Detection of HPV16 in Esophageal Cancer in a High-Incidence Region of Malawi


Abstract
:
1. Introduction
2. Results
2.1. Multiplex Real-Time PCR Assay
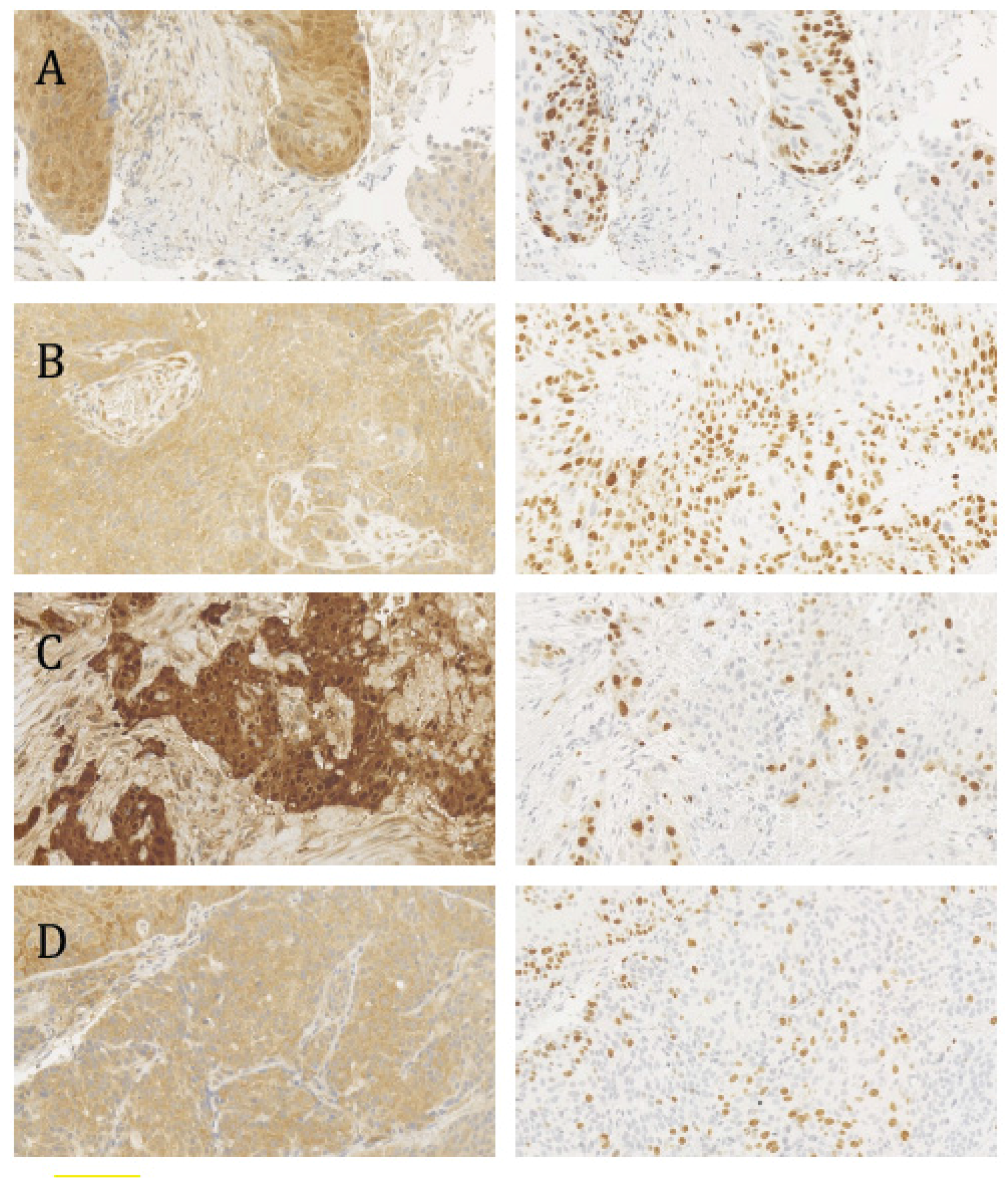
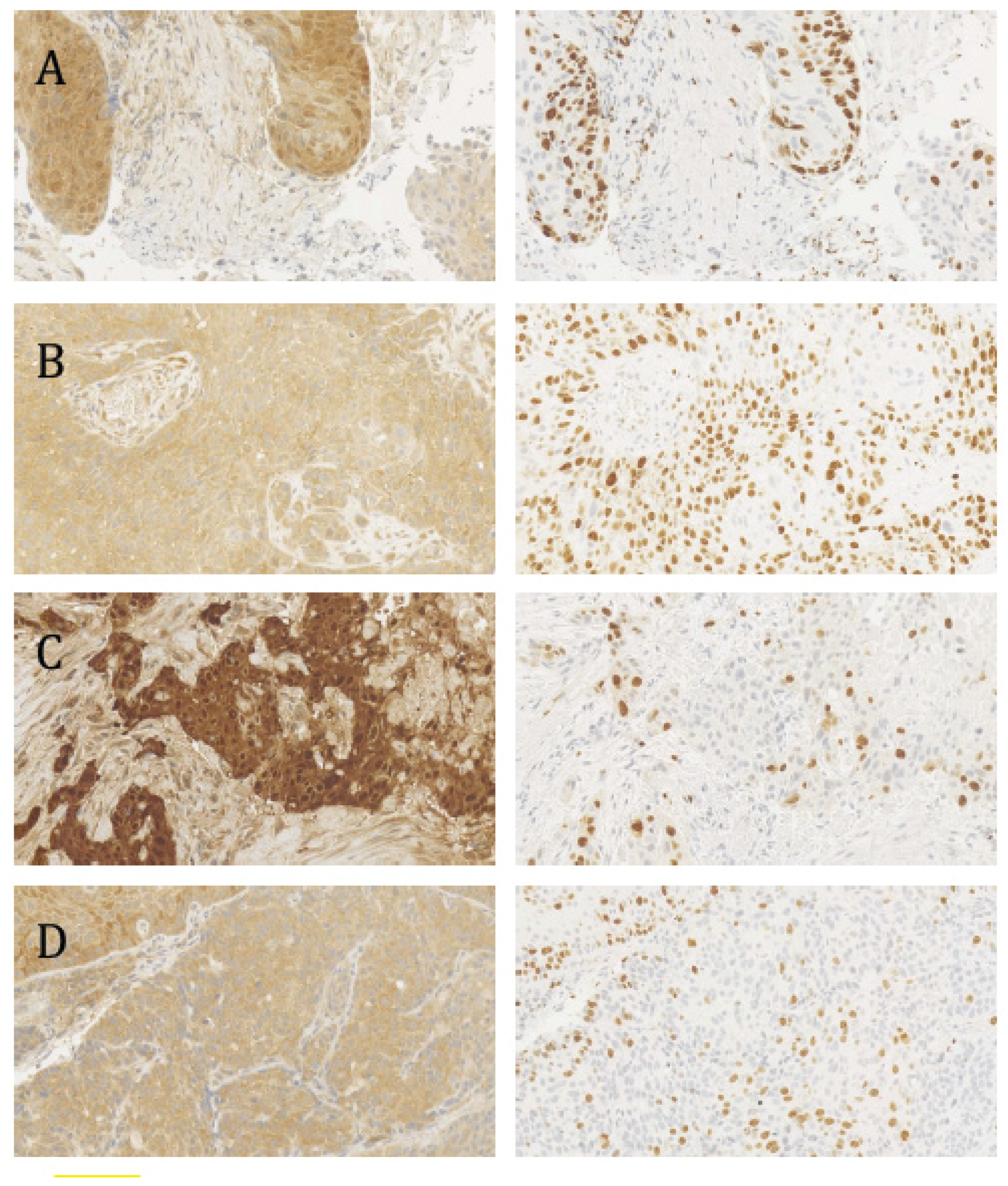
2.2. Histopathological Analysis, p16INK4a Status, and the Ki-67 Proliferation Index
2.3. In Situ Hybridization
2.4. Association between HPV Status, Clinical Data and Risk Factors
3. Discussion
4. Material and Methods
4.1. DNA Extraction
4.2. Multiplex Real-Time PCR Assay (TaqMan Format)
4.3. In Situ Hybridization
4.4. p16INK4a
4.5. Ki-67
4.6. Serological Analysis (ELISA)
4.7. Data on Alcohol Consumption and Smoking, and Clinical Data
4.8. Pathological Analysis
4.9. Statistical Analysis
Supplementary Materials
Author Contribution
Conflict of Interests
Abbreviations
| HPV | Human papillomavirus |
| ESCC | Esophageal squamous cell carcinoma |
| Rb | Retinoblastoma |
| qPCR | Quantitative PCR |
| TBC | Tuberculosis |
| HIV | Human immunodeficiency virus |
References
- The International Agency for Research on Cancer (IARC). GLOBOCAN 2012: Estimated Cancer Incidence, Mortality and Prevalence Worldwide in 2012. Available online: http://globocan.iarc.fr/Default.aspx (accessed on 27 January 2018).
