. 2019, 8, 73. https://doi.org/10.3390/jcm8010073
Lin E-S, Chang W-A, Chen Y-Y, Wu L-Y, Chen Y-J, Kuo P-L. Deduction of Novel Genes Potentially Involved in Keratinocytes of Type 2 Diabetes Using Next-Generation Sequencing and Bioinformatics Approaches. Journal of Clinical Medicine. 2019; 8(1):73. https://doi.org/10.3390/jcm8010073
Chicago/Turabian StyleLin, En-Shyh, Wei-An Chang, Yang-Yi Chen, Ling-Yu Wu, Yi-Jen Chen, and Po-Lin Kuo. 2019. "Deduction of Novel Genes Potentially Involved in Keratinocytes of Type 2 Diabetes Using Next-Generation Sequencing and Bioinformatics Approaches" Journal of Clinical Medicine 8, no. 1: 73. https://doi.org/10.3390/jcm8010073