Molecular Targets in Hepatocarcinogenesis and Implications for Therapy




Abstract
:1. Introduction
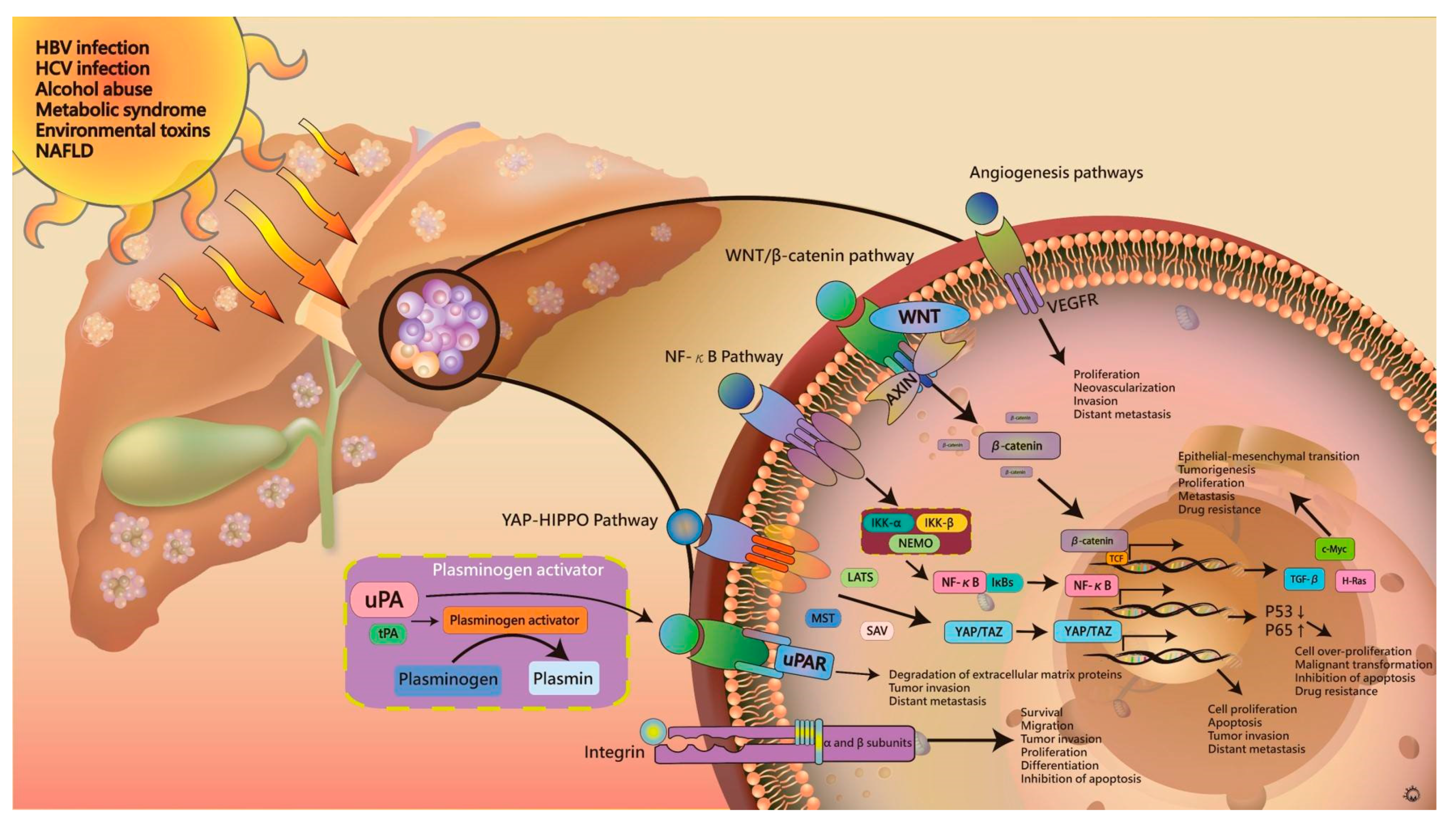
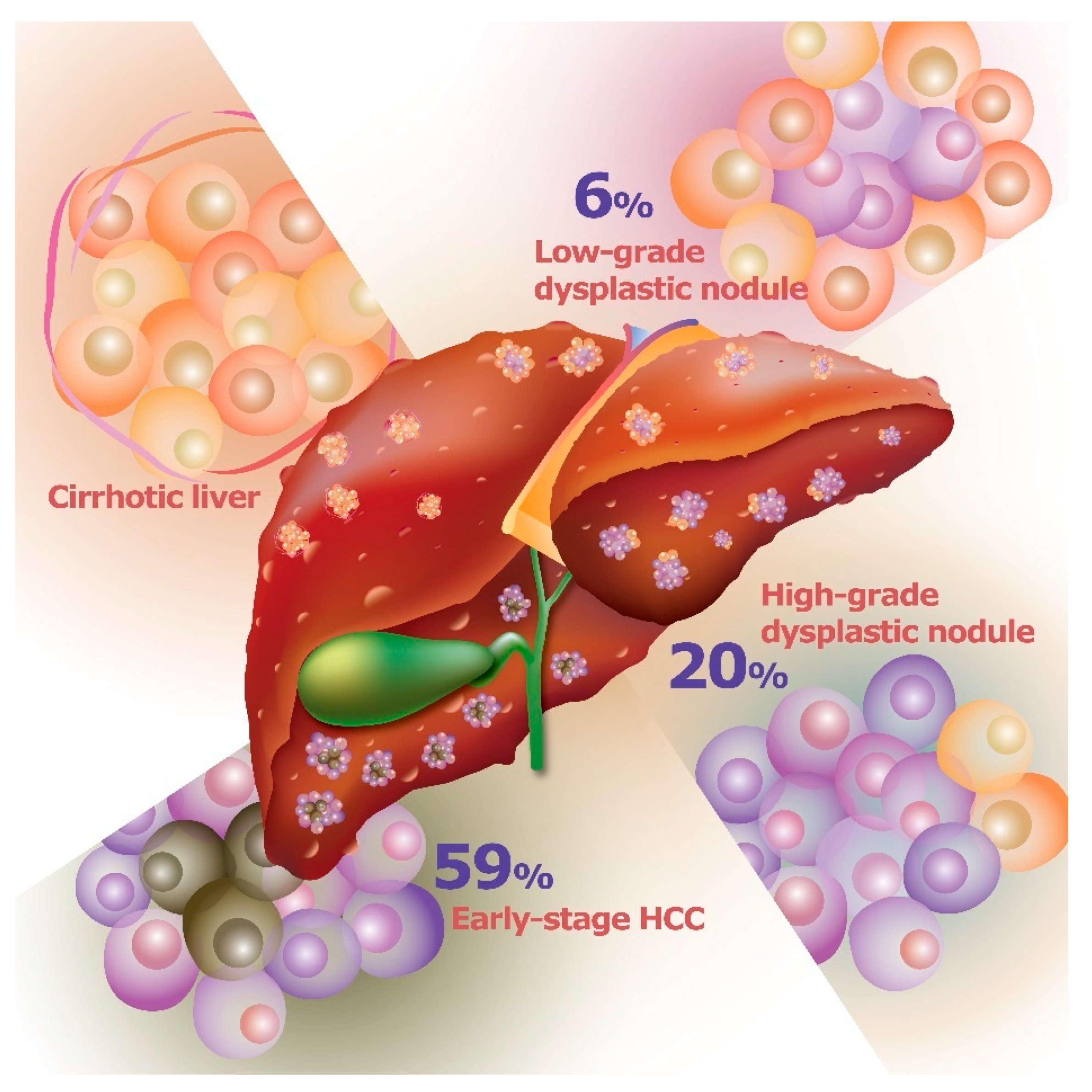
2. Pathogenesis of Hepatocellular Carcinoma
3. Telomere Maintenance
4. HBV and HCV Infections
5. Signaling Pathways in the Tumor Microenvironment
5.1. WNT/β-Catenin Signaling Pathway
5.2. NF-κB Pathway
5.3. Yes-Associated Protein (YAP)-Hippo Pathway
5.4. Angiogenesis Pathways
5.5. Plasminogen Activator (PA)
5.6. Integrin
6. Farnesoid X Receptor in Hepatocarcinogenesis
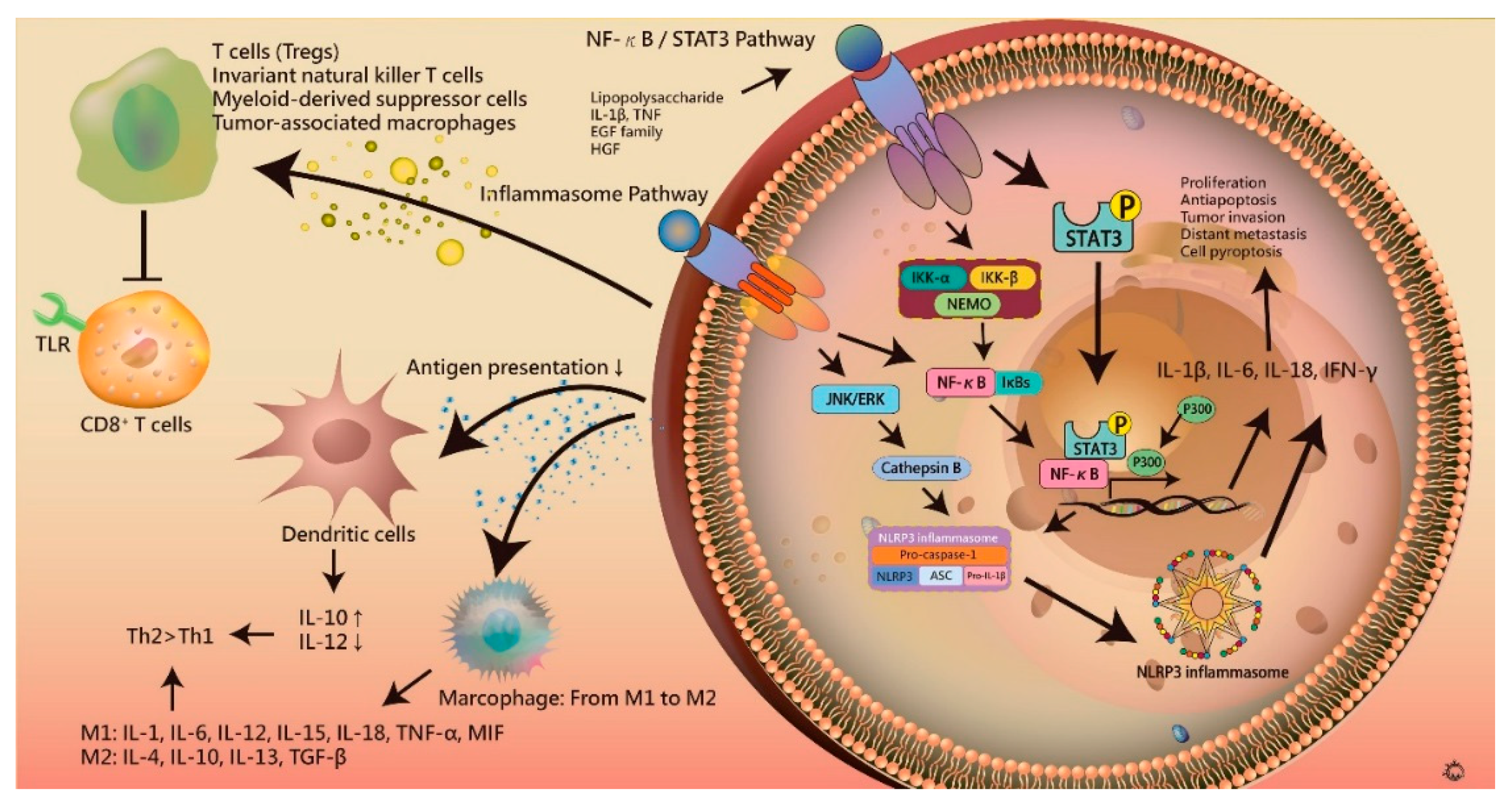
7. Inflammation in Hepatocarcinogenesis
7.1. Inflammasome
7.2. NF-κB and STAT3 Signaling in Hepatocarcinogenesis
7.3. Immune Escape
8. The Microbiota and HCC
8.1. Targeting Probiotics in HCC
8.2. Targeting Gut Microbiota in HCC
9. Major Therapeutic Strategies for Hepatocarcinogenesis
9.1. Anti-Angiogenesis
9.1.1. Bevacizumab
9.1.2. Sunitinib
9.1.3. Apatinib
9.1.4. Linifanib
9.1.5. Thalidomide
9.2. Ras/Raf/Mek/Erk Signaling
9.3. PI3K/Akt/mTOR Signaling
9.3.1. Sirolimus
9.3.2. Everolimus
9.4. Wnt/β-Catenin Signaling
9.5. NF-κB Signaling
9.6. Immunotargeting Drugs
9.7. Inflammasome-Targeting Therapy
9.8. Immunotherapy for HCC
10. Conclusions and Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Torre, L.A.; Siegel, R.L.; Ward, E.M.; Jemal, A. Global cancer incidence and mortality rates and trends—An update. Cancer Epidemiol. Biomark. Prev. 2016, 25, 16–27. [Google Scholar] [CrossRef] [PubMed]
- Davila, J.A.; Morgan, R.O.; Shaib, Y.; McGlynn, K.A.; El-Serag, H.B. Hepatitis C infection and the increasing incidence of hepatocellular carcinoma: A population-based study. Gastroenterology 2004, 127, 1372–1380. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.D.; Harmsen, W.S.; Slettedahl, S.W.; Chaiteerakij, R.; Enders, F.T.; Therneau, T.M.; Orsini, L.; Kim, W.R.; Roberts, L.R. Factors that affect risk for hepatocellular carcinoma and effects of surveillance. Clin. Gastroenterol. Hepatol. 2011, 9, 617–623. [Google Scholar] [CrossRef] [PubMed]
- Beasley, R.P. Hepatitis B virus. The major etiology of hepatocellular carcinoma. Cancer 1988, 61, 1942–1956. [Google Scholar] [CrossRef]
- Yu, S.Z. Primary prevention of hepatocellular carcinoma. J. Gastroenterol. Hepatol. 1995, 10, 674–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghebranious, N.; Sell, S. Hepatitis B injury, male gender, aflatoxin, and p53 expression each contribute to hepatocarcinogenesis in transgenic mice. Hepatology 1998, 27, 383–391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, J.F.; Jeng, J.E.; Chuang, L.Y.; Ho, M.S.; Ko, Y.C.; Lin, Z.Y.; Hsieh, M.Y.; Chen, S.C.; Chuang, W.L.; Wang, L.Y.; et al. Habitual betel quid chewing as a risk factor for cirrhosis: A case-control study. Medicine 2003, 82, 365–372. [Google Scholar] [CrossRef] [PubMed]
- Tsai, J.F.; Chuang, L.Y.; Jeng, J.E.; Ho, M.S.; Hsieh, M.Y.; Lin, Z.Y.; Wang, L.Y. Betel quid chewing as a risk factor for hepatocellular carcinoma: A case-control study. Br. J. Cancer 2001, 84, 709–713. [Google Scholar] [CrossRef] [PubMed]
- Chu, Y.J.; Yang, H.I.; Wu, H.C.; Liu, J.; Wang, L.Y.; Lu, S.N.; Lee, M.H.; Jen, C.L.; You, S.L.; Santella, R.M.; et al. Aflatoxin B1 exposure increases the risk of cirrhosis and hepatocellular carcinoma in chronic hepatitis B virus carriers. Int. J. Cancer 2017, 141, 711–720. [Google Scholar] [CrossRef] [PubMed]
- Bressac, B.; Kew, M.; Wands, J.; Ozturk, M. Selective G to T mutations of p53 gene in hepatocellular carcinoma from southern Africa. Nature 1991, 350, 429–431. [Google Scholar] [CrossRef] [PubMed]
- Unsal, H.; Yakicier, C.; Marcais, C.; Kew, M.; Volkmann, M.; Zentgraf, H.; Isselbacher, K.J.; Ozturk, M. Genetic heterogeneity of hepatocellular carcinoma. Proc. Natl. Acad. Sci. USA 1994, 91, 822–826. [Google Scholar] [CrossRef] [PubMed]
- Yan, R.Q.; Su, J.J.; Huang, D.R.; Gan, Y.C.; Yang, C.; Huang, G.H. Human hepatitis B virus and hepatocellular carcinoma. II. Experimental induction of hepatocellular carcinoma in tree shrews exposed to hepatitis B virus and aflatoxin B1. J. Cancer Res. Clin. Oncol. 1996, 122, 289–295. [Google Scholar] [CrossRef] [PubMed]
- Marquardt, J.U.; Andersen, J.B.; Thorgeirsson, S.S. Functional and genetic deconstruction of the cellular origin in liver cancer. Nat. Rev. Cancer 2015, 15, 653–667. [Google Scholar] [CrossRef] [PubMed]
- Alizadeh, A.A.; Aranda, V.; Bardelli, A.; Blanpain, C.; Bock, C.; Borowski, C.; Caldas, C.; Califano, A.; Doherty, M.; Elsner, M.; et al. Toward understanding and exploiting tumor heterogeneity. Nat. Med. 2015, 21, 846–853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schulze, K. Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nat. Genet. 2015, 47, 505–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guichard, C.; Amaddeo, G.; Imbeaud, S.; Ladeiro, Y.; Pelletier, L.; Maad, I.B.; Calderaro, J.; Bioulac-Sage, P.; Letexier, M.; Degos, F.; et al. Integrated analysis of somatic mutations and focal copy-number changes identifies key genes and pathways in hepatocellular carcinoma. Nat. Genet. 2012, 44, 694–698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartmann, D.; Srivastava, U.; Thaler, M.; Kleinhans, K.N.; N’Kontchou, G.; Scheffold, A.; Bauer, K.; Kratzer, R.F.; Kloos, N.; Katz, S.F.; et al. Telomerase gene mutations are associated with cirrhosis formation. Hepatology 2011, 53, 1608–1617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calado, R.T.; Brudno, J.; Mehta, P.; Kovacs, J.J.; Wu, C.; Zago, M.A.; Chanock, S.J.; Boyer, T.D.; Young, N.S. Constitutional telomerase mutations are genetic risk factors for cirrhosis. Hepatology 2011, 53, 1600–1607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rudolph, K.L.; Chang, S.; Millard, M.; Schreiber-Agus, N.; DePinho, R.A. Inhibition of experimental liver cirrhosis in mice by telomerase gene delivery. Science 2000, 287, 1253–1258. [Google Scholar] [CrossRef] [PubMed]
- Wege, H.; Brummendorf, T.H. Telomerase activation in liver regeneration and hepatocarcinogenesis: Dr. Jekyll or Mr. Hyde? Curr. Stem Cell Res. Ther. 2007, 2, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Wiemann, S.U.; Satyanarayana, A.; Tsahuridu, M.; Tillmann, H.L.; Zender, L.; Klempnauer, J.; Flemming, P.; Franco, S.; Blasco, M.A.; Manns, M.P.; et al. Hepatocyte telomere shortening and senescence are general markers of human liver cirrhosis. FASEB J. 2002, 16, 935–942. [Google Scholar] [CrossRef] [PubMed]
- Farazi, P.A.; Glickman, J.; Jiang, S.; Yu, A.; Rudolph, K.L.; DePinho, R.A. Differential impact of telomere dysfunction on initiation and progression of hepatocellular carcinoma. Cancer Res. 2003, 63, 5021–5027. [Google Scholar] [PubMed]
