Self-Seeding Microwells to Isolate and Assess the Viability of Single Circulating Tumor Cells

 , , ,
, , ,
Abstract
:
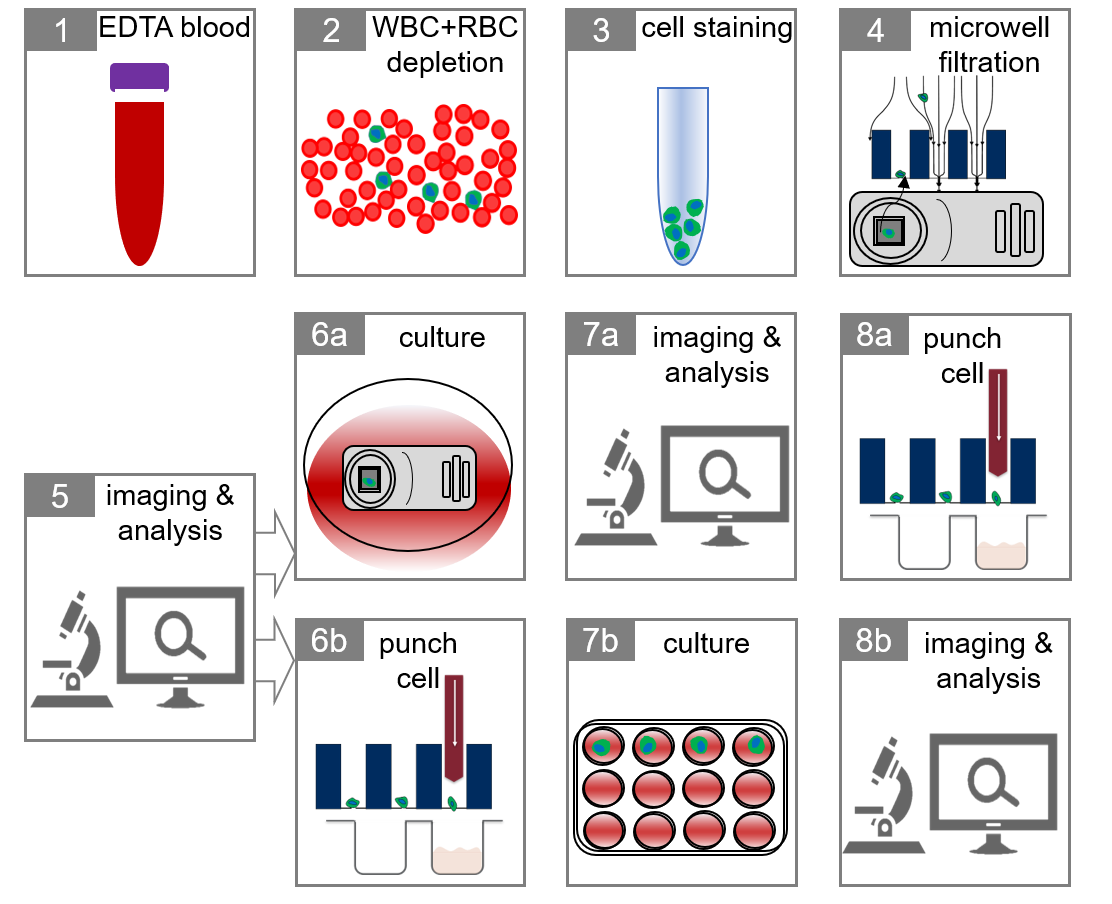
1. Introduction
2. Results
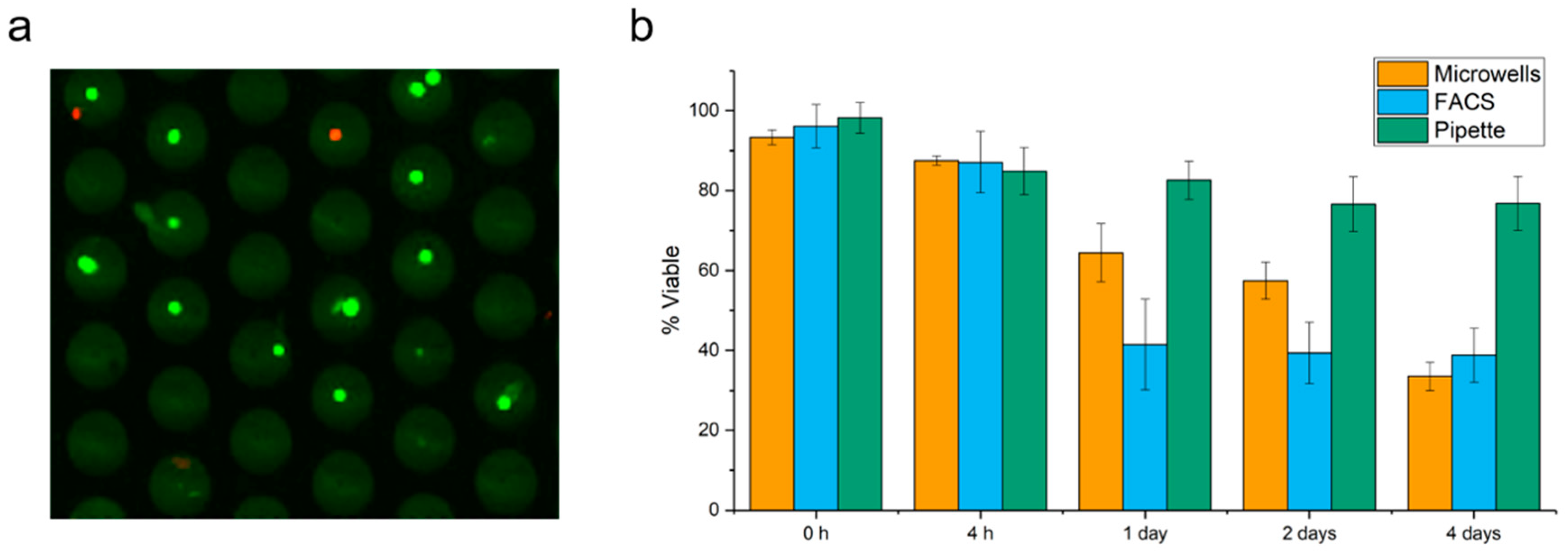
2.1. Cell Viability in Microwells
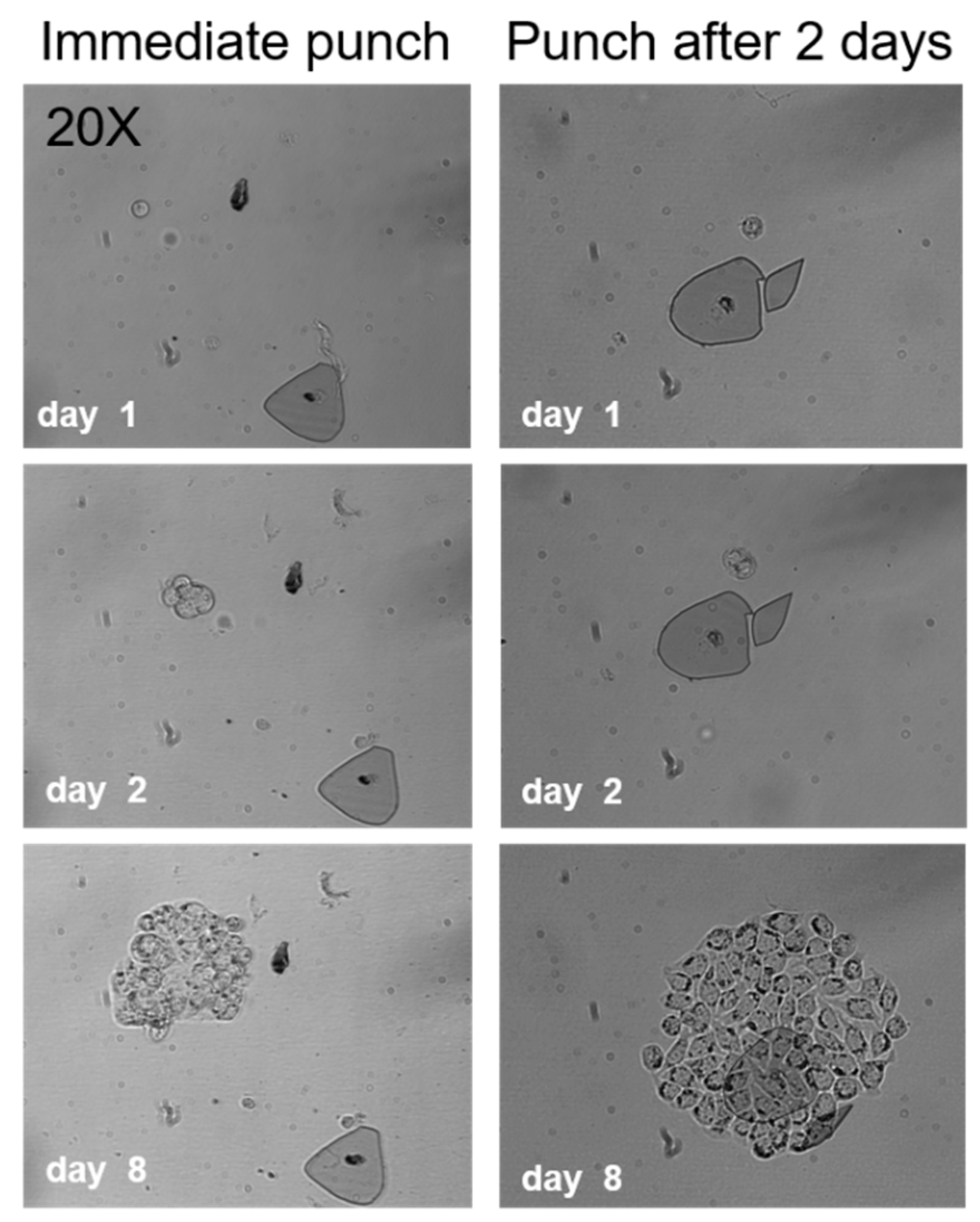
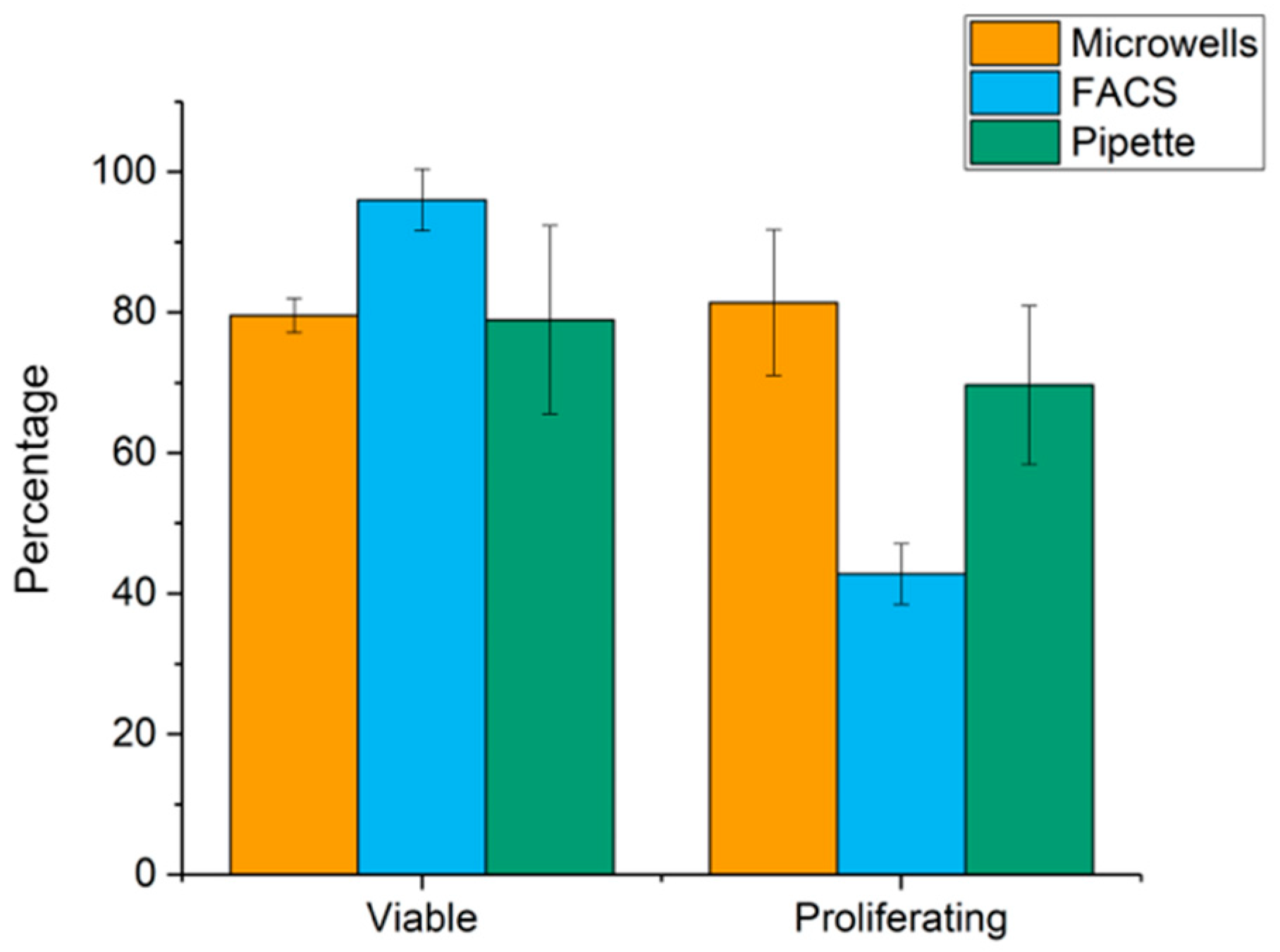
2.2. Cell Viability after Punching
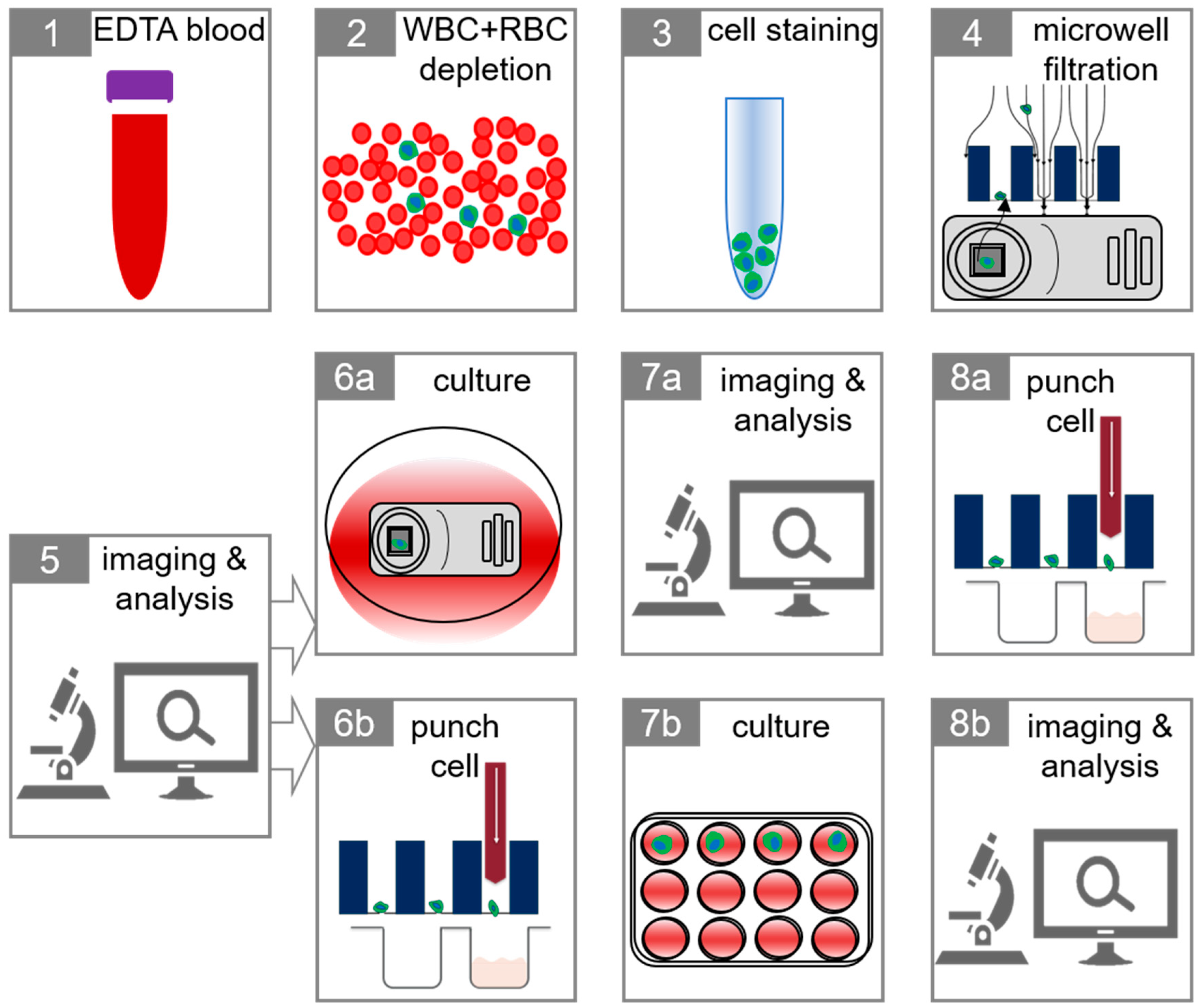
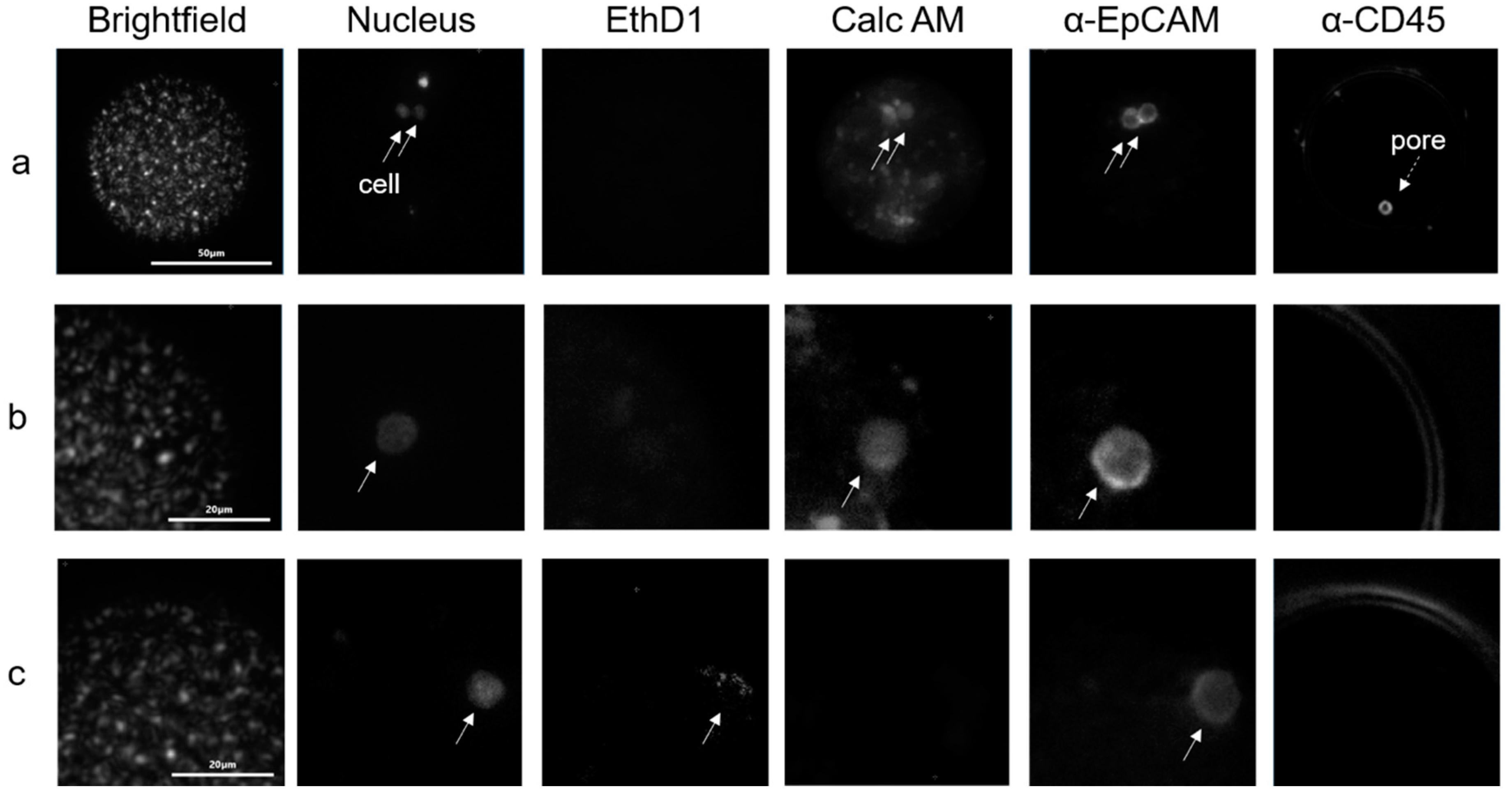
2.3. Isolation of Tumor Cells in Whole Blood
2.4. Tumor Cells in Blood of Metastatic Breast and Prostate Cancer Patients
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Viability Assay
4.3. Microwell Degassing and Sterilization
4.4. Cell Punching
4.5. FACS Sorting
4.6. Manual Pipetting
4.7. Spiking and Enrichment
4.8. Patient Samples
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| CTC | Circulating tumor cells |
| Calc AM | Calcein AM |
| EthD1 | Ethidium homodimer-1 |
| FACS | Fluorescent activated cells sorting |
| RBC | Red blood cells |
| WBC | White blood cells |
References
- Diaz-Cano, S.J. Tumor Heterogeneity: Mechanisms and Bases for a Reliable Application of Molecular Marker Design. Int. J. Mol. Sci. 2012, 13, 1951–2011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crowley, E.; Di Nicolantonio, F.; Loupakis, F.; Bardelli, A. Liquid biopsy: Monitoring cancer-genetics in the blood. Nat. Rev. Clin. Oncol. 2013, 10, 472–484. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andree, K.C.; van Dalum, G.; Terstappen, L.W.M.M. Challenges in circulating tumor cell detection by the CellSearch system. Mol. Oncol. 2016, 10, 395–407. [Google Scholar] [CrossRef] [PubMed]
