1. Introduction
Programmable nucleases like Clustered regularly interspaced short palindromic repeats/Cas9 (CRSIPR/Cas9), transcription activator-like effector nuclease (TALEn) and Zinc-finger nucleases (ZFN) have made considerable contributions to the development of precision gene editing and transgenics, but up to date they still face limitations due to a shortage of known sites for reliable and sustained transgene expression in transgenic livestock [
1]. The pig in particular, represents an important animal model with significance to both agriculture and biomedicine, yet the predefined genetic loci available for robust and durable transgene expression within the pig genome are very limited. To date only a handful of such loci in the pig have been studied, informed by comparative genomics of rodents, i.e., Rosa26, H11 and
COL1A sites [
2,
3,
4,
5]. There is an ever-increasing need to identify novel safe harbors in the pig genome so as to expand the toolbox to generate transgenic pigs for robust and site-specific transgene expression for economic trait improvement, disease modeling and mass production of therapeutic proteins [
6]. But up to now there has been no powerful approach available to identify novel safe sites in vivo at genome-wide level.
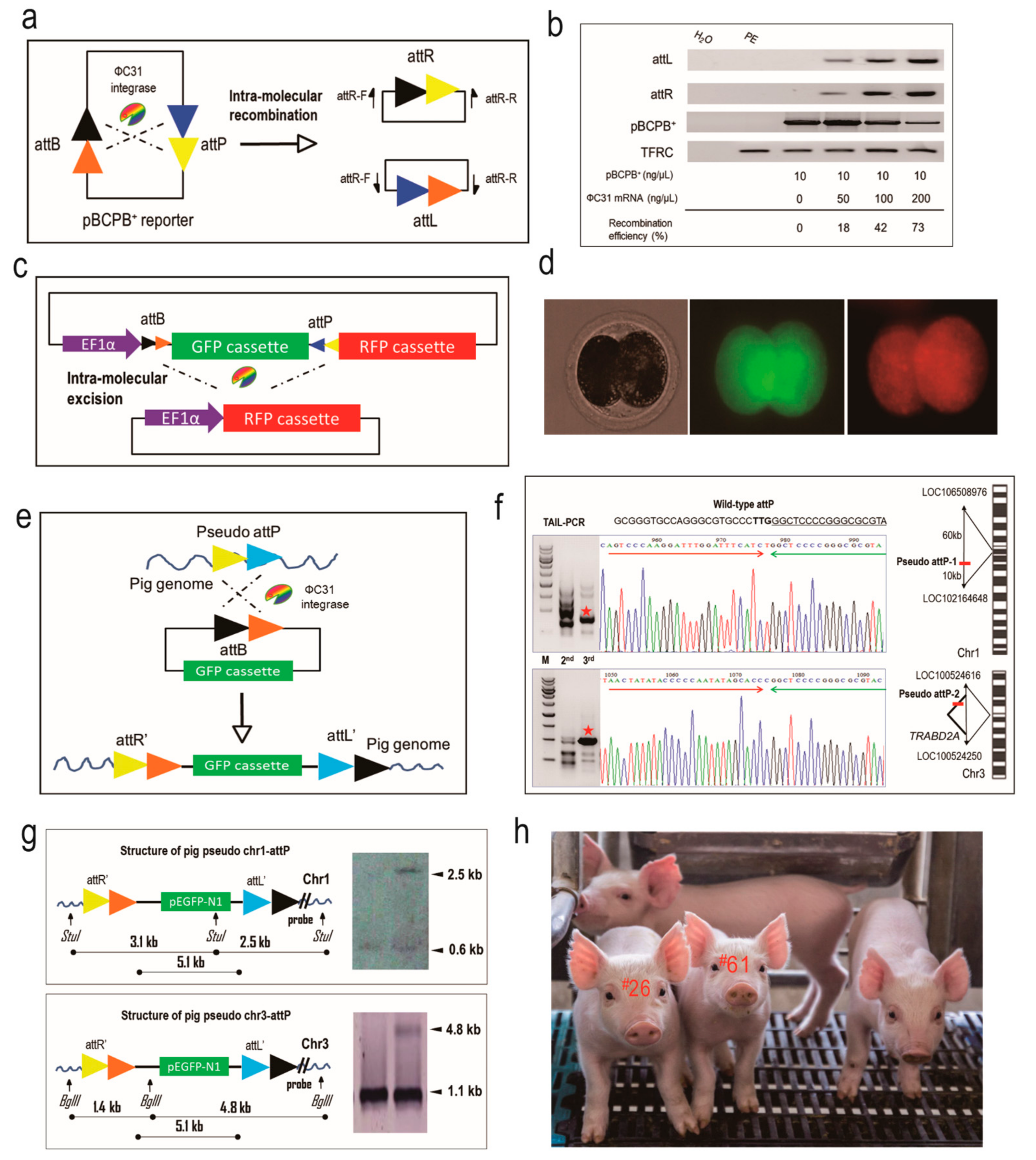
To address this issue, we took advantage of ФC31 integrase to integrate a reporter gene into pseudo attP sites of the pig genome so as to assess the expression potential of these sites. As shown in previous work [
7], ФC31 integrase is capable of catalyzing site-specific DNA recombination between an attB-containing donor plasmid and a pseudo attP site in mammalian genomes to form a single-copy and unidirectional genetic structure, which is believed to be beneficial to transgene expression [
8,
9,
10]. More importantly, the profiling of the pseudo attP sites in a given mammalian genome would help determine candidate sites for transgene integration and expression [
11]. Therefore, ФC31 integrase can be harnessed to locate potential safe harbors in the pig genome. Here, we isolated two novel pseudo attP sites in the pig genome using ФC31 integrase and characterized them as safe harbors for site-specific transgene integration and robust transgene expression. These sites add to our repertoire and provide new options to stably integrate a wide range of transgenes for robust and predictable expression. We envision that these novel safe harbors will facilitate the creation of transgenic pigs for agricultural and biomedical applications.
3. Discussion
Transgenic pigs hold promise for applications in the fields of agriculture and medicine. Tremendous effort has been invested in modifying and altering functional genes within the porcine genome to produce various lines of transgenic pigs. Nonetheless, the intricate interplays between exogenously integrated DNA sequences and the endogenous elements of the porcine chromosomal locus that can affect the phenotypes restrict the reliability of swine transgenics for basic, medical and agricultural applications. Although precision gene editing has made considerable progress, the issue of where to integrate a transgene into the porcine genome to minimize the risk of transgene expression silencing and maximize its expression efficacy has received little attention. Addressing this question would greatly accelerate the study and application of transgenic pigs.