- Nault, J.C.; Calderaro, J.; Di Tommaso, L.; Balabaud, C.; Zafrani, E.S.; Bioulac-Sage, P.; Roncalli, M.; Zucman-Rossi, J. Telomerase reverse transcriptase promoter mutation is an early somatic genetic alteration in the transformation of premalignant nodules in hepatocellular carcinoma on cirrhosis. Hepatology 2014, 60, 1983–1992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahn, S.M.; Jang, S.J.; Shim, J.H.; Kim, D.; Hong, S.M.; Sung, C.O.; Baek, D.; Haq, F.; Ansari, A.A.; Lee, S.Y.; et al. Genomic portrait of resectable hepatocellular carcinomas: Implications of RB1 and FGF19 aberrations for patient stratification. Hepatology 2014, 60, 1972–1982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nault, J.C.; Mallet, M.; Pilati, C.; Calderaro, J.; Bioulac-Sage, P.; Laurent, C.; Laurent, A.; Cherqui, D.; Balabaud, C.; Zucman-Rossi, J. High frequency of telomerase reverse-transcriptase promoter somatic mutations in hepatocellular carcinoma and preneoplastic lesions. Nat. Commun. 2013, 4, 2218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Block, T.M.; Mehta, A.S.; Fimmel, C.J.; Jordan, R. Molecular viral oncology of hepatocellular carcinoma. Oncogene 2003, 22, 5093–5107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tokino, T.; Tamura, H.; Hori, N.; Matsubara, K. Chromosome deletions associated with hepatitis B virus integration. Virology 1991, 185, 879–882. [Google Scholar] [CrossRef]
- Murakami, Y.; Saigo, K.; Takashima, H.; Minami, M.; Okanoue, T.; Brechot, C.; Paterlini-Brechot, P. Large scaled analysis of hepatitis B virus (HBV) DNA integration in HBV related hepatocellular carcinomas. Gut 2005, 54, 1162–1168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarn, C.; Lee, S.; Hu, Y.; Ashendel, C.; Andrisani, O.M. Hepatitis B virus X protein differentially activates RAS-RAF-MAPK and JNK pathways in X-transforming versus non-transforming AML12 hepatocytes. J. Biol. Chem. 2001, 276, 34671–34680. [Google Scholar] [CrossRef] [PubMed]
- Nijhara, R.; Jana, S.S.; Goswami, S.K.; Rana, A.; Majumdar, S.S.; Kumar, V.; Sarkar, D.P. Sustained activation of mitogen-activated protein kinases and activator protein 1 by the hepatitis B virus X protein in mouse hepatocytes in vivo. J. Virol. 2001, 75, 10348–10358. [Google Scholar] [CrossRef] [PubMed]
- Nijhara, R.; Jana, S.S.; Goswami, S.K.; Kumar, V.; Sarkar, D.P. An internal segment (residues 58–119) of the hepatitis B virus X protein is sufficient to activate MAP kinase pathways in mouse liver. FEBS Lett. 2001, 504, 59–64. [Google Scholar] [CrossRef]
- Feitelson, M.A.; Sun, B.; Satiroglu Tufan, N.L.; Liu, J.; Pan, J.; Lian, Z. Genetic mechanisms of hepatocarcinogenesis. Oncogene 2002, 21, 2593–2604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, D.Y.; Moon, H.B.; Son, J.K.; Jeong, S.; Yu, S.L.; Yoon, H.; Han, Y.M.; Lee, C.S.; Park, J.S.; Lee, C.H.; et al. Incidence of hepatocellular carcinoma in transgenic mice expressing the hepatitis B virus X-protein. J. Hepatol. 1999, 31, 123–132. [Google Scholar] [CrossRef]
- Kim, C.-M.; Koike, K.; Saito, I.; Miyamura, T.; Jay, G. HBx gene of hepatitis B virus induces liver cancer in transgenic mice. Nature 1991, 351, 317–320. [Google Scholar] [CrossRef] [PubMed]
- Wieland, S.; Thimme, R.; Purcell, R.H.; Chisari, F.V. Genomic analysis of the host response to hepatitis B virus infection. Proc. Natl. Acad. Sci. USA 2004, 101, 6669–6674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rehermann, B.; Nascimbeni, M. Immunology of hepatitis B virus and hepatitis C virus infection. Nat. Rev. Immunol. 2005, 5, 215–229. [Google Scholar] [CrossRef] [PubMed]
- Farazi, P.A.; DePinho, R.A. Hepatocellular carcinoma pathogenesis: From genes to environment. Nat. Rev. Immunol. 2006, 6, 674–687. [Google Scholar] [CrossRef] [PubMed]
- Lindenbach, B.D.; Rice, C.M. Unravelling hepatitis C virus replication from genome to function. Nature 2005, 436, 933–938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindenbach, B.D.; Evans, M.J.; Syder, A.J.; Wolk, B.; Tellinghuisen, T.L.; Liu, C.C.; Maruyama, T.; Hynes, R.O.; Burton, D.R.; McKeating, J.A.; et al. Complete replication of hepatitis C virus in cell culture. Science 2005, 309, 623–626. [Google Scholar] [CrossRef] [PubMed]
- Wakita, T.; Pietschmann, T.; Kato, T.; Date, T.; Miyamoto, M.; Zhao, Z.; Murthy, K.; Habermann, A.; Krausslich, H.G.; Mizokami, M.; et al. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat. Med. 2005, 11, 791–796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vilchez, V.; Turcios, L.; Marti, F.; Gedaly, R. Targeting Wnt/beta-catenin pathway in hepatocellular carcinoma treatment. World J. Gastroenterol. 2016, 22, 823–832. [Google Scholar] [CrossRef] [PubMed]
- Harada, N.; Oshima, H.; Katoh, M.; Tamai, Y.; Oshima, M.; Taketo, M.M. Hepatocarcinogenesis in mice with beta-catenin and Ha-ras gene mutations. Cancer Res. 2004, 64, 48–54. [Google Scholar] [CrossRef] [PubMed]
- Clevers, H.; Nusse, R. Wnt/beta-catenin signaling and disease. Cell 2012, 149, 1192–1205. [Google Scholar] [CrossRef] [PubMed]
- Qiao, L.; Zhang, H.; Yu, J.; Francisco, R.; Dent, P.; Ebert, M.P.; Rocken, C.; Farrell, G. Constitutive activation of NF-kappaB in human hepatocellular carcinoma: Evidence of a cytoprotective role. Hum. Gene Ther. 2006, 17, 280–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papa, S.; Bubici, C.; Zazzeroni, F.; Franzoso, G. Mechanisms of liver disease: Cross-talk between the NF-kappaB and JNK pathways. Biol. Chem. 2009, 390, 965–976. [Google Scholar] [CrossRef] [PubMed]
- Barre, B.; Perkins, N.D. A cell cycle regulatory network controlling NF-kappaB subunit activity and function. EMBO J. 2007, 26, 4841–4855. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.F.; Witzel, I.I.; Perkins, N.D. p53-dependent regulation of mitochondrial energy production by the RelA subunit of NF-kappaB. Cancer Res. 2011, 71, 5588–5597. [Google Scholar] [CrossRef] [PubMed]
- Alves, A.; Mamede, A.C.; Alves, M.; Oliveira, P.F.; Rocha, S.M.; Botelho, F.; Maia, C.J. Glycolysis inhibition as a strategy for hepatocellular carcinoma treatment? Curr. Cancer Drug Targets 2018. [Google Scholar] [CrossRef] [PubMed]
- Lei, S.; Yang, J.; Chen, C.; Sun, J.; Yang, L.; Tang, H.; Yang, T.; Chen, A.; Zhao, H.; Li, Y.; et al. FLIP(L) is critical for aerobic glycolysis in hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2016, 35, 79. [Google Scholar] [CrossRef] [PubMed]
- Asgharzadeh, M.R.; Barar, J.; Pourseif, M.M.; Eskandani, M.; Jafari Niya, M.; Mashayekhi, M.R.; Omidi, Y. Molecular machineries of pH dysregulation in tumor microenvironment: Potential targets for cancer therapy. Bioimpacts 2017, 7, 115–133. [Google Scholar] [CrossRef] [PubMed]
- Kretzmann, N.A.; Chiela, E.; Matte, U.; Marroni, N.; Marroni, C.A. N-acetylcysteine improves antitumoural response of Interferon alpha by NF-kB downregulation in liver cancer cells. Comp. Hepatol. 2012, 11, 4. [Google Scholar] [CrossRef] [PubMed]