- Pantel, K.; Alix-Panabières, C. Liquid biopsy: Potential and challenges. Mol. Oncol. 2016, 10, 371–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allard, W.J.; Matera, J.; Miller, M.C.; Repollet, M.; Connelly, M.C.; Rao, C.; Tibbe, A.G.J.; Uhr, J.W.; Terstappen, L.W.M.M. Tumor cells circulate in the peripheral blood of all major carcinomas but not in healthy subjects or patients with nonmalignant diseases. Clin. Cancer Res. 2004, 10, 6897–6904. [Google Scholar] [CrossRef] [PubMed]
- Soler, A.; Cayrefourcq, L.; Mazel, M.; Alix-Panabières, C. EpCAM-Independent Enrichment and Detection of Viable Circulating Tumor Cells Using the EPISPOT Assay. In Methods in Molecular Biology (Clifton, N.J.); Humana Press: New York, NY, USA, 2017; Volume 1634, pp. 263–276. [Google Scholar]
- Haber, D.A.; Gray, N.S.; Baselga, J. The Evolving War on Cancer. Cell 2011, 145, 19–24. [Google Scholar] [CrossRef] [Green Version]
- Yu, M.; Bardia, A.; Aceto, N.; Bersani, F.; Madden, M.W.; Donaldson, M.C.; Desai, R.; Zhu, H.; Comaills, V.; Zheng, Z.; et al. Cancer therapy. Ex vivo culture of circulating breast tumor cells for individualized testing of drug susceptibility. Science 2014, 345, 216–220. [Google Scholar] [CrossRef]
- Drost, J.; Karthaus, W.R.; Gao, D.; Driehuis, E.; Sawyers, C.L.; Chen, Y.; Clevers, H. Organoid culture systems for prostate epithelial and cancer tissue. Nat. Protoc. 2016, 11, 347–358. [Google Scholar] [CrossRef] [Green Version]
