Prevalence of Antibiotic Resistance Genes in Air-Conditioning Systems in Hospitals, Farms, and Residences

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
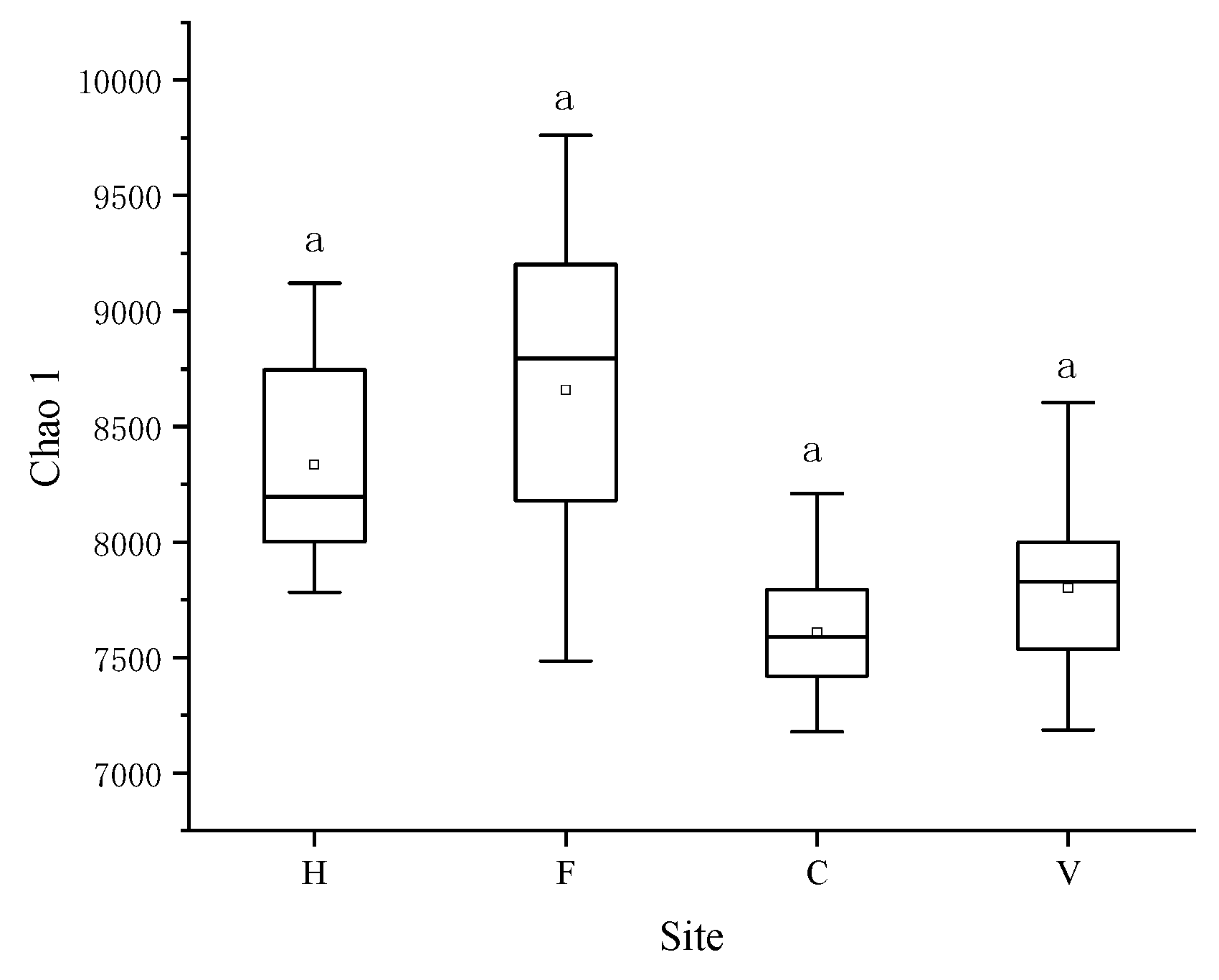{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Site Selection and Sample Collection