- Eltzschig, H.K.; Carmeliet, P. Hypoxia and inflammation. N. Engl. J. Med. 2011, 364, 656–665. [Google Scholar] [CrossRef] [PubMed]
- Nizet, V.; Johnson, R.S. Interdependence of hypoxic and innate immune responses. Nat. Rev. Immunol. 2009, 9, 609–617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DiGiacomo, J.W.; Gilkes, D.M. Tumor hypoxia as an enhancer of inflammation-mediated metastasis: Emerging therapeutic strategies. Target. Oncol. 2018, 13, 157–173. [Google Scholar] [CrossRef] [PubMed]
- Barry, E.R.; Morikawa, T.; Butler, B.L.; Shrestha, K.; de la Rosa, R.; Yan, K.S.; Fuchs, C.S.; Magness, S.T.; Smits, R.; Ogino, S.; et al. Restriction of intestinal stem cell expansion and the regenerative response by YAP. Nature 2013, 493, 106–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizvi, S.; Fischbach, S.R.; Bronk, S.F.; Hirsova, P.; Krishnan, A.; Dhanasekaran, R.; Smadbeck, J.B.; Smoot, R.L.; Vasmatzis, G.; Gores, G.J. YAP-associated chromosomal instability and cholangiocarcinoma in mice. Oncotarget 2018, 9, 5892–5905. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, H.; Higashi, T.; Yokoyama, N.; Kaida, T.; Sakamoto, K.; Fukushima, Y.; Ishimoto, T.; Kuroki, H.; Nitta, H.; Hashimoto, D.; et al. An imbalance in TAZ and YAP expression in hepatocellular carcinoma confers cancer stem cell-like behaviors contributing to disease progression. Cancer Res. 2015, 75, 4985–4997. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Liu, Y.; Jiang, X.W.; Li, W.F.; Guo, G.; Gong, J.P.; Ding, X. Clinicopathological and prognostic significance of Yes-associated protein expression in hepatocellular carcinoma and hepatic cholangiocarcinoma. Tumor Biol. 2016, 37, 13499–13508. [Google Scholar] [CrossRef] [PubMed]
- Camargo, F.D.; Gokhale, S.; Johnnidis, J.B.; Fu, D.; Bell, G.W.; Jaenisch, R.; Brummelkamp, T.R. YAP1 increases organ size and expands undifferentiated progenitor cells. Curr. Biol. 2007, 17, 2054–2060. [Google Scholar] [CrossRef] [PubMed]
- Yin, F.; Yu, J.; Zheng, Y.; Chen, Q.; Zhang, N.; Pan, D. Spatial organization of Hippo signaling at the plasma membrane mediated by the tumor suppressor Merlin/NF2. Cell 2013, 154, 1342–1355. [Google Scholar] [CrossRef] [PubMed]
- Kerbel, R.S. Tumor angiogenesis: Past, present and the near future. Carcinogenesis 2000, 21, 505–515. [Google Scholar] [CrossRef] [PubMed]
- Andisheh-Tadbir, A.; Hamzavi, M.; Rezvani, G.; Ashraf, M.J.; Fattahi, M.J.; Khademi, B.; Kamali, F. Tissue expression, serum and salivary levels of vascular endothelial growth factor in patients with HNSCC. Braz. J. Otorhinolaryngol. 2014, 80, 503–507. [Google Scholar] [CrossRef] [PubMed]
- Rahbari, N.N.; Kedrin, D.; Incio, J.; Liu, H.; Ho, W.W.; Nia, H.T.; Edrich, C.M.; Jung, K.; Daubriac, J.; Chen, I.; et al. Anti-VEGF therapy induces ECM remodeling and mechanical barriers to therapy in colorectal cancer liver metastases. Sci. Transl. Med. 2016, 8, 360ra135. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Zhang, Y.; Iwamoto, H.; Hosaka, K.; Seki, T.; Andersson, P.; Lim, S.; Fischer, C.; Nakamura, M.; Abe, M.; et al. Discontinuation of anti-VEGF cancer therapy promotes metastasis through a liver revascularization mechanism. Nat. Commun. 2016, 7, 12680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kutluer, G.; Cicek, N.M.; Moraloglu, O.; Ertargin, P.; Sarikaya, E.; Artar, I.; Erdem, O. Low VEGF expression in conceptus material and maternal serum AFP and beta-hCG levels as indicators of defective angiogenesis in first-trimester miscarriages. J. Turk. Ger. Gynecol. Assoc. 2012, 13, 111–117. [Google Scholar] [PubMed]
- Ye, S.; Mao, B.; Yang, L.; Fu, W.; Hou, J. Thrombosis recanalization by paeoniflorin through the upregulation of urokinasetype plasminogen activator via the MAPK signaling pathway. Mol. Med. Rep. 2016, 13, 4593–4598. [Google Scholar] [CrossRef] [PubMed]
- Santibanez, J.F. Urokinase type plasminogen activator and the molecular mechanisms of its regulation in cancer. Protein Pept. Lett. 2017, 24, 936–946. [Google Scholar] [CrossRef] [PubMed]
- Mauro, C.D.; Pesapane, A.; Formisano, L.; Rosa, R.; D’Amato, V.; Ciciola, P.; Servetto, A.; Marciano, R.; Orsini, R.C.; Monteleone, F.; et al. Urokinase-type plasminogen activator receptor (uPAR) expression enhances invasion and metastasis in RAS mutated tumors. Sci. Rep. 2017, 7, 9388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pavon, M.A.; Arroyo-Solera, I.; Cespedes, M.V.; Casanova, I.; Leon, X.; Mangues, R. uPA/uPAR and SERPINE1 in head and neck cancer: Role in tumor resistance, metastasis, prognosis and therapy. Oncotarget 2016, 7, 57351–57366. [Google Scholar] [CrossRef] [PubMed]
- Markl, B.; Hardt, J.; Franz, S.; Schaller, T.; Schenkirsch, G.; Kriening, B.; Hoffmann, R.; Ruth, S. Tumor budding, uPA, and PAI-1 in colorectal cancer: Update of a prospective study. Gastroenterol. Res. Pract. 2017, 2017, 6504960. [Google Scholar] [CrossRef] [PubMed]
- Andres, S.A.; Edwards, A.B.; Wittliff, J.L. Expression of urokinase-type plasminogen activator (uPA), its receptor (uPAR), and inhibitor (PAI-1) in human breast carcinomas and their clinical relevance. J. Clin. Lab. Anal. 2012, 26, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Rubina, K.A.; Sysoeva, V.Y.; Zagorujko, E.I.; Tsokolaeva, Z.I.; Kurdina, M.I.; Parfyonova, Y.V.; Tkachuk, V.A. Increased expression of uPA, uPAR, and PAI-1 in psoriatic skin and in basal cell carcinomas. Arch. Dermatol. Res. 2017, 309, 433–442. [Google Scholar] [CrossRef] [PubMed]
- Montuori, N.; Ragno, P. Role of uPA/uPAR in the modulation of angiogenesis. Chem. Immunol. Allergy 2014, 99, 105–122. [Google Scholar] [PubMed]
- Wu, Y.; Qiao, X.; Qiao, S.; Yu, L. Targeting integrins in hepatocellular carcinoma. Expert Opin. Ther. Targets 2011, 15, 421–437. [Google Scholar] [CrossRef] [PubMed]
- Weis, S.M.; Cheresh, D.A. αV integrins in angiogenesis and cancer. Cold Spring Harb. Perspect. Med. 2011, 1, a006478. [Google Scholar] [CrossRef] [PubMed]
- Soejima, Y.; Inoue, M.; Takahashi, Y.; Uozaki, H.; Sawabe, M.; Fukusato, T. Integrins αvβ6, α6β4 and α3β1 are down-regulated in cholangiolocellular carcinoma but not cholangiocarcinoma. Hepatol. Res. 2014, 44, 320–334. [Google Scholar] [CrossRef] [PubMed]
- Lijuan, W.; Liu, X.; Yu, H.; Zhou, F.; Chen, H.; Liu, Q. Integrins mediate the migration of HepG2 cells induced by low shear stress. Sheng Wu Yi Xue Gong Cheng Xue Za Zhi 2014, 31, 336–340. [Google Scholar] [PubMed]
- Tian, T.; Li, C.L.; Fu, X.; Wang, S.H.; Lu, J.; Guo, H.; Yao, Y.; Nan, K.J.; Yang, Y.J. beta1 integrin-mediated multicellular resistance in hepatocellular carcinoma through activation of the FAK/Akt pathway. J. Int. Med. Res. 2018, 46, 1311–1325. [Google Scholar] [CrossRef] [PubMed]