- Fatehullah, A.; Tan, S.H.; Barker, N. Organoids as an in vitro model of human development and disease. Nat. Cell Biol. 2016, 18, 246–254. [Google Scholar] [CrossRef]
- Gao, D.; Vela, I.; Sboner, A.; Iaquinta, P.J.; Karthaus, W.R.; Gopalan, A.; Dowling, C.; Wanjala, J.N.; Undvall, E.A.; Arora, V.K.; et al. Organoid Cultures Derived from Patients with Advanced Prostate Cancer. Cell 2014, 159, 176–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolostova, K.; Cegan, M.; Bobek, V. Circulating tumour cells in patients with urothelial tumours: Enrichment and in vitro culture. Can. Urol. Assoc. J. 2014, 8, E715–E720. [Google Scholar] [CrossRef]
- Cayrefourcq, L.; Mazard, T.; Joosse, S.; Solassol, J.; Ramos, J.; Assenat, E.; Schumacher, U.; Costes, V.; Maudelonde, T.; Pantel, K.; et al. Establishment and characterization of a cell line from human circulating colon cancer cells. Cancer Res. 2015, 75, 892–901. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Shiratsuchi, H.; Lin, J.; Chen, G.; Reddy, R.M.; Azizi, E.; Fouladdel, S.; Chang, A.C.; Lin, L.; Jiang, H.; et al. Expansion of CTCs from early stage lung cancer patients using a microfluidic co-culture model. Oncotarget 2014, 5, 12383–12397. [Google Scholar] [CrossRef] [Green Version]
- Lambros, M.B.; Seed, G.; Sumanasuriya, S.; Gil, V.; Crespo, M.; Fontes, M.S.; Chandler, R.; Mehra, N.; Fowler, G.; Ebbs, B.; et al. Single Cell Analyses of Prostate Cancer Liquid Biopsies Acquired by Apheresis. Clin. Cancer Res. 2018. [Google Scholar] [CrossRef] [PubMed]