- Arnold, M.; Soerjomataram, I.; Ferlay, J.; Forman, D. Global incidence of oesophageal cancer by histological subtype in 2012. Gut 2015, 64, 381–387. [Google Scholar] [CrossRef] [PubMed]
- Napier, K.J.; Scheerer, M.; Misra, S. Esophageal cancer: A Review of epidemiology, pathogenesis, staging workup and treatment modalities. World J. Gastrointest. Oncol. 2014, 6, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Syrjanen, K.J. HPV infections and oesophageal cancer. J. Clin. Pathol. 2002, 55, 721–728. [Google Scholar] [CrossRef] [PubMed]
- Forman, D.; de Martel, C.; Lacey, C.J.; Soerjomataram, I.; Lortet-Tieulent, J.; Bruni, L.; Vignat, J.; Ferlay, J.; Bray, F.; Plummer, M.; et al. Global burden of human papillomavirus and related diseases. Vaccine 2012, 30 (Suppl. 5), F12–F23. [Google Scholar] [CrossRef] [PubMed]
- Durst, M.; Gissmann, L.; Ikenberg, H.; zur Hausen, H. A papillomavirus DNA from a cervical carcinoma and its prevalence in cancer biopsy samples from different geographic regions. Proc. Natl. Acad. Sci. USA 1983, 80, 3812–3815. [Google Scholar] [CrossRef] [PubMed]
- Zur Hausen, H. Papillomaviruses and cancer: From basic studies to clinical application. Nat. Rev. Cancer 2002, 2, 342–350. [Google Scholar] [CrossRef] [PubMed]
- Gillison, M.L.; Shah, K.V. Human papillomavirus-associated head and neck squamous cell carcinoma: Mounting evidence for an etiologic role for human papillomavirus in a subset of head and neck cancers. Curr. Opin. Oncol. 2001, 13, 183–188. [Google Scholar] [CrossRef] [PubMed]
- Gillison, M.L.; Shah, K.V. Chapter 9: Role of mucosal human papillomavirus in nongenital cancers. J. Natl. Cancer Inst. Monogr. 2003, 2003, 57–65. [Google Scholar] [CrossRef]
- Zur Hausen, H. Papillomaviruses causing cancer: Evasion from host-cell control in early events in carcinogenesis. J. Natl. Cancer Inst. 2000, 92, 690–698. [Google Scholar] [CrossRef] [PubMed]
- Durst, M.; Glitz, D.; Schneider, A.; zur Hausen, H. Human papillomavirus type 16 (HPV 16) gene expression and DNA replication in cervical neoplasia: Analysis by in situ hybridization. Virology 1992, 189, 132–140. [Google Scholar] [CrossRef]
- Snijders, P.J.; Steenbergen, R.D.; Heideman, D.A.; Meijer, C.J. HPV-mediated cervical carcinogenesis: Concepts and clinical implications. J. Pathol. 2006, 208, 152–164. [Google Scholar] [CrossRef] [PubMed]
- Syrjanen, K.J. Histological changes identical to those of condylomatous lesions found in esophageal squamous cell carcinomas. Arch. Geschwulstforsch. 1982, 52, 283–292. [Google Scholar] [PubMed]
- The International Agency for Research on Cancer (IARC). Monographs on the Evaluation of Carcinogenic Risks to Humans; IARC: Lyon, France, 2012; Volume 100B. [Google Scholar]
- Halec, G.; Schmitt, M.; Egger, S.; Abnet, C.C.; Babb, C.; Dawsey, S.M.; Flechtenmacher, C.; Gheit, T.; Hale, M.; Holzinger, D.; et al. Mucosal α-papillomaviruses are not associated with esophageal squamous cell carcinomas: Lack of mechanistic evidence from South Africa, China and Iran and from a world-wide meta-analysis. Int. J. Cancer 2016, 139, 85–98. [Google Scholar] [CrossRef] [PubMed]
- Hubbard, R.A. Human papillomavirus testing methods. Arch. Pathol. Lab. Med. 2003, 127, 940–945. [Google Scholar] [PubMed]