- Feng, T.; Yu, H.; Xia, Q.; Ma, Y.; Yin, H.; Shen, Y.; Liu, X. Cross-talk mechanism between endothelial cells and hepatocellular carcinoma cells via growth factors and integrin pathway promotes tumor angiogenesis and cell migration. Oncotarget 2017, 8, 69577–69593. [Google Scholar] [CrossRef] [PubMed]
- Kainuma, M.; Takada, I.; Makishima, M.; Sano, K. Farnesoid X receptor activation enhances transforming growth factor beta-induced epithelial-mesenchymal transition in hepatocellular carcinoma cells. Int. J. Mol. Sci. 2018, 19, 1898. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Ma, K.; Zhang, J.; Qatanani, M.; Cuvillier, J.; Liu, J.; Dong, B.; Huang, X.; Moore, D.D. Nuclear receptor-dependent bile acid signaling is required for normal liver regeneration. Science 2006, 312, 233–236. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.F.; Zhao, W.Y.; Huang, W.D. FXR and liver carcinogenesis. Acta Pharmacol. Sin. 2015, 36, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Deuschle, U.; Schuler, J.; Schulz, A.; Schluter, T.; Kinzel, O.; Abel, U.; Kremoser, C. FXR controls the tumor suppressor NDRG2 and FXR agonists reduce liver tumor growth and metastasis in an orthotopic mouse xenograft model. PLoS ONE 2012, 7, e43044. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Huang, X.; Yi, T.; Yen, Y.; Moore, D.D.; Huang, W. Spontaneous development of liver tumors in the absence of the bile acid receptor farnesoid X receptor. Cancer Res. 2007, 67, 863–867. [Google Scholar] [CrossRef] [PubMed]
- Fujino, T.; Takeuchi, A.; Maruko-Ohtake, A.; Ohtake, Y.; Satoh, J.; Kobayashi, T.; Tanaka, T.; Ito, H.; Sakamaki, R.; Kashimura, R.; et al. Critical role of farnesoid X receptor for hepatocellular carcinoma cell proliferation. J. Biochem. 2012, 152, 577–586. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.; Morimura, K.; Shah, Y.; Yang, Q.; Ward, J.M.; Gonzalez, F.J. Spontaneous hepatocarcinogenesis in farnesoid X receptor-null mice. Carcinogenesis 2007, 28, 940–946. [Google Scholar] [CrossRef] [PubMed]
- Niu, Y.; Xu, M.; Slagle, B.L.; Huang, H.; Li, S.; Guo, G.L.; Shi, G.; Qin, W.; Xie, W. Farnesoid X receptor ablation sensitizes mice to hepatitis b virus X protein-induced hepatocarcinogenesis. Hepatology 2017, 65, 893–906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Llovet, J.M.; Zucman-Rossi, J.; Pikarsky, E.; Sangro, B.; Schwartz, M.; Sherman, M.; Gores, G. Hepatocellular carcinoma. Nat. Rev. Dis. Prim. 2016, 2, 16018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gores, G.J. Apoptosis and hepatic necroinflammation. Gastroenterol. Hepatol. 2008, 4, 394–395. [Google Scholar]
- Ringelhan, M.; Pfister, D.; O’Connor, T.; Pikarsky, E.; Heikenwalder, M. The immunology of hepatocellular carcinoma. Nat. Immunol. 2018, 19, 222–232. [Google Scholar] [CrossRef] [PubMed]
- Linkermann, A.; Stockwell, B.R.; Krautwald, S.; Anders, H.J. Regulated cell death and inflammation: An auto-amplification loop causes organ failure. Nat. Rev. Immunol. 2014, 14, 759–767. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Callaway, J.B.; Ting, J.P.Y. Inflammasomes: Mechanism of action, role in disease, and therapeutics. Nat. Med. 2015, 21, 677. [Google Scholar] [CrossRef] [PubMed]
- Ting, J.P.; Duncan, J.A.; Lei, Y. How the noninflammasome NLRs function in the innate immune system. Science 2010, 327, 286–290. [Google Scholar] [CrossRef] [PubMed]
- Gross, O.; Yazdi, A.S.; Thomas, C.J.; Masin, M.; Heinz, L.X.; Guarda, G.; Quadroni, M.; Drexler, S.K.; Tschopp, J. Inflammasome activators induce interleukin-1α secretion via distinct pathways with differential requirement for the protease function of caspase-1. Immunity 2012, 36, 388–400. [Google Scholar] [CrossRef] [PubMed]
- Ghiringhelli, F.; Apetoh, L.; Tesniere, A.; Aymeric, L.; Ma, Y.; Ortiz, C.; Vermaelen, K.; Panaretakis, T.; Mignot, G.; Ullrich, E.; et al. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1beta-dependent adaptive immunity against tumors. Nat. Med. 2009, 15, 1170–1178. [Google Scholar] [CrossRef] [PubMed]
- Cassel, S.L.; Joly, S.; Sutterwala, F.S. The NLRP3 inflammasome: A sensor of immune danger signals. Semin. Immunol. 2009, 21, 194–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lukens, J.R.; Kanneganti, T.D. Beyond canonical inflammasomes: Emerging pathways in IL-1-mediated autoinflammatory disease. Semin. Immunopathol. 2014, 36, 595–609. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Fu, S.; Sun, S.; Li, Z.; Guo, B. Inflammasome activation has an important role in the development of spontaneous colitis. Mucosal Immunol. 2014, 7, 1139–1150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamkanfi, M.; Dixit, V.M. Mechanisms and functions of inflammasomes. Cell 2014, 157, 1013–1022. [Google Scholar] [CrossRef] [PubMed]
- Zitvogel, L.; Kepp, O.; Galluzzi, L.; Kroemer, G. Inflammasomes in carcinogenesis and anticancer immune responses. Nat. Immunol. 2012, 13, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity 2011, 34, 637–650. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, T.; Kawai, T. Toll-like receptor signaling pathways. Front. Immunol. 2014, 5, 461. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F.; Burns, K.; Tschopp, J. The inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef]
- Martinon, F.; Mayor, A.; Tschopp, J. The inflammasomes: Guardians of the body. Ann. Rev. Immunol. 2009, 27, 229–265. [Google Scholar] [CrossRef] [PubMed]
- Ye, Z.; Ting, J.P. NLR, the nucleotide-binding domain leucine-rich repeat containing gene family. Curr. Opin. Immunol. 2008, 20, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Bauernfeind, F.; Ablasser, A.; Bartok, E.; Kim, S.; Schmid-Burgk, J.; Cavlar, T.; Hornung, V. Inflammasomes: Current understanding and open questions. Cell. Mol. Life Sci. CMLS 2011, 68, 765–783. [Google Scholar] [CrossRef] [PubMed]
- Cayrol, C.; Girard, J.P. The IL-1-like cytokine IL-33 is inactivated after maturation by caspase-1. Proc. Natl. Acad. Sci. USA 2009, 106, 9021–9026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schroder, K.; Tschopp, J. The inflammasomes. Cell 2010, 140, 821–832. [Google Scholar] [CrossRef] [PubMed]
- Csak, T.; Ganz, M.; Pespisa, J.; Kodys, K.; Dolganiuc, A.; Szabo, G. Fatty acid and endotoxin activate inflammasomes in mouse hepatocytes that release danger signals to stimulate immune cells. Hepatology 2011, 54, 133–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boaru, S.G.; Borkham-Kamphorst, E.; Tihaa, L.; Haas, U.; Weiskirchen, R. Expression analysis of inflammasomes in experimental models of inflammatory and fibrotic liver disease. J. Inflamm. 2012, 9, 49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, G.; Karin, M. NF-κB and STAT3–key players in liver inflammation and cancer. Cell Res. 2011, 21, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Luedde, T.; Schwabe, R.F. NF-κB in the liver—Linking injury, fibrosis and hepatocellular carcinoma. Nat. Rev. Gastroenterol. Hepatol. 2011, 8, 108–118. [Google Scholar] [CrossRef] [PubMed]