- Faugeroux, V.; Deas, O.; Catelain, C.; Pailler, E.; Andree, K.; Lucibello, F.; Alexandrova, E.; Judde, J.; Stoecklein, N.; Scoazec, J.; et al. Establishment and characterization of a circulating tumor cell-derived xenograft (CDX) in prostate cancer. In Proceedings of the ACTC, Rhodes, Greece, 4–7 October 2017; p. 106. [Google Scholar]
- Swennenhuis, J.F.; Tibbe, A.G.J.; Stevens, M.; Katika, M.R.; van Dalum, J.; Duy Tong, H.; van Rijn, C.J.M.; Terstappen, L.W.M.M. Self-seeding microwell chip for the isolation and characterization of single cells. Lab Chip 2015, 15, 3039–3046. [Google Scholar] [CrossRef]
- Stevens, M.; Oomens, L.; Broekmaat, J.; Weersink, J.; Abali, F.; Swennenhuis, J.; Tibbe, A. VyCAP’s puncher technology for single cell identification, isolation, and analysis. Cytom. Part A 2018, 93, 1255–1259. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, H.M. Flow systems. In Practical Flow Cytometry; Wiley-Liss: New York, NY, USA, 1994; pp. 133–134. [Google Scholar]
- Shapiro, H.M. Chapter 6. Flow sorting. In Practical Flow Cytometry; Wiley-Liss: New York, NY, USA, 1994; pp. 217–228. [Google Scholar]
- Coumans, F.A.W.; van Dalum, G.; Beck, M.; Terstappen, L.W.M.M. Filter Characteristics Influencing Circulating Tumor Cell Enrichment from Whole Blood. PLoS ONE 2013, 8, e61770. [Google Scholar] [CrossRef]
- Coumans, F.; Dalum, G.; Terstappen, L.W.M.M. CTC Technologies and Tools. Cytom. Part A 2018, 93, 1197–1201. [Google Scholar] [CrossRef]