In the pig, only a handful of studies have investigated the utility of just a few genomic loci for transgene expression, deduced from evolutionary comparative analysis. The Rosa26 locus was widely used in rodents, rats and human cells for transgene integration, and the porcine Rosa26 locus was identified recently [
2]. Another porcine transgene safe harbor was H11, which was identified as the orthologue to the mouse H11 site and proved to be a reliable locus for ectopic gene expression [
3]. The
COL1A locus was recently utilized for gene knock in in pigs [
4,
5]. However, all three loci are located near endogenous genes that can possibly be disregulated by the addition of a transgene and thus their expression efficacy and safety profiles remain uncertain. As a result, the identification of novel porcine safe harbors is highly desirable to provide suitable solutions for the specific requirements of different transgenic applications.
To date, there have been no specialized tools developed to identify safe harbors in the mammalian genome. The ФC31 integrase, which originates from a Streptomyces phage, has been utilized to integrate transgenes into the genomes of a variety of eukaryotes, ranging from yeast to human. The integrase is capable of recognizing the pseudo attP sites in the mammalian genomes and integrating a single-copy and unidirectional transgene into the pseudo attP sites, resulting in a transgenic structure more favorable for efficient transgene expression, than when compared to random integration [
14]. Accordingly, this advantage enables the direct comparison of the transgene expression efficacy at different pseudo attP sites and identifies the ideal integration locus as a safe harbor. In fact, ФC31 integrase has been applied to identify safe harbors from several mammalian species. In the bovine genome, Ou et al. discovered a safe harbor named BF4 at 4q31, where the transgene expression was as high as ~328 μg/mL, more than twice that of the other pseudo attP site [
15]. Olivares et al. integrated
Factor IX gene into the mouse genome by ФC31 integrase and identified mpsL1 as a preferential safe harbor which does not lie within coding sequences and which supports the
Factor IX ectopic expression as high as ~4 μg/mL in the serum [
8]. Bi et al. identified four pseudo attP sites in the porcine genome from cultured porcine cells and experimentally analyzed their expression efficacy in a reporter system, two of which were more potent than the other pseudo attP sites [
12]. However, many these studies merely detected pseudo attP sites in cultured cell lines, which still need further research to test their availability and in vivo potential for transgene expression.