- Takeda, K.; Akira, S. STAT family of transcription factors in cytokine-mediated biological responses. Cytokine Growth Factor Rev. 2000, 11, 199–207. [Google Scholar] [CrossRef]
- Hirano, T.; Ishihara, K.; Hibi, M. Roles of STAT3 in mediating the cell growth, differentiation and survival signals relayed through the IL-6 family of cytokine receptors. Oncogene 2000, 19, 2548–2556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.J.; Liao, W.T.; Wu, M.Y.; Chu, P.Y. New insights into the role of autophagy in tumor immune microenvironment. Int. J. Mol. Sci. 2017, 18, 1566. [Google Scholar] [CrossRef] [PubMed]
- Matsui, M.; Machida, S.; Itani-Yohda, T.; Akatsuka, T. Downregulation of the proteasome subunits, transporter, and antigen presentation in hepatocellular carcinoma, and their restoration by interferon-gamma. J. Gastroenterol. Hepatol. 2002, 17, 897–907. [Google Scholar] [CrossRef] [PubMed]
- Ormandy, L.A.; Hillemann, T.; Wedemeyer, H.; Manns, M.P.; Greten, T.F.; Korangy, F. Increased populations of regulatory T cells in peripheral blood of patients with hepatocellular carcinoma. Cancer Res. 2005, 65, 2457–2464. [Google Scholar] [CrossRef] [PubMed]
- Unitt, E.; Rushbrook, S.M.; Marshall, A.; Davies, S.; Gibbs, P.; Morris, L.S.; Coleman, N.; Alexander, G.J. Compromised lymphocytes infiltrate hepatocellular carcinoma: The role of T-regulatory cells. Hepatology 2005, 41, 722–730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cariani, E.; Pilli, M.; Zerbini, A.; Rota, C.; Olivani, A.; Pelosi, G.; Schianchi, C.; Soliani, P.; Campanini, N.; Silini, E.M.; et al. Immunological and molecular correlates of disease recurrence after liver resection for hepatocellular carcinoma. PLoS ONE 2012, 7, e32493. [Google Scholar] [CrossRef] [PubMed]
- Hoechst, B.; Ormandy, L.A.; Ballmaier, M.; Lehner, F.; Kruger, C.; Manns, M.P.; Greten, T.F.; Korangy, F. A new population of myeloid-derived suppressor cells in hepatocellular carcinoma patients induces CD4(+)CD25(+)Foxp3(+) T cells. Gastroenterology 2008, 135, 234–243. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.D.; Zhang, J.B.; Zhuang, P.Y.; Zhu, H.G.; Zhang, W.; Xiong, Y.Q.; Wu, W.Z.; Wang, L.; Tang, Z.Y.; Sun, H.C. High expression of macrophage colony-stimulating factor in peritumoral liver tissue is associated with poor survival after curative resection of hepatocellular carcinoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2008, 26, 2707–2716. [Google Scholar] [CrossRef] [PubMed]
- Yeung, O.W.; Lo, C.M.; Ling, C.C.; Qi, X.; Geng, W.; Li, C.X.; Ng, K.T.; Forbes, S.J.; Guan, X.Y.; Poon, R.T.; et al. Alternatively activated (M2) macrophages promote tumour growth and invasiveness in hepatocellular carcinoma. J. Hepatol. 2015, 62, 607–616. [Google Scholar] [CrossRef] [PubMed]
- Ding, T.; Xu, J.; Wang, F.; Shi, M.; Zhang, Y.; Li, S.P.; Zheng, L. High tumor-infiltrating macrophage density predicts poor prognosis in patients with primary hepatocellular carcinoma after resection. Hum. Pathol. 2009, 40, 381–389. [Google Scholar] [CrossRef] [PubMed]
- Behboudi, S.; Alisa, A.; Boswell, S.; Anastassiou, J.; Pathan, A.A.; Williams, R. Expansion of anti-AFP Th1 and Tc1 responses in hepatocellular carcinoma occur in different stages of disease. Br. J. Cancer 2010, 102, 748–753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, M.-Y.; Li, C.-J.; Hou, M.-F.; Chu, P.-Y. New insights into the role of inflammation in the pathogenesis of atherosclerosis. Int. J. Mol. Sci. 2017, 18, 2034. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.-X.; Ling, Y.; Wang, H.-Y. Role of nonresolving inflammation in hepatocellular carcinoma development and progression. NPJ Precis. Oncol. 2018, 2, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Budhu, A.; Forgues, M.; Ye, Q.H.; Jia, H.L.; He, P.; Zanetti, K.A.; Kammula, U.S.; Chen, Y.; Qin, L.X.; Tang, Z.Y.; et al. Prediction of venous metastases, recurrence, and prognosis in hepatocellular carcinoma based on a unique immune response signature of the liver microenvironment. Cancer Cell 2006, 10, 99–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hattori, E.; Okumoto, K.; Adachi, T.; Takeda, T.; Ito, J.; Sugahara, K.; Watanabe, H.; Saito, K.; Saito, T.; Togashi, H.; et al. Possible contribution of circulating interleukin-10 (IL-10) to anti-tumor immunity and prognosis in patients with unresectable hepatocellular carcinoma. Hepatol. Res. Off. J. Jpn Soc. Hepatol. 2003, 27, 309–314. [Google Scholar] [CrossRef]
- Kim, C.; Chung, S.; Yuchun, L.; Kim, M.C.; Chan, J.K.; Asada, H.H.; Kamm, R.D. In vitro angiogenesis assay for the study of cell-encapsulation therapy. Lab Chip 2012, 12, 2942–2950. [Google Scholar] [CrossRef] [PubMed]
- Won, C.; Kim, B.H.; Yi, E.H.; Choi, K.J.; Kim, E.K.; Jeong, J.M.; Lee, J.H.; Jang, J.J.; Yoon, J.H.; Jeong, W.I.; et al. Signal transducer and activator of transcription 3-mediated CD133 up-regulation contributes to promotion of hepatocellular carcinoma. Hepatology 2015, 62, 1160–1173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ray, K. Diagnosis: Programmed probiotics light up liver cancer in urine. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 429. [Google Scholar] [CrossRef] [PubMed]
- Dapito, D.H.; Mencin, A.; Gwak, G.Y.; Pradere, J.P.; Jang, M.K.; Mederacke, I.; Caviglia, J.M.; Khiabanian, H.; Adeyemi, A.; Bataller, R.; et al. Promotion of hepatocellular carcinoma by the intestinal microbiota and TLR4. Cancer Cell 2012, 21, 504–516. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; Chang, L.; Yuan, L.; Duan, Z. Nutrition status and small intestinal bacterial overgrowth in patients with virus-related cirrhosis. Asia Pac. J. Clin. Nutr. 2016, 25, 283–291. [Google Scholar] [PubMed]
- Lin, A.; Wang, G.; Zhao, H.; Zhang, Y.; Han, Q.; Zhang, C.; Tian, Z.; Zhang, J. TLR4 signaling promotes a COX-2/PGE2/STAT3 positive feedback loop in hepatocellular carcinoma (HCC) cells. Oncoimmunology 2016, 5, e1074376. [Google Scholar] [CrossRef] [PubMed]