- Coumans, F.A.W.; van Dalum, G.; Beck, M.; Terstappen, L.W.M.M. Filtration Parameters Influencing Circulating Tumor Cell Enrichment from Whole Blood. PLoS ONE 2013, 8, e61774. [Google Scholar] [CrossRef] [PubMed]
- Mulherin, C.; Larson, C.J.; Horton, A.; Hermann, M.; Repollet, M.; Terstappen, L.W. CellSave (TM) a Blood Collection Tube for the Preservation of Circulating Tumor Cells and Lymphocyte Subsets; Div John Wiley & Sons Inc.: Hoboken, NJ, USA, 2004. [Google Scholar]
- Mu, Z.; Benali-Furet, N.; Uzan, G.; Znaty, A.; Ye, Z.; Paolillo, C.; Wang, C.; Austin, L.; Rossi, G.; Fortina, P.; et al. Detection and Characterization of Circulating Tumor Associated Cells in Metastatic Breast Cancer. Int. J. Mol. Sci. 2016, 17, 1665. [Google Scholar] [CrossRef] [PubMed]
- Smirnov, D.A.; Zweitzig, D.R.; Foulk, B.W.; Miller, M.C.; Doyle, G.V.; Pienta, K.J.; Meropol, N.J.; Weiner, L.M.; Cohen, S.J.; Moreno, J.G.; et al. Global gene expression profiling of circulating tumor cells. Cancer Res. 2005, 65, 4993–4997. [Google Scholar] [CrossRef] [PubMed]
- Talasaz, A.H.; Powell, A.A.; Huber, D.E.; Berbee, J.G.; Roh, K.-H.; Yu, W.; Xiao, W.; Davis, M.M.; Pease, R.F.; Mindrinos, M.N.; et al. Isolating highly enriched populations of circulating epithelial cells and other rare cells from blood using a magnetic sweeper device. Proc. Natl. Acad. Sci. USA 2009, 106, 3970–3975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leong, K.G.; Niessen, K.; Kulic, I.; Raouf, A.; Eaves, C.; Pollet, I.; Karsan, A. Jagged1-mediated Notch activation induces epithelial-to-mesenchymal transition through Slug-induced repression of E-cadherin. J. Exp. Med. 2007, 204, 2935–2948. [Google Scholar] [CrossRef] [Green Version]