- Gerdes, J.; Lemke, H.; Baisch, H.; Wacker, H.H.; Schwab, U.; Stein, H. Cell cycle analysis of a cell proliferation-associated human nuclear antigen defined by the monoclonal antibody Ki-67. J. Immunol. 1984, 133, 1710–1715. [Google Scholar] [PubMed]
- Keating, J.T.; Cviko, A.; Riethdorf, S.; Riethdorf, L.; Quade, B.J.; Sun, D.; Duensing, S.; Sheets, E.E.; Munger, K.; Crum, C.P. Ki-67, cyclin E, and p16INK4 are complimentary surrogate biomarkers for human papilloma virus-related cervical neoplasia. Am. J. Surg. Pathol. 2001, 25, 884–891. [Google Scholar] [CrossRef] [PubMed]
- Dyson, N.; Howley, P.M.; Munger, K.; Harlow, E. The human papilloma virus-16 E7 oncoprotein is able to bind to the retinoblastoma gene product. Science 1989, 243, 934–937. [Google Scholar] [CrossRef] [PubMed]
- Petrick, J.L.; Wyss, A.B.; Butler, A.M.; Cummings, C.; Sun, X.; Poole, C.; Smith, J.S.; Olshan, A.F. Prevalence of human papillomavirus among oesophageal squamous cell carcinoma cases: Systematic review and meta-analysis. Br. J. Cancer 2014, 110, 2369–2377. [Google Scholar] [CrossRef] [PubMed]
- Guo, F.; Liu, Y.; Wang, X.; He, Z.; Weiss, N.S.; Madeleine, M.M.; Liu, F.; Tian, X.; Song, Y.; Pan, Y.; et al. Human papillomavirus infection and esophageal squamous cell carcinoma: A case-control study. Cancer Epidemiol. Biomark. Prev. 2012, 21, 780–785. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.B.; Guo, M.; Quan, L.P.; Zhang, W.; Lu, Z.M.; Wang, Q.H.; Ke, Y.; Xu, N.Z. Detection of human papillomavirus in Chinese esophageal squamous cell carcinoma and its adjacent normal epithelium. World J. Gastroenterol. 2003, 9, 1170–1173. [Google Scholar] [CrossRef] [PubMed]
- Kayamba, V.; Bateman, A.C.; Asombang, A.W.; Shibemba, A.; Zyambo, K.; Banda, T.; Soko, R.; Kelly, P. HIV infection and domestic smoke exposure, but not human papillomavirus, are risk factors for esophageal squamous cell carcinoma in Zambia: A case-control study. Cancer Med. 2015, 4, 588–595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liyanage, S.S.; Rahman, B.; Ridda, I.; Newall, A.T.; Tabrizi, S.N.; Garland, S.M.; Segelov, E.; Seale, H.; Crowe, P.J.; Moa, A.; et al. The aetiological role of human papillomavirus in oesophageal squamous cell carcinoma: A meta-analysis. PLoS ONE 2013, 8, e69238. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Zhang, Q.; Zhou, L.; Huo, L.; Zhang, Y.; Shen, Z.; Zhu, Y. Comparison of prevalence, viral load, physical status and expression of human papillomavirus-16, -18 and -58 in esophageal and cervical cancer: A case-control study. BMC Cancer 2010, 10, 650. [Google Scholar] [CrossRef] [PubMed]
- Schneider, A.; Oltersdorf, T.; Schneider, V.; Gissmann, L. Distribution pattern of human papilloma virus 16 genome in cervical neoplasia by molecular in situ hybridization of tissue sections. Int. J. Cancer 1987, 39, 717–721. [Google Scholar] [CrossRef] [PubMed]
- Evans, M.F.; Mount, S.L.; Beatty, B.G.; Cooper, K. Biotinyl-tyramide-based in situ hybridization signal patterns distinguish human papillomavirus type and grade of cervical intraepithelial neoplasia. Mod. Pathol. 2002, 15, 1339–1347. [Google Scholar] [CrossRef] [PubMed]
- Si, H.X.; Tsao, S.W.; Poon, C.S.; Wang, L.D.; Wong, Y.C.; Cheung, A.L. Viral load of HPV in esophageal squamous cell carcinoma. Int. J. Cancer 2003, 103, 496–500. [Google Scholar] [CrossRef] [PubMed]
- Si, H.X.; Tsao, S.W.; Poon, C.S.; Wong, Y.C.; Cheung, A.L. Physical status of HPV-16 in esophageal squamous cell carcinoma. J. Clin. Virol. 2005, 32, 19–23. [Google Scholar] [CrossRef] [PubMed]