- Tampaki, E.C.; Tampakis, A.; Alifieris, C.E.; Krikelis, D.; Pazaiti, A.; Kontos, M.; Trafalis, D.T. Efficacy and Safety of neoadjuvant treatment with bevacizumab, liposomal doxorubicin, cyclophosphamide and paclitaxel combination in locally/regionally advanced, HER2-negative, Grade III at premenopausal status breast cancer: A Phase II Study. Clin. Drug Investig. 2018. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Vacarezza, N.; Alonso, I.; Arroyo, G.; Martinez, J.; De Andres, F.; LLerena, A.; Estevez-Carrizo, F. Predictive biomarkers candidates for patients with metastatic colorectal cancer treated with bevacizumab-containing regimen. Drug Metab. Pers. Ther. 2016, 31, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Pinter, M.; Ulbrich, G.; Sieghart, W.; Kolblinger, C.; Reiberger, T.; Li, S.; Ferlitsch, A.; Muller, C.; Lammer, J.; Peck-Radosavljevic, M. Hepatocellular carcinoma: A Phase II randomized controlled double-blind trial of transarterial chemoembolization in combination with biweekly intravenous administration of bevacizumab or a placebo. Radiology 2015, 277, 903–912. [Google Scholar] [CrossRef] [PubMed]
- Pokuri, V.K.; Tomaszewski, G.M.; Ait-Oudhia, S.; Groman, A.; Khushalani, N.I.; Lugade, A.A.; Thanavala, Y.; Ashton, E.A.; Grande, C.; Fetterly, G.J.; et al. Efficacy, safety, and potential biomarkers of sunitinib and transarterial chemoembolization (TACE) combination in advanced hepatocellular carcinoma (HCC): Phase II trial. Am. J. Clin. Oncol. 2018, 41, 332–338. [Google Scholar] [CrossRef] [PubMed]
- Cheng, A.L.; Kang, Y.K.; Lin, D.Y.; Park, J.W.; Kudo, M.; Qin, S.; Chung, H.C.; Song, X.; Xu, J.; Poggi, G.; et al. Sunitinib versus sorafenib in advanced hepatocellular cancer: Results of a randomized phase III trial. J. Clin. Oncol. 2013, 31, 4067–4075. [Google Scholar] [CrossRef] [PubMed]
- Yau, T.; Chan, P.; Pang, R.; Ng, K.; Fan, S.T.; Poon, R.T. Phase 1-2 trial of PTK787/ZK222584 combined with intravenous doxorubicin for treatment of patients with advanced hepatocellular carcinoma: Implication for antiangiogenic approach to hepatocellular carcinoma. Cancer 2010, 116, 5022–5029. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.C.; Zhang, K.Z.; Chen, S.G.; Liu, W.F. Efficacy and Safety of apatinib in patients with intermediate/advanced hepatocellular carcinoma: A prospective observation study. Medicine 2018, 97, e9704. [Google Scholar] [CrossRef] [PubMed]
- Kong, Y.; Sun, L.; Hou, Z.; Zhang, Y.; Chen, P.; Cui, Y.; Zhu, X.; Song, T.; Li, Q.; Li, H.; et al. Apatinib is effective for treatment of advanced hepatocellular carcinoma. Oncotarget 2017, 8, 105596–105605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cainap, C.; Qin, S.; Huang, W.T.; Chung, I.J.; Pan, H.; Cheng, Y.; Kudo, M.; Kang, Y.K.; Chen, P.J.; Toh, H.C.; et al. Linifanib versus Sorafenib in patients with advanced hepatocellular carcinoma: Results of a randomized phase III trial. J. Clin. Oncol. 2015, 33, 172–179. [Google Scholar] [CrossRef] [PubMed]
- Chiu, Y.L.; Carlson, D.M.; Pradhan, R.S.; Ricker, J.L. Exposure-response (safety) analysis to identify linifanib dose for a Phase III study in patients with hepatocellular carcinoma. Clin. Ther. 2013, 35, 1770–1777. [Google Scholar] [CrossRef] [PubMed]
- Hatemi, I.; Hatemi, G.; Pamuk, O.N.; Erzin, Y.; Celik, A.F. TNF-alpha antagonists and thalidomide for the management of gastrointestinal Behcet’s syndrome refractory to the conventional treatment modalities: A case series and review of the literature. Clin. Exp. Rheumatol. 2015, 33 (Suppl. 94), S129–S137. [Google Scholar] [PubMed]
- Segarra, M.; Lozano, E.; Corbera-Bellalta, M.; Vilardell, C.; Cibeira, M.T.; Esparza, J.; Izco, N.; Blade, J.; Cid, M.C. Thalidomide decreases gelatinase production by malignant B lymphoid cell lines through disruption of multiple integrin-mediated signaling pathways. Haematologica 2010, 95, 456–463. [Google Scholar] [CrossRef] [PubMed]
- Cao, D.D.; Xu, H.L.; Liu, L.; Zheng, Y.F.; Gao, S.F.; Xu, X.M.; Ge, W. Thalidomide combined with transcatheter artierial chemoembolzation for primary hepatocellular carcinoma: A systematic review and meta-analysis. Oncotarget 2017, 8, 44976–44993. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, Y.; Fukumoto, M. Sorafenib: Complexities of Raf-dependent and Raf-independent signaling are now unveiled. Med. Mol. Morphol. 2011, 44, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, A.; Ho, M. Role of sorafenib in the treatment of advanced hepatocellular carcinoma: An update. Hepatol. Res. 2013, 43, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Palmer, D.H. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar]
- Abdel-Rahman, O. Impact of baseline characteristics on outcomes of advanced HCC patients treated with sorafenib: A secondary analysis of a phase III study. J. Cancer Res. Clin. Oncol. 2018, 144, 901–908. [Google Scholar] [CrossRef] [PubMed]
- Villanueva, A.; Chiang, D.Y.; Newell, P.; Peix, J.; Thung, S.; Alsinet, C.; Tovar, V.; Roayaie, S.; Minguez, B.; Sole, M.; et al. Pivotal role of mTOR signaling in hepatocellular carcinoma. Gastroenterology 2008, 135, 1972–1983. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.N.; Wan, R.Z.; Liu, Z.P. Recent developments of small molecule PI3K/mTOR dual inhibitors. Mini Rev. Med. Chem. 2013, 13, 2047–2059. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhou, J.; Fan, J.; Tan, C.J.; Qiu, S.J.; Yu, Y.; Huang, X.W.; Tang, Z.Y. Sirolimus inhibits the growth and metastatic progression of hepatocellular carcinoma. J. Cancer Res. Clin. Oncol. 2009, 135, 715–722. [Google Scholar] [CrossRef] [PubMed]
- Geissler, E.K.; Schnitzbauer, A.A.; Zulke, C.; Lamby, P.E.; Proneth, A.; Duvoux, C.; Burra, P.; Jauch, K.W.; Rentsch, M.; Ganten, T.M.; et al. Sirolimus use in liver transplant recipients with hepatocellular carcinoma: A randomized, multicenter, open-label Phase 3 trial. Transplantation 2016, 100, 116–125. [Google Scholar] [CrossRef] [PubMed]
- Koeberle, D.; Dufour, J.F.; Demeter, G.; Li, Q.; Ribi, K.; Samaras, P.; Saletti, P.; Roth, A.D.; Horber, D.; Buehlmann, M.; et al. Sorafenib with or without everolimus in patients with advanced hepatocellular carcinoma (HCC): A randomized multicenter, multinational phase II trial (SAKK 77/08 and SASL 29). Ann. Oncol. 2016, 27, 856–861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, C.M.; Fan, S.T.; Ng, I.O. β-Catenin mutation and overexpression in hepatocellular carcinoma: Clinicopathologic and prognostic significance. Cancer 2001, 92, 136–145. [Google Scholar] [CrossRef]
- Hwang, S.Y.; Deng, X.; Byun, S.; Lee, C.; Lee, S.J.; Suh, H.; Zhang, J.; Kang, Q.; Zhang, T.; Westover, K.D.; et al. Direct targeting of beta-catenin by a small molecule stimulates proteasomal degradation and suppresses oncogenic Wnt/beta-catenin signaling. Cell Rep. 2016, 16, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Arsura, M.; Cavin, L.G. Nuclear factor-kappaB and liver carcinogenesis. Cancer Lett. 2005, 229, 157–169. [Google Scholar] [CrossRef] [PubMed]