- Agerbæk, M.Ø.; Bang-Christensen, S.R.; Yang, M.-H.; Clausen, T.M.; Pereira, M.A.; Sharma, S.; Ditlev, S.B.; Nielsen, M.A.; Choudhary, S.; Gustavsson, T.; et al. The VAR2CSA malaria protein efficiently retrieves circulating tumor cells in an EpCAM-independent manner. Nat. Commun. 2018, 9, 3279. [Google Scholar] [CrossRef] [PubMed]
- Rossi, E.; Basso, U.; Celadin, R.; Zilio, F.; Pucciarelli, S.; Aieta, M.; Barile, C.; Sava, T.; Bonciarelli, G.; Tumolo, S.; et al. M30 neoepitope expression in epithelial cancer: Quantification of apoptosis in circulating tumor cells by CellSearch analysis. Clin. Cancer Res. 2010, 16, 5233–5243. [Google Scholar] [CrossRef] [PubMed]
- Smerage, J.B.; Budd, G.T.; Doyle, G.V.; Brown, M.; Paoletti, C.; Muniz, M.; Miller, M.C.; Repollet, M.I.; Chianese, D.A.; Connelly, M.C.; et al. Monitoring apoptosis and Bcl-2 on circulating tumor cells in patients with metastatic breast cancer. Mol. Oncol. 2013, 7, 680–692. [Google Scholar] [CrossRef]
- Larson, C.J.; Moreno, J.G.; Pienta, K.J.; Gross, S.; Repollet, M.; O’Hara, S.M.; Russell, T.; Terstappen, L.W.M.M. Apoptosis of circulating tumor cells in prostate cancer patients. Cytom. Part A 2004, 62, 46–53. [Google Scholar] [CrossRef] [Green Version]
- Abali, F.; Stevens, M.; Tibbe, A.G.J.; Terstappen, L.W.M.M.; van der Velde, P.N.; Schasfoort, R.B.M. Isolation of single cells for protein therapeutics using microwell selection and Surface Plasmon Resonance imaging. Anal. Biochem. 2017, 531, 45–47. [Google Scholar] [CrossRef]