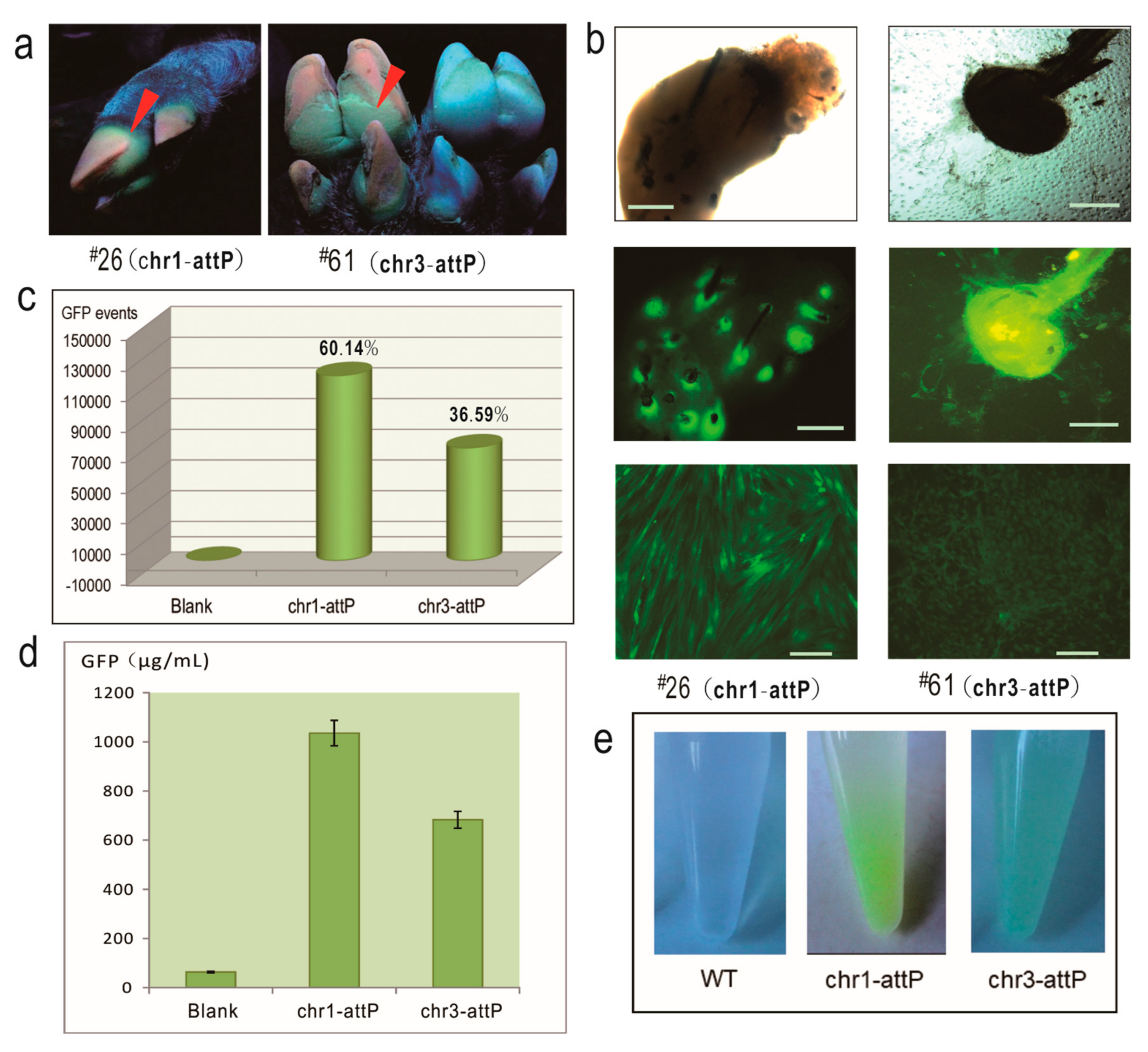
Here we extended the research to porcine fertilized embryos and identified safe harbors in the pig genome. Two microinjection routes were tested and assessed for their integration efficiency. For ICI, we failed to produce EGFP-positive piglets. We reasoned that the reporter DNA in the cytoplasm might not be efficiently transported into the nucleus for integration. For PNI, seven piglets were demonstrated to contain EGFP reporter, and two of them were identified to have EGFP integrated at pseudo attP sites. We speculated that for the other five piglets, integration was not mediated by ФC31 integrase but occurred through random integration by pronuclear microinjection. The two pseudo attP sites identified in this study recapitulated the previous work [
12], demonstrating that both the two pseudo attP sites are hot spots for transgene integration and expression. In particular, chr1-attP is an ideal transgene safe harbor as it is located in an intergenic region, with very low probability for any interference with endogenous gene expression. In contrast, chr3-attP is located in the intron of an endogenous gene, which could result in potential adverse effects on the physiology of the transgenic pigs. But due to the fact that the piglets grow and feed normally and are healthy, we conclude that chr3-attP may still be considered as safe harbor for reliable transgene expression in pigs.