2.2. DNA Extraction and Sequencing
2.3. Phylotype Analysis
2.4. ARGs Determination
2.5. Analysis of ARGs
2.6. Statistical and Network Analysis
3. Results
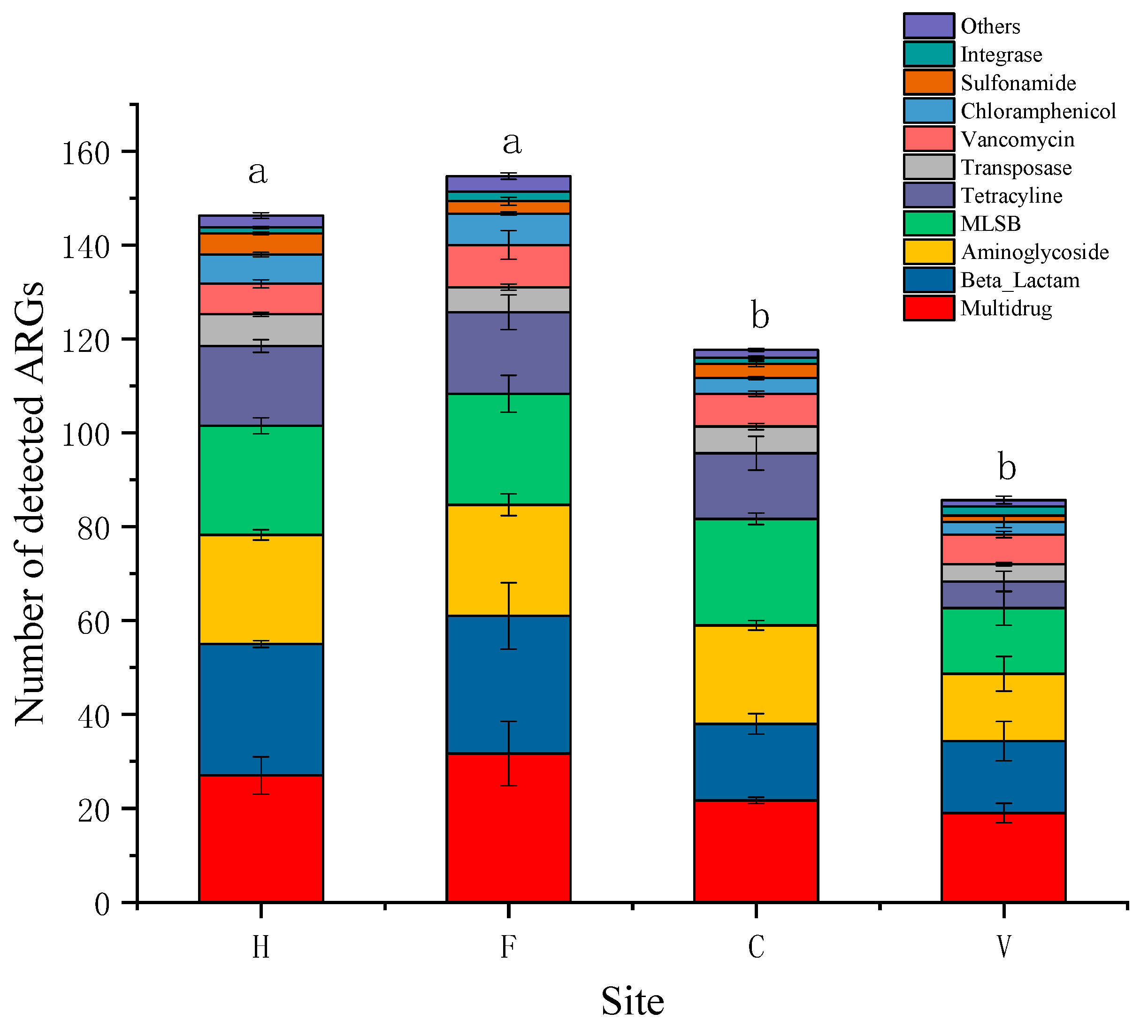
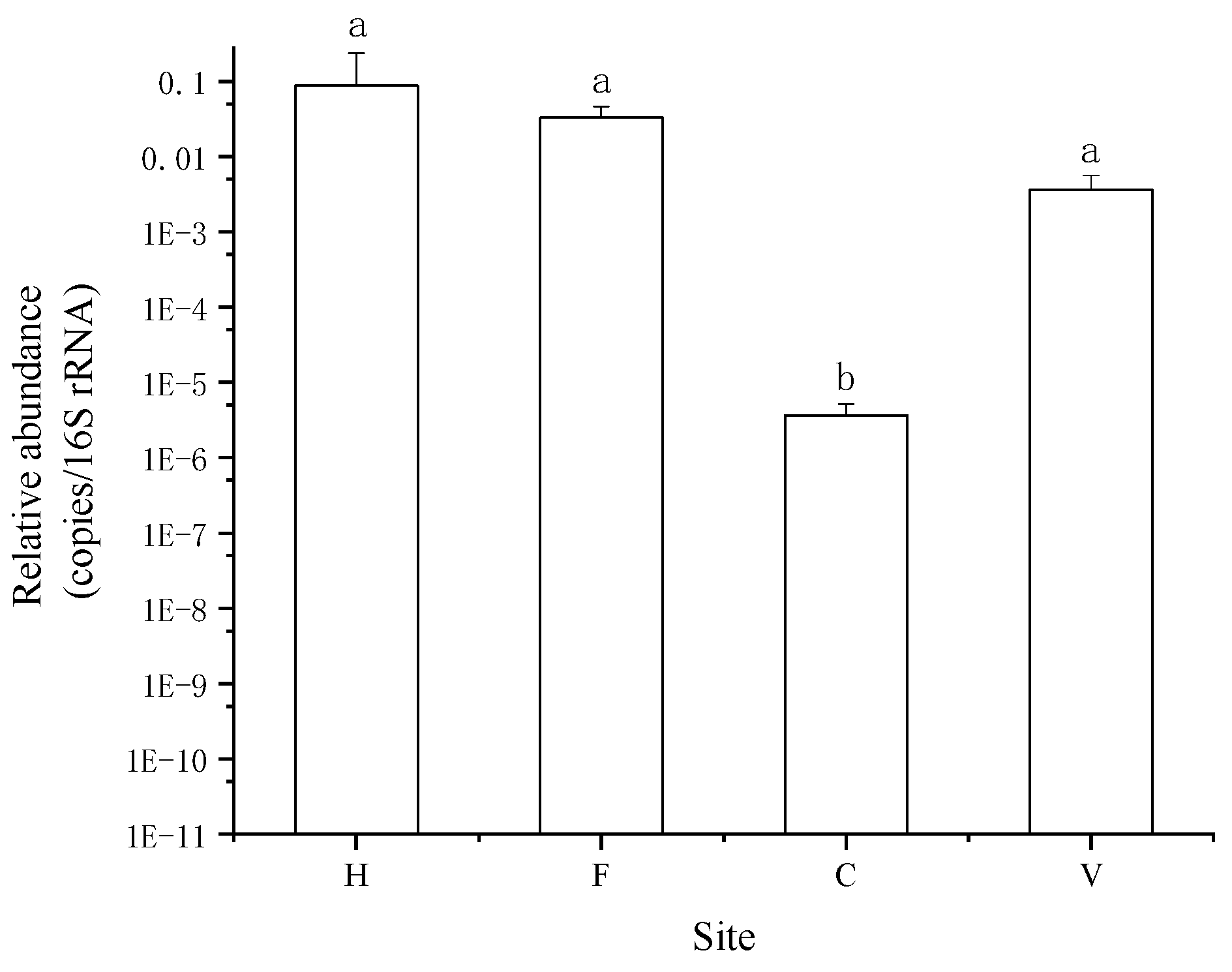
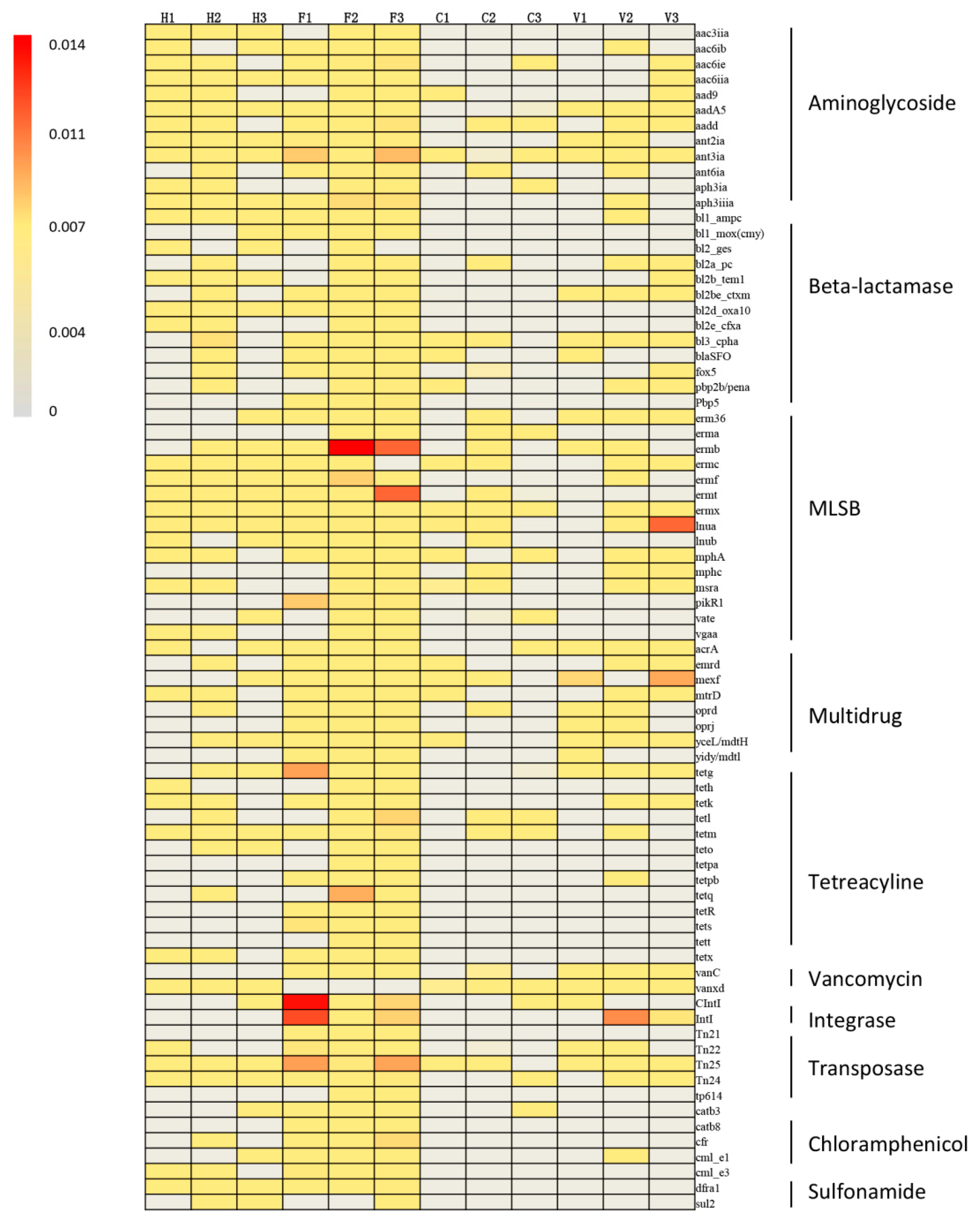