- Thurgood, P.; Baratchi, S.; Szydzik, C.; Zhu, J.Y.; Nahavandi, S.; Mitchell, A.; Khoshmanesh, K. A self-sufficient micro-droplet generation system using highly porous elastomeric sponges: A versatile tool for conducting cellular assays. Sens. Actuators B Chem. 2018, 274, 645–653. [Google Scholar] [CrossRef]
- Di Trapani, M.; Manaresi, N.; Medoro, G. DEPArray™ system: An automatic image-based sorter for isolation of pure circulating tumor cells. Cytom. Part A 2018. [Google Scholar] [CrossRef] [PubMed]
- Nelep, C.; Eberhardt, J. Automated rare single cell picking with the ALS cellcelectorTM. Cytom. Part A 2018. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Sa, S.; Wang, L.; Dulmage, K.; Bhagwat, N.; Yee, S.S.; Sen, M.; Pletcher, C.H.; Moore, J.S.; Saksena, S.; et al. An integrated enrichment system to facilitate isolation and molecular characterization of single cancer cells from whole blood. Cytom. Part A 2018, 93, 1226–1233. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| % Recovery | % Viable | % Grow | ||||
|---|---|---|---|---|---|---|
| MCF-7 | MDA-MB-231 | MCF-7 | MDA-MB-231 | MCF-7 | MDA-MB-231 | |
| 1 | 61 | 67 | 63 | 78 | 55 | 49 |
| 2 | 47 | 51 | 61 | 67 | 55 | 48 |
| 3 | 43 | 39 | 74 | 65 | 64 | inf |
| 4 | 45 | 49 | 75 | 65 | 61 | 44 |
| 5 | 33 | 38 | 72 | 77 | 59 | inf |
| 6 | 30 | 41 | 58 | 65 | 69 | 54 |
| mean | 43 | 48 | 67 | 70 | 59 | 49 |
| SD | 11 | 11 | 7 | 6 | 4 | 4 |