We also observed that only a proportion of the cells we derived from our piglets were GFP-positive. This indicated that both EGFP-positive piglets were mosaic animals and that the integrase-mediated integration must have occurred not at the one-cell stage but when the embryo had already undergone the first cleavage division. The observed range for EGFP-positive cell populations further indicates that there could be a broad time window for the activation of ФC31 integrase in individual injected embryos. Injection of the integrase as a recombinant protein might allow an immediate and narrower time window for integration events following injection and minimize the level of mosaicism.
Although CRISPR/Cas9 technology is a promising tool for the editing of mammalian genomes, applications are presently limited due to a shortage of well characterized safe harbor sites and the low efficiencies for knockins of transgenes with the CRISPR technology. Our study identified novel safe harbors in the pig genome that can be utilized to allow site-specific integration and knockin of transgenes for reliable expression while not interfering with endogenous gene functions. We also demonstrated the utility of the ФC31 integrase for the efficient knockin of transgenes in pig, implying that it is possible to directly use it in other species. Considering the advantage of ФC31 integrase-catalyzing recombination, it could be modified to further improve the gene delivery efficiency by re-targeting or repeated use of pseudo attP sites. For the upcoming studies of efficient and repeated access of these safe harbor sites, our strategy would be extended from knockin of reporter plasmid to the knockin of an attP site or recombination sites such as loxP or FRT to fully harness the enzymatic efficiencies of ФC31 integrase or recombinases. This manipulation will also make most use of the potential of these porcine pseudo attP sites to allow for robust transgene expression.
4. Materials and Methods
4.1. Animal Ethics
All experimental procedures involving animals were reviewed, approved and supervised by the Animal Care Committee of the Institute of Animal Science and Veterinary Medicine, Hubei Academy of AgroSciences, Wuhan, China (Ethics code: 2013-14. Approved date: 8 October 2013). All wild-type and EGFP-positive pigs were raised with same diet and under identical conditions.
4.2. Plasmids and Strains
The ФC31 integrase expression plasmid (pCMV-Int) and reporter pBCPB
+ were obtained from Addgene (Cambridge, MA, USA). The reporter donor pEGFP-N1-attB was constructed as previously described [
10]. The reporter plasmid pT2Kmin-XIpGbR was kindly provided by Dr. James A. Lister (Virginia Commonwealth University, Richmond, VA, USA). TA cloning plasmid was purchased from Takara (Dalian, China). Competent
E. coli DH5α cells were prepared according to standard protocols. PureLink
® HiPure Plasmid Filter Purification Kits were used for midi and maxi preparation of all plasmid DNA (Invitrogen, Carlsbad, CA, USA). A P-class nanophotometer was used to measure the quality and quantity of DNA (Implen, Munich, Germany).