- Jeon, S.; Allen-Hoffmann, B.L.; Lambert, P.F. Integration of human papillomavirus type 16 into the human genome correlates with a selective growth advantage of cells. J. Virol. 1995, 69, 2989–2997. [Google Scholar] [PubMed]
- Malik, S.M.; Nevin, D.T.; Cohen, S.; Hunt, J.L.; Palazzo, J.P. Assessment of immunohistochemistry for p16INK4 and high-risk HPV DNA by in situ hybridization in esophageal squamous cell carcinoma. Int. J. Surg. Pathol. 2011, 19, 31–34. [Google Scholar] [CrossRef] [PubMed]
- Herbster, S.; Ferraro, C.T.; Koff, N.K.; Rossini, A.; Kruel, C.D.; Andreollo, N.A.; Rapozo, D.C.; Blanco, T.C.; Faria, P.A.; Santos, P.T.; et al. HPV infection in Brazilian patients with esophageal squamous cell carcinoma: Interpopulational differences, lack of correlation with surrogate markers and clinicopathological parameters. Cancer Lett. 2012, 326, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Cooper, K.; Taylor, L.; Govind, S. Human papillomavirus DNA in oesophageal carcinomas in South Africa. J. Pathol. 1995, 175, 273–277. [Google Scholar] [CrossRef] [PubMed]
- Cooper, K.; Herrington, C.S.; Stickland, J.E.; Evans, M.F.; McGee, J.O. Episomal and integrated human papillomavirus in cervical neoplasia shown by non-isotopic in situ hybridisation. J. Clin. Pathol. 1991, 44, 990–996. [Google Scholar] [CrossRef] [PubMed]
- Sano, T.; Oyama, T.; Kashiwabara, K.; Fukuda, T.; Nakajima, T. Immunohistochemical overexpression of p16 protein associated with intact retinoblastoma protein expression in cervical cancer and cervical intraepithelial neoplasia. Pathol. Int. 1998, 48, 580–585. [Google Scholar] [CrossRef] [PubMed]
- Cao, F.; Zhang, W.; Zhang, F.; Han, H.; Xu, J.; Cheng, Y. Prognostic significance of high-risk human papillomavirus and p16(INK4A) in patients with esophageal squamous cell carcinoma. Int. J. Clin. Exp. Med. 2014, 7, 3430–3438. [Google Scholar] [PubMed]
- Nogueira, M.C.; Guedes Neto Ede, P.; Rosa, M.W.; Zettler, E.; Zettler, C.G. Immunohistochemical expression of p16 and p53 in vulvar intraepithelial neoplasia and squamous cell carcinoma of the vulva. Pathol. Oncol. Res. 2006, 12, 153–157. [Google Scholar] [CrossRef] [PubMed]
- Gronhoj Larsen, C.; Gyldenlove, M.; Jensen, D.H.; Therkildsen, M.H.; Kiss, K.; Norrild, B.; Konge, L.; von Buchwald, C. Correlation between human papillomavirus and p16 overexpression in oropharyngeal tumours: A systematic review. Br. J. Cancer 2014, 110, 1587–1594. [Google Scholar] [CrossRef] [PubMed]
- Serup-Hansen, E.; Linnemann, D.; Skovrider-Ruminski, W.; Hogdall, E.; Geertsen, P.F.; Havsteen, H. Human papillomavirus genotyping and p16 expression as prognostic factors for patients with American Joint Committee on Cancer stages I to III carcinoma of the anal canal. J. Clin. Oncol. 2014, 32, 1812–1817. [Google Scholar] [CrossRef] [PubMed]
- Zhao, P.; Hu, Y.C.; Talbot, I.C. Expressing patterns of p16 and CDK4 correlated to prognosis in colorectal carcinoma. World J. Gastroenterol. 2003, 9, 2202–2206. [Google Scholar] [CrossRef] [PubMed]
- Tokugawa, T.; Sugihara, H.; Tani, T.; Hattori, T. Modes of silencing of p16 in development of esophageal squamous cell carcinoma. Cancer Res. 2002, 62, 4938–4944. [Google Scholar] [PubMed]