| Cancer | CellSearch® CTC | Microwell CTC | Recovery% | % Viable | % Viable after 2 Days | |
|---|---|---|---|---|---|---|
| 1 | Breast | 102 | 1 | 1 | 0 | 0 |
| 2 | Breast | >100 | 0 | 0 | 0 | 0 |
| 3 | Prostate | 498 | 22 | 4 | 36 | 25 |
| 4 | Breast | 94 | 3 | 3 | 33 | 0 |
| 5 | Breast | 22 | 0 | 0 | 0 | 0 |
| 6 | Breast | 111 | 5 | 5 | 20 | 100 |
| 7a | Prostate | 55 | 1 | 2 | 0 | 0 |
| mean | 140 | 5 | 2 | 13 | 18 | |
| SD | 161 | 8 | 2 | 17 | 37 | |
| 7b * | Prostate | 55 | 15 | 27 | NA | NA |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Andree, K.C.; Abali, F.; Oomens, L.; Passanha, F.R.; Broekmaat, J.J.; Kraan, J.; Mendelaar, P.A.J.; Sleijfer, S.; Terstappen, L.W.M.M. Self-Seeding Microwells to Isolate and Assess the Viability of Single Circulating Tumor Cells. Int. J. Mol. Sci. 2019, 20, 477. https://doi.org/10.3390/ijms20030477
Andree KC, Abali F, Oomens L, Passanha FR, Broekmaat JJ, Kraan J, Mendelaar PAJ, Sleijfer S, Terstappen LWMM. Self-Seeding Microwells to Isolate and Assess the Viability of Single Circulating Tumor Cells. International Journal of Molecular Sciences. 2019; 20(3):477. https://doi.org/10.3390/ijms20030477
Chicago/Turabian StyleAndree, Kiki C., Fikri Abali, Lisa Oomens, Fiona R. Passanha, Joska J. Broekmaat, Jaco Kraan, Pauline A.J. Mendelaar, Stefan Sleijfer, and Leon W.M.M. Terstappen. 2019. "Self-Seeding Microwells to Isolate and Assess the Viability of Single Circulating Tumor Cells" International Journal of Molecular Sciences 20, no. 3: 477. https://doi.org/10.3390/ijms20030477