- Wahl, K.; Siegemund, M.; Lehner, F.; Vondran, F.; Nussler, A.; Langer, F.; Krech, T.; Kontermann, R.; Manns, M.P.; Schulze-Osthoff, K.; et al. Increased apoptosis induction in hepatocellular carcinoma by a novel tumor-targeted TRAIL fusion protein combined with bortezomib. Hepatology 2013, 57, 625–636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, G.P.; Mahoney, M.R.; Szydlo, D.; Mok, T.S.; Marshke, R.; Holen, K.; Picus, J.; Boyer, M.; Pitot, H.C.; Rubin, J.; et al. An international, multicenter phase II trial of bortezomib in patients with hepatocellular carcinoma. Investig. New Drugs 2012, 30, 387–394. [Google Scholar] [CrossRef] [PubMed]
- Pardee, A.D.; Butterfield, L.H. Immunotherapy of hepatocellular carcinoma: Unique challenges and clinical opportunities. Oncoimmunology 2012, 1, 48–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sangro, B.; Gomez-Martin, C.; de la Mata, M.; Inarrairaegui, M.; Garralda, E.; Barrera, P.; Riezu-Boj, J.I.; Larrea, E.; Alfaro, C.; Sarobe, P.; et al. A clinical trial of CTLA-4 blockade with tremelimumab in patients with hepatocellular carcinoma and chronic hepatitis C. J. Hepatol. 2013, 59, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Juliana, C.; Fernandes-Alnemri, T.; Wu, J.; Datta, P.; Solorzano, L.; Yu, J.W.; Meng, R.; Quong, A.A.; Latz, E.; Scott, C.P.; et al. Anti-inflammatory compounds parthenolide and Bay 11-7082 are direct inhibitors of the inflammasome. J. Biol. Chem. 2010, 285, 9792–9802. [Google Scholar] [CrossRef] [PubMed]
- Honda, H.; Nagai, Y.; Matsunaga, T.; Okamoto, N.; Watanabe, Y.; Tsuneyama, K.; Hayashi, H.; Fujii, I.; Ikutani, M.; Hirai, Y.; et al. Isoliquiritigenin is a potent inhibitor of NLRP3 inflammasome activation and diet-induced adipose tissue inflammation. J. Leukoc. Biol. 2014, 96, 1087–1100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, A.H.; Liu, W.; Jiang, N.; Xu, Q.; Tan, R.X. Spirodalesol, an NLRP3 Inflammasome Activation Inhibitor. Org. Lett. 2016, 18, 6496–6499. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.X.; Chen, W.; Hu, X.Z.; Feng, S.Y.; Li, K.Y.; Qi, S.; Lei, Q.Q.; Hu, G.Q.; Li, N.; Zhou, F.H.; et al. Liver X receptors agonists suppress NLRP3 inflammasome activation. Cytokine 2017, 91, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Lee, H.E.; Lee, J.Y. A pharmacological inhibitor of NLRP3 inflammasome prevents non-alcoholic fatty liver disease in a mouse model induced by high fat diet. Sci. Rep. 2016, 6, 24399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Youm, Y.H.; Nguyen, K.Y.; Grant, R.W.; Goldberg, E.L.; Bodogai, M.; Kim, D.; D’Agostino, D.; Planavsky, N.; Lupfer, C.; Kanneganti, T.D.; et al. The ketone metabolite beta-hydroxybutyrate blocks NLRP3 inflammasome-mediated inflammatory disease. Nat. Med. 2015, 21, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Coll, R.C.; Robertson, A.A.; Chae, J.J.; Higgins, S.C.; Munoz-Planillo, R.; Inserra, M.C.; Vetter, I.; Dungan, L.S.; Monks, B.G.; Stutz, A.; et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat. Med. 2015, 21, 248–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gris, D.; Ye, Z.; Iocca, H.A.; Wen, H.; Craven, R.R.; Gris, P.; Huang, M.; Schneider, M.; Miller, S.D.; Ting, J.P. NLRP3 plays a critical role in the development of experimental autoimmune encephalomyelitis by mediating Th1 and Th17 responses. J. Immunol. 2010, 185, 974–981. [Google Scholar] [CrossRef] [PubMed]
- Inoue, M.; Williams, K.L.; Gunn, M.D.; Shinohara, M.L. NLRP3 inflammasome induces chemotactic immune cell migration to the CNS in experimental autoimmune encephalomyelitis. Proc. Natl. Acad. Sci. USA 2012, 109, 10480–10485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inoue, M.; Williams, K.L.; Oliver, T.; Vandenabeele, P.; Rajan, J.V.; Miao, E.A.; Shinohara, M.L. Interferon-beta therapy against EAE is effective only when development of the disease depends on the NLRP3 inflammasome. Sci. Signal. 2012, 5, ra38. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Sala, M.; Castells, L.; Suarez, Y.; Vilana, R.; Bianchi, L.; Ayuso, C.; Vargas, V.; Rodes, J.; Bruix, J. Randomized controlled trial of interferon treatment for advanced hepatocellular carcinoma. Hepatology 2000, 31, 54–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikeda, K.; Arase, Y.; Saitoh, S.; Kobayashi, M.; Suzuki, Y.; Suzuki, F.; Tsubota, A.; Chayama, K.; Murashima, N.; Kumada, H. Interferon beta prevents recurrence of hepatocellular carcinoma after complete resection or ablation of the primary tumor-A prospective randomized study of hepatitis C virus-related liver cancer. Hepatology 2000, 32, 228–232. [Google Scholar] [CrossRef] [PubMed]
- Sakon, M.; Nagano, H.; Dono, K.; Nakamori, S.; Umeshita, K.; Yamada, A.; Kawata, S.; Imai, Y.; Iijima, S.; Monden, M. Combined intraarterial 5-fluorouracil and subcutaneous interferon-alpha therapy for advanced hepatocellular carcinoma with tumor thrombi in the major portal branches. Cancer 2002, 94, 435–442. [Google Scholar] [CrossRef] [PubMed]
- Kubo, S.; Nishiguchi, S.; Hirohashi, K.; Tanaka, H.; Shuto, T.; Yamazaki, O.; Shiomi, S.; Tamori, A.; Oka, H.; Igawa, S.; et al. Effects of long-term postoperative interferon-alpha therapy on intrahepatic recurrence after resection of hepatitis C virus-related hepatocellular carcinoma. A randomized, controlled trial. Ann. Intern. Med. 2001, 134, 963–967. [Google Scholar] [CrossRef] [PubMed]
- Reinisch, W.; Holub, M.; Katz, A.; Herneth, A.; Lichtenberger, C.; Schoniger-Hekele, M.; Waldhoer, T.; Oberhuber, G.; Ferenci, P.; Gangl, A.; et al. Prospective pilot study of recombinant granulocyte-macrophage colony-stimulating factor and interferon-gamma in patients with inoperable hepatocellular carcinoma. J. Immunother. 2002, 25, 489–499. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, G.; Montella, L.; Milo, M.; Fiore, R.; Biondi, E.; Bianco, A.R.; Martignetti, A. Ultra-low-dose interleukin-2 in unresectable hepatocellular carcinoma. Am. J. Clin. Oncol. 2002, 25, 224–226. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Wang, N.; Zhao, H.; Jin, H.; Wang, G.; Niu, C.; Terunuma, H.; He, H.; Li, W. Combination of radiofrequency ablation and sequential cellular immunotherapy improves progression-free survival for patients with hepatocellular carcinoma. Int. J. Cancer 2014, 134, 342–351. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.; Xu, M.B.; Yun, M.M.; Wang, Y.Z.; Zhang, R.M.; Meng, X.K.; Ou-Yang, X.H.; Yun, S. Hepatocellular carcinoma-specific immunotherapy with synthesized alpha1,3-galactosyl epitope-pulsed dendritic cells and cytokine-induced killer cells. World J. Gastroenterol. 2011, 17, 5260–5266. [Google Scholar] [CrossRef] [PubMed]
- Kamiya, T.; Chang, Y.H.; Campana, D. Expanded and activated natural killer cells for immunotherapy of hepatocellular carcinoma. Cancer Immunol. Res. 2016, 4, 574–581. [Google Scholar] [CrossRef] [PubMed]