4.3. Nucleic Acid Manipulation
Pig genomic DNA was extracted by a Purelink Genomic DNA Mini kit (Invitrogen) and quantified using P-class nanophotometer. Probes used for Southern blotting were generated by PCR primer pairs PPP1-F/R and PPP2-F/R (
Table S1) and labeled using a DIG High Prime DNA Labeling and Detection Starter Kit I (Roche, Basel, Switzerland) in accordance with the manufacturer’s instructions. For Southern blotting, 20 μg of genomic DNA was digested by StuI or BglII restriction enzymes and the resultant fragments were separated by electrophoresis in an 0.8% agarose gel run at 20 V overnight. The gel was soaked in alkali solution (0.5 M NaOH, 1.5 M NaCl) for 2 × 15 min and the denatured DNA was transferred to a nylon membrane (Amersham, Piscataway, NJ, USA) using the sandwich method [
16]. The DNA was fixed by baking at 120 °C for 30 min. Hybridization was carried out at 42 °C overnight in transfer buffer (0.5 M NaOH, 1.5 M NaCl). The membrane was washed with 2 × Saline Sodium Citrate (SSC)/0.5% Sodium dodecyl sulfate (SDS) washing buffer to remove the non-specifically bound probes. Hybridization signals were detected by color development using Nitro-Blue-Tetrazolium/5-Bromo-4-Chloro-3-Indolyl Phosphate (NBT/BCIP) solution included in the kit.
The DNA sequence encoding the ФC31 integrase mRNA was amplified using PCR with T7-promoter-containing primers (
Table S1) and its mRNA was in vitro synthesized using a mMESSAGE mMACHINE mRNA transcription synthesis kit (Ambion, Austin, TX, USA). The ФC31 integrase mRNA was purified using phenol:chloroform extraction and isopropanol precipitation, and resuspended in RNase-free water.
4.4. Pig Embryo Engineering and Microinjection
For preparation of porcine parthenogenetic embryos, selected cumulus oocyte complexes (COCs) were washed three times in Dulbecco’s Phosphate Buffered Saline (DPBS) supplemented with 5% fetal bovine serum and three times in maturation medium consisting of Medium 199 (GIBCO) supplemented with 10% (
v/
v) pig follicular fluid, 0.1% (
w/
v) polyvinyl alcohol, 3.05 mM glucose, 0.91 mM sodium pyruvate, 0.57 mM
l-Cysteine, 100 IU/mL streptomycin sulphate (GIBCO), 100 IU/mL potassium penicillin G (GIBCO), 10 IU/mL PMSG (Ningbo Second Hormone Factory, Ningbo, China) and 10 IU/mL hCG (Ningbo Second Hormone Factory) [
17]. The COCs were incubated for 42–44 h in a 5-well dish at 39 °C in the Submarine Incubation System with 100% humidity and 5% CO
2. After the incubation, oocytes were denuded from cumulus cells by gent pipetting in DPBS with hyaluronidase (1 mg/mL). Oocytes with polar body I (pb I) were selected and washed three times with activation solution (0.3 M mannitol, 1 mM CaCl
2, 0.5 mM MgSO
4 and 0.05 mg/mL bovine serum albumin), then activated by a single DC pulse of 1.5 kV/cm for 30 μ s using a BTX Electro-Cell Manipulator 2001 (BTX Inc., San Diego, CA, USA).
The estrus conditions of all the sows (Landrace breed) were observed for 40 days (two estrus cycles) before beginning with the injection experiment. Sows (
n = 89) exhibiting normal estrus cycles were used for surgery and embryo transfer (
Figure S3). Sows showing signs of estrus were artificially inseminated twice with Large White boar semen (2 billion spermatozoa in 50 mL of semen extender) with an interval of 12 h between inseminations. Twelve hours after the second insemination, sows were anesthetized by 2.5% pentobarbital. Both of their oviducts were surgically exposed and in vivo fertilized embryos were flushed out into DPBS solution using a syringe of a 12-gauge needle. One-cell stage embryos were centrifuged in DPBS at 400×
g for 5 min to expose the pronucleus, and then microinjected, either into the pronucleus or the cytoplasm, with the ФC31 integrase mRNA and reporter plasmid and transferred back into the recipients. For the optimization of ФC31 integrase mRNA dosage, three concentrations (50, 100 and 200 ng/μL) were tested using parthenogenetic embryos. The optimal dosage turned out to be 200 ng/μL. A penicillin-streptomycin combination mixture was injected intraperitoneally into the recipients to protect against surgical wound infections.