- Koshiol, J.; Wei, W.Q.; Kreimer, A.R.; Chen, W.; Gravitt, P.; Ren, J.S.; Abnet, C.C.; Wang, J.B.; Kamangar, F.; Lin, D.M.; et al. No role for human papillomavirus in esophageal squamous cell carcinoma in China. Int. J. Cancer 2010, 127, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Teng, H.; Li, X.; Liu, X.; Wu, J.; Zhang, J. The absence of human papillomavirus in esophageal squamous cell carcinoma in East China. Int. J. Clin. Exp. Pathol. 2014, 7, 4184–4193. [Google Scholar] [PubMed]
- Klaes, R.; Friedrich, T.; Spitkovsky, D.; Ridder, R.; Rudy, W.; Petry, U.; Dallenbach-Hellweg, G.; Schmidt, D.; von Knebel, D. Overexpression of p16(INK4A) as a specific marker for dysplastic and neoplastic epithelial cells of the cervix uteri. Int. J. Cancer 2001, 92, 276–284. [Google Scholar] [CrossRef] [PubMed]
- Witkiewicz, A.K.; Knudsen, K.E.; Dicker, A.P.; Knudsen, E.S. The meaning of p16(ink4a) expression in tumors: Functional significance, clinical associations and future developments. Cell Cycle 2011, 10, 2497–2503. [Google Scholar] [CrossRef] [PubMed]
- Bai, P.; Xiao, X.; Zou, J.; Cui, L.; Bui Nguyen, T.M.; Liu, J.; Xiao, J.; Chang, B.; Wu, J.; Wang, H. Expression of p14(ARF), p15(INK4b), p16(INK4a) and skp2 increases during esophageal squamous cell cancer progression. Exp. Ther. Med. 2012, 3, 1026–1032. [Google Scholar] [CrossRef] [PubMed]
- Boffetta, P.; Hashibe, M. Alcohol and cancer. Lancet Oncol. 2006, 7, 149–156. [Google Scholar] [CrossRef]
- Shephard, G.S.; van der Westhuizen, L.; Gatyeni, P.M.; Somdyala, N.I.; Burger, H.M.; Marasas, W.F. Fumonisin mycotoxins in traditional Xhosa maize beer in South Africa. J. Agric. Food Chem. 2005, 53, 9634–9637. [Google Scholar] [CrossRef] [PubMed]
- Hendricks, D.; Parker, M.I. Oesophageal cancer in Africa. IUBMB Life 2002, 53, 263–268. [Google Scholar] [CrossRef] [PubMed]
- Niller, H.H.; Wolf, H.; Minarovits, J. Viral hit and run-oncogenesis: Genetic and epigenetic scenarios. Cancer Lett. 2011, 305, 200–217. [Google Scholar] [CrossRef] [PubMed]
- Campo, M.S.; Moar, M.H.; Sartirana, M.L.; Kennedy, I.M.; Jarrett, W.F. The presence of bovine papillomavirus type 4 DNA is not required for the progression to, or the maintenance of, the malignant state in cancers of the alimentary canal in cattle. EMBO J. 1985, 4, 1819–1825. [Google Scholar] [PubMed]
- Quint, K.D.; Genders, R.E.; de Koning, M.N.; Borgogna, C.; Gariglio, M.; Bouwes Bavinck, J.N.; Doorbar, J.; Feltkamp, M.C. Human β-papillomavirus infection and keratinocyte carcinomas. J. Pathol. 2015, 235, 342–354. [Google Scholar] [CrossRef] [PubMed]
- Viarisio, D.; Gissmann, L.; Tommasino, M. Human papillomaviruses and carcinogenesis: Well-established and novel models. Curr. Opin. Virol. 2017, 26, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Howley, P.M.; Pfister, H.J. β genus papillomaviruses and skin cancer. Virology 2015, 479–480, 290–296. [Google Scholar] [CrossRef] [PubMed]
- Weissenborn, S.J.; Nindl, I.; Purdie, K.; Harwood, C.; Proby, C.; Breuer, J.; Majewski, S.; Pfister, H.; Wieland, U. Human papillomavirus-DNA loads in actinic keratoses exceed those in non-melanoma skin cancers. J. Investig. Dermatol. 2005, 125, 93–97. [Google Scholar] [CrossRef] [PubMed]
- Kusters-Vandevelde, H.V.; de Koning, M.N.; Melchers, W.J.; Quint, W.G.; de Wilde, P.C.; de Jong, E.M.; van de Kerkhof, P.C.; Blokx, W.A. Expression of p14ARF, p16INK4a and p53 in relation to HPV in (pre-)malignant squamous skin tumours. J. Cell. Mol. Med. 2009, 13, 2148–2157. [Google Scholar] [CrossRef] [PubMed]