- Sawada, Y.; Yoshikawa, T.; Nobuoka, D.; Shirakawa, H.; Kuronuma, T.; Motomura, Y.; Mizuno, S.; Ishii, H.; Nakachi, K.; Konishi, M.; et al. Phase I trial of a glypican-3-derived peptide vaccine for advanced hepatocellular carcinoma: Immunologic evidence and potential for improving overall survival. Clin. Cancer Res. 2012, 18, 3686–3696. [Google Scholar] [CrossRef] [PubMed]
- Voron, T.; Colussi, O.; Marcheteau, E.; Pernot, S.; Nizard, M.; Pointet, A.L.; Latreche, S.; Bergaya, S.; Benhamouda, N.; Tanchot, C.; et al. VEGF-A modulates expression of inhibitory checkpoints on CD8+ T cells in tumors. J. Exp. Med. 2015, 212, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Chan, A.W.; Tong, J.H.; Chan, S.L.; Lai, P.B.; To, K.F. Expression of stemness markers (CD133 and EpCAM) in prognostication of hepatocellular carcinoma. Histopathology 2014, 64, 935–950. [Google Scholar] [CrossRef] [PubMed]
- Vesely, M.D.; Kershaw, M.H.; Schreiber, R.D.; Smyth, M.J. Natural innate and adaptive immunity to cancer. Ann. Rev. Immunol. 2011, 29, 235–271. [Google Scholar] [CrossRef] [PubMed]
- Collins, A.V.; Brodie, D.W.; Gilbert, R.J.; Iaboni, A.; Manso-Sancho, R.; Walse, B.; Stuart, D.I.; van der Merwe, P.A.; Davis, S.J. The interaction properties of costimulatory molecules revisited. Immunity 2002, 17, 201–210. [Google Scholar] [CrossRef]
- Duffy, A.G.; Ulahannan, S.V.; Makorova-Rusher, O.; Rahma, O.; Wedemeyer, H.; Pratt, D.; Davis, J.L.; Hughes, M.S.; Heller, T.; ElGindi, M.; et al. Tremelimumab in combination with ablation in patients with advanced hepatocellular carcinoma. J. Hepatol. 2017, 66, 545–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, L.; Chen, S.; Yang, L.; Li, Y. The role of PD-1 and PD-L1 in T-cell immune suppression in patients with hematological malignancies. J. Hematol. Oncol. 2013, 6, 74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, S.; Jia, R.; Zhang, X.; Fang, Q.; Huang, L. The PD-1/PD-Ls pathway and autoimmune diseases. Cell. Immunol. 2014, 290, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Hamanishi, J.; Mandai, M.; Ikeda, T.; Minami, M.; Kawaguchi, A.; Murayama, T.; Kanai, M.; Mori, Y.; Matsumoto, S.; Chikuma, S.; et al. Safety and antitumor activity of anti-PD-1 antibody, nivolumab, in patients with platinum-resistant ovarian cancer. J. Clin. Oncol. 2015, 33, 4015–4022. [Google Scholar] [CrossRef] [PubMed]
- El-Khoueiry, A.B.; Sangro, B.; Yau, T.; Crocenzi, T.S.; Kudo, M.; Hsu, C.; Kim, T.Y.; Choo, S.P.; Trojan, J.; Welling, T.H.R.; et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): An open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet 2017, 389, 2492–2502. [Google Scholar] [CrossRef]
- Iwai, Y.; Ishida, M.; Tanaka, Y.; Okazaki, T.; Honjo, T.; Minato, N. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc. Natl. Acad. Sci. USA 2002, 99, 12293–12297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, F.; Cheng, X.; Sun, S.; Zhou, J. Transcriptional activation of PD-L1 by Sox2 contributes to the proliferation of hepatocellular carcinoma cells. Oncol. Rep. 2017, 37, 3061–3067. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Liu, Y.; Meng, L.; Liu, K.; Ji, B. Targeting the PD-L1/DNMT1 axis in acquired resistance to sorafenib in human hepatocellular carcinoma. Oncol. Rep. 2017, 38, 899–907. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Liu, J.; Wu, Y.; Xiao, F.; Wang, Y.; Wang, R.; Yang, H.; Wang, G.; Yang, J.; Deng, H.; et al. Immunotherapy of hepatocellular carcinoma with a vaccine based on xenogeneic homologous alpha fetoprotein in mice. Biochem. Biophys. Res. Commun. 2008, 376, 10–14. [Google Scholar] [CrossRef] [PubMed]
- Sawada, Y.; Yoshikawa, T.; Ofuji, K.; Yoshimura, M.; Tsuchiya, N.; Takahashi, M.; Nobuoka, D.; Gotohda, N.; Takahashi, S.; Kato, Y.; et al. Phase II study of the GPC3-derived peptide vaccine as an adjuvant therapy for hepatocellular carcinoma patients. Oncoimmunol. 2016, 5, e1129483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takakura, K.; Kajihara, M.; Ito, Z.; Ohkusa, T.; Gong, J.; Koido, S. Dendritic-tumor fusion cells in cancer immunotherapy. Discov. Med. 2015, 19, 169–174. [Google Scholar] [PubMed]
- Zhao, T.; Jia, H.; Cheng, Q.; Xiao, Y.; Li, M.; Ren, W.; Li, C.; Feng, Y.; Feng, Z.; Wang, H.; et al. Nifuroxazide prompts antitumor immune response of TCL-loaded DC in mice with orthotopically-implanted hepatocarcinoma. Oncol. Rep. 2017, 37, 3405–3414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Bayer, M.E.; Chen, X.; Fredrickson, C.; Cornforth, A.N.; Liang, G.; Cannon, J.; He, J.; Fu, Q.; Liu, J.; et al. Phase I trial of active specific immunotherapy with autologous dendritic cells pulsed with autologous irradiated tumor stem cells in hepatitis B-positive patients with hepatocellular carcinoma. J. Surg. Oncol. 2015, 111, 862–867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, Z.J.; Yu, S.J.; Heinrich, B.; Ma, C.; Fu, Q.; Sandhu, M.; Agdashian, D.; Zhang, Q.; Korangy, F.; Greten, T.F. Indoleamine 2,3-dioxygenase provides adaptive resistance to immune checkpoint inhibitors in hepatocellular carcinoma. Cancer Immunol. Immunother. 2018, 67, 1305–1315. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Strategies | Subsets | Targets and Applications | Refs. |
|---|---|---|---|
| Cytokine therapy | IFN IL-2 | INF-α2b, INF-β, INF-α, INF-α + 5-FU, IFN-γ + GM-CSF | [175,176,177,178,179,180] |
| Cell transfer immunotherapy | CIK cells NK cells CAR T cells | CIK with RFA and/or TACE, TAA-pulsed DC and CIK RFA + NK, sorafenib, NKG2D GPC3, EpCAM, VEGF-A + Osteopontin | [181,182,183,184,185,186] |
| Immune checkpoint inhibitors | PD-1 inhibitors PDL-1 inhibitors CTL-A4 inhibitors | Nivolumab, Decitabine, c-Met inhibitor, Pidilizumab Durvalumab, Tremelimumab, Ipilimumab, DNMT1 inhibitor Anti-CTL-A4 antibody, Tremelimumab with RFA or TACE | [164,187,188,189,190,191,192,193,194,195,196] |
| Vaccine strategy | Antigen peptide vaccines DC vaccines | AFP, GPC3, NY-ESO-1, SSX-2, HCA587, MAGE-A3, TERT DC + CIK, DC + radiation, DC + AFP peptide | [197,198,199,200,201] |
| IDO inhibitor | Indoximod | L-tryptophan (Trp) into L-kynurenine (Kyn) | [202] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, M.-Y.; Yiang, G.-T.; Cheng, P.-W.; Chu, P.-Y.; Li, C.-J. Molecular Targets in Hepatocarcinogenesis and Implications for Therapy. J. Clin. Med. 2018, 7, 213. https://doi.org/10.3390/jcm7080213
Wu M-Y, Yiang G-T, Cheng P-W, Chu P-Y, Li C-J. Molecular Targets in Hepatocarcinogenesis and Implications for Therapy. Journal of Clinical Medicine. 2018; 7(8):213. https://doi.org/10.3390/jcm7080213
Chicago/Turabian StyleWu, Meng-Yu, Giuo-Teng Yiang, Pei-Wen Cheng, Pei-Yi Chu, and Chia-Jung Li. 2018. "Molecular Targets in Hepatocarcinogenesis and Implications for Therapy" Journal of Clinical Medicine 7, no. 8: 213. https://doi.org/10.3390/jcm7080213