4.5. End Point Real-Time and TAIL-PCR
An Long and Accurate (LA) Taq (Takara) DNA polymerase was used in the end-point PCR testing. A typical LA Taq PCR reaction mixture contained 5 μL 10 × LA PCR buffer (Mg
++ plus), 8 μL dNTP mixture (2.5 mM each), LA Taq polymerase 0.5 μL (5 units/μL), forward and reverse primer 1 μL (20 μM, respectively), 0.5 μg template and PCR-grade water added to 50 μL in total volume. Cycling conditions were 94 °C for 2 min, followed by 30 cycles of 94 °C 30 s, 55 °C 30 s and 72 °C 1 min/kb, followed by final extension for 5 min at 72 °C. A fifth of the volume of the PCR reaction was analyzed on either a 1% or 2% agarose gel and documented using a ChemDOC™ XRS
+ (Biorad, Berkeley, CA, USA). A SYBR Green I real-time PCR master mix from Toyobo (Osaka, Japan) was used in all quantitative PCR tests performed in a Rotor Gene6000 real-time rotary analyzer (Corbette Lifescience, Eppendorf, Hamburg, Germany). Briefly, the 20 μL reaction mixture included 10 μL 2× master mix, 0.5 μL primer mix (5 μM each), 1 μL template DNA (100 ng for genomic DNA or 5 ng for plasmid DNA) and 8.5 μL PCR-grade water. A two-step amplification protocol was used with the following parameters: a 2-min incubation at 95 °C was used to fully denature the template and activate the Taq DNA polymerase followed by 40 cycles, each of denaturation at 95 °C for 8 s, annealing and extension at 60 °C for 25 s. A final melting temperature analysis from 50 to 99 °C was used to ensure amplicon uniformity. Fluorescence was acquired at annealing and extension steps. All PCR amplifications were performed in triplicate. Relative signal intensities were calculated by the
ΔΔCt method with the built-in software RG6000 series 1.7. Copy number of the transgene was calculated using the single-copy
transferrin receptor protein 1 gene (TFRC) as an internal control with the following formula: Transgene copy number/pig genome = molecules of transgene/molecules of
TFRC × 2. For the quantitative expression analysis, cDNAs were assessed using the
ΔΔCt method and normalized against the reference gene
GAPDH. TAIL-PCR was conducted essentially as previously described [
18]. All primer sequences are listed in
Table S1.
4.6. Cell Culture
Primary fibroblast cells of the two piglets were isolated from ear tissues when notching. The ear tissues were immediately stored in sterile DPBS with 250 U/mL penicillin and 250 U/mL streptomycin and transported to the laboratory. After rinsing 3 to 5 times in the DPBS and then 70% alcohol, the ear biopsies were cut into pieces of 1–3 mm3 in size, and digested in 0.25% trypsin and 0.04% EDTA solution at 4 °C for 1–2 h and then at 39 °C for additional 30–60 min. The treatment was stopped by adding 1 mL of 39 °C pre-warmed DMEM/F12 (GIBCO) culture medium (containing 20% fetal bovine serum, 2 mmol/L glutamine, 100 U/mL penicillin and 100 U/mL streptomycin). The cell density was adjusted to 5 × 105 to 1 × 106 cells/mL with culture medium. The flask was incubated in a 39 °C, saturated humidity, 5% CO2 incubator, and the culture medium was replaced every two days.
4.7. FACS and ELISA
The FACS analyses were performed using a BD FACS Aria III (BD Biosciences, San Jose, CA, USA). Excitation and emission wavelengths for EGFP were 488 nm and 520 nm, respectively. Briefly, the cells were filtered by a 100-well net and resuspended in DPBS with a total of 2 × 105 cells in each sample for sorting. Average EGFP fluorescence (GFP-A) was measured and the EGFP-positive cell population was collected. The sorted cells were further cultured until they reach 90% confluence and then 1 × 106 cells were harvested and lysed to measure the concentration of EGFP in the cell lysate by an ELISA kit (Abcam, San Francisco, CA, USA). All artwork was created using CorelDRAW Graphics (Corel Corporation, Ottawa, ON, Canada). All data generated or analyzed during this study are included in this manuscript. All materials in this study are available to non-commercial uses.



{kind=link}
{kind=link}
{kind=link}