- Abedi-Ardekani, B.; Hainaut, P. Cancers of the upper gastro-intestinal tract: A review of somatic mutation distributions. Arch. Iran. Med. 2014, 17, 286–292. [Google Scholar] [PubMed]
- Holland, P.M.; Abramson, R.D.; Watson, R.; Gelfand, D.H. Detection of specific polymerase chain reaction product by utilizing the 5′—3′ exonuclease activity of Thermus aquaticus DNA polymerase. Proc. Natl. Acad. Sci. USA 1991, 88, 7276–7280. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, M.; Scheungraber, C.; Herrmann, J.; Teller, K.; Gajda, M.; Runnebaum, I.B.; Durst, M. Quantitative multiplex PCR assay for the detection of the seven clinically most relevant high-risk HPV types. J. Clin. Virol. 2009, 44, 302–307. [Google Scholar] [CrossRef] [PubMed]
- Kleiboeker, S.B. Quantitative assessment of the effect of uracil-DNA glycosylase on amplicon DNA degradation and RNA amplification in reverse transcription-PCR. Virol. J. 2005, 2, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
| Study Number | Hist. Diagn. 1 | Grading | Ki-67 Index Overall 2 | p16INK4a Staining Pattern 3 | p16INK4a Stained Tumor Cell Areas 4 | % of Ki-67 Stained Cells within p16INK4a Stained Areas | HPV qPCR 5 | HPV ISH 6 | HIV Status 7 |
|---|---|---|---|---|---|---|---|---|---|
| HV090610-44 | 1 | 1 | 3 | 2 | 1 | 71 | 1 | 0 | 0 |
| DV280610-68 | 1 | 1 | 3 | 1 | 2 | 88 | 1 | 0 | 0 |
| EC070710-73 | 2 | - | 3 | 1 | 2 | 51 | 1 | 0 | 0 |
| FN280710-105 | 1 | 1 | 3 | 1 | 2 | 67 | 1 | 0 | - |
| AM240510-22 | 1 | 2 | 3 | 1 | 2 | 61 | 1 | 0 | 0 |
| JS070710-72 | 1 | 1 | 3 | 1 | 2 | 72 | 1 | 0 | 0 |
| MRD160610-57 | 1 | 2 | - | - | - | - | 1 | - | 0 |
| PD120510-1 | 1 | 2 | 3 | 0 | 0 | - | 0 | 0 | 0 |
| CC050710-70 | 2 | - | 3 | 0 | 0 | - | 0 | 0 | 1 |
| AM14071087 | 1 | 2 | 2 | 2 | 2 | 26 | 0 | 0 | 0 |
| EK070610-40 | 1 | 2 | 3 | 0 | 0 | - | 0 | - | 0 |
| SN210710-101 | 1 | 2 | 3 | 0 | 0 | - | 0 | - | - |
| SN210710-96 | 1 | 2 | 3 | 0 | 0 | - | 0 | - | - |
| HM070710-74 | 1 | 3 | - | - | - | - | 0 | - | 0 |
| LI190710-91 | 1 | 2 | - | - | - | - | 0 | - | - |
| DM090610-41 | 1 | 2 | 3 | 1 | 2 | 60 | 0 | - | 0 |
| AOM160610-55 | 1 | 2 | 3 | 1 | 2 | 53 | 0 | 0 | 0 |
| MG260510-29 | 1 | 2 | 3 | 1 | 2 | 55 | 0 | - | 0 |
| EO160610-59 | 1 | 3 | 3 | 1 | 1 | 60 | 0 | - | 0 |
| LC150610-97 | 1 | 2 | 2 | 1 | 2 | 25 | 0 | - | - |
| LM180510-10 | 1 | 2 | 3 | 0 | 0 | - | 0 | - | 1 |
| PL130710-81 | 1 | 2 | 2 | 1 | 2 | 46 | 0 | 0 | 0 |
| RC170510-7 | 1 | 2 | 2 | 1 | 2 | 45 | 0 | - | 0 |
| EM280610-69 | 2 | - | 3 | 1 | 2 | 67 | 0 | - | 0 |
| LN180510-12 | 1 | 3 | 2 | 0 | 0 | - | 0 | - | - |
| ESL190510-17 | 1 | 3 | 2 | 1 | 2 | 48 | 0 | - | 0 |
| JC260710-102 | 1 | 2 | 3 | 0 | 0 | - | 0 | - | - |
| DL160610-56 | 1 | 3 | 2 | 1 | 2 | 49 | 0 | - | 1 |
| JC120510-2 | 1 | 2 | 3 | 0 | 0 | - | 0 | - | 0 |
| JN140610-54 | 1 | 2 | 0 | 1 | 1 | 0 | 0 | 0 | 0 |
| RM170510-8 | 1 | 2 | 2 | 1 | 2 | 47 | 0 | - | 0 |
| ZV160610-58 | 1 | 2 | 3 | 1 | 1 | 62 | 0 | - | 0 |
| SF270710-103 | 1 | - | 2 | 0 | 0 | - | 0 | - | - |
| FM140710-86 | 1 | 3 | 3 | 1 | 2 | 54 | 0 | - | 0 |
| OD120510-3 | 1 | 2 | 2 | 1 | 2 | 45 | 0 | - | 0 |
| DK280610-66 | 1 | 3 | 3 | 1 | 1 | 78 | 0 | - | 0 |
| MJ020610-34 | 1 | 3 | 2 | 0 | 0 | - | 0 | - | 1 |
| JK140710-89 | 1 | 2 | 3 | 1 | 2 | 65 | 0 | - | 0 |
| RM210710-93 | 1 | 2 | 3 | 0 | 0 | - | 0 | - | - |
| AM140710-84 | 1 | 3 | 3 | 0 | 0 | - | 0 | - | 0 |
| BZ020610-35 | 1 | 2 | 0 | 1 | 2 | 8 | 0 | 0 | 0 |
| GP140710-90 | 1 | 2 | 2 | 1 | 1 | 41 | 0 | - | - |
| JC070710-77 | 1 | 2 | 3 | 2 | 2 | 28 | 0 | - | 0 |
| RM150610-98 | 0 | - | 2 | 0 | 0 | - | 0 | - | 0 |
| MJ240510-23 | 0 | - | 3 | 0 | 0 | - | 0 | - | 1 |
| EC070710-73 | 0 | - | 2 | 0 | 0 | - | 0 | - | - |
| DD170510-9 | 0 | - | 3 | 0 | 0 | - | 0 | - | 0 |
| FN280610-62 | 0 | - | 2 | 0 | 0 | - | 0 | - | 0 |
| AM140610-50 | 0 | - | 2 | 0 | 0 | - | 0 | - | 1 |
| KG170510-4 | 0 | - | 1 | 0 | 0 | - | 0 | - | 1 |
| AD130710-83 | 0 | - | 2 | 0 | 0 | - | 0 | - | 0 |
| AT090610-45 | 0 | - | 1 | 0 | 0 | - | 0 | - | 0 |
| GN280710-107 | 0 | - | 2 | 0 | 0 | - | 0 | - | - |
| MB140610-51 | 0 | - | 3 | 0 | 0 | - | 0 | - | 0 |
| JM130710-82 | 0 | - | 0 | 1 | 1 | 6 | 0 | - | - |
| Patients’ Characteristics | HPV-Positive Tumor Patients (n = 6) | HPV-Negative Tumor Patients (n = 34) | P Value * | Non-Tumor Patients (n = 12) | P Value ** |
|---|---|---|---|---|---|
| General | |||||
| Age (years) | 50.0 (13.0) | 50.5 (28.0) | 0.7 | 43.5 (38.0) | 0.7 |
| Male gender | 5 (83.3%) | 22 (64.7%) | 0.6 | 9 (81.8%) | 1.0 |
| Risk factors | |||||
| Oral thrush in history | 1 (20.0%) | 4 (14.3%) | 1.0 | 1 (10.0%) | 1.0 |
| Tuberculosis in history | 0 (0.0%) | 4 (14.3%) | 1.0 | 0 (0.0%) | a |
| Herpes zoster in history | 0 (0.0%) | 2 (7.1%) | 1.0 | 0 (0.0%) | a |
| HIV-positive | 0 (0.0%) | 4 (12.9%) | 1.0 | 2 (20.0%) | 0.5 |
| Smoker | 4 (80.0%) | 13 (46.4%) | 0.3 | 4 (40.0%) | 0.3 |
| Pack years | 3.0 (12.0) | 0.0 (4.0) | 0.1 | 0.0 (1.0) | 0.05 |
| Duration of smoking (years) | 15.0 (30.0) | 0.0 (9.0) | 0.08 | 0.0 (4.0) | 0.04 |
| Number of cigarettes | 6.0 (7.0) | 0.0 (6.0) | 0.2 | 0.0 (5.0) | 0.05 |
| Alcohol | 4 (80.0%) | 10 (35.7%) | 0.1 | 2 (20.0%) | 0.09 |
| Locally brewed alcohol | 4 (80.0%) | 8 (28.6%) | 0.047 | 1 (10.0%) | 0.02 |
| Duration of drinking alcohol (years) | 17.0 (21.0) | 0.0 (5.0) | 0.1 | 0.0 (1.0) | 0.02 |
| PCR Primer Mixture | Run | Number of Tested Samples | |
|---|---|---|---|
| Mixture 1 | HPV16, 18, 31, 45 | 1 | 55 |
| 2 | 55 | ||
| Duplex only | HPV16 and β-globin | 1 | 55 |
| 2 | 30 | ||
| 3 | 30 | ||
| Mixture 1 and 2 | HPV16, 18, 31, 45 and HPV33, 52, 58 and β-globin | 1 | 15 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Geßner, A.L.; Borkowetz, A.; Baier, M.; Göhlert, A.; Wilhelm, T.J.; Thumbs, A.; Borgstein, E.; Jansen, L.; Beer, K.; Mothes, H.; et al. Detection of HPV16 in Esophageal Cancer in a High-Incidence Region of Malawi. Int. J. Mol. Sci. 2018, 19, 557. https://doi.org/10.3390/ijms19020557
Geßner AL, Borkowetz A, Baier M, Göhlert A, Wilhelm TJ, Thumbs A, Borgstein E, Jansen L, Beer K, Mothes H, et al. Detection of HPV16 in Esophageal Cancer in a High-Incidence Region of Malawi. International Journal of Molecular Sciences. 2018; 19(2):557. https://doi.org/10.3390/ijms19020557
Chicago/Turabian StyleGeßner, Anja Lidwina, Angelika Borkowetz, Michael Baier, Angela Göhlert, Torsten J. Wilhelm, Alexander Thumbs, Eric Borgstein, Lars Jansen, Katrin Beer, Henning Mothes, and et al. 2018. "Detection of HPV16 in Esophageal Cancer in a High-Incidence Region of Malawi" International Journal of Molecular Sciences 19, no. 2: 557. https://doi.org/10.3390/ijms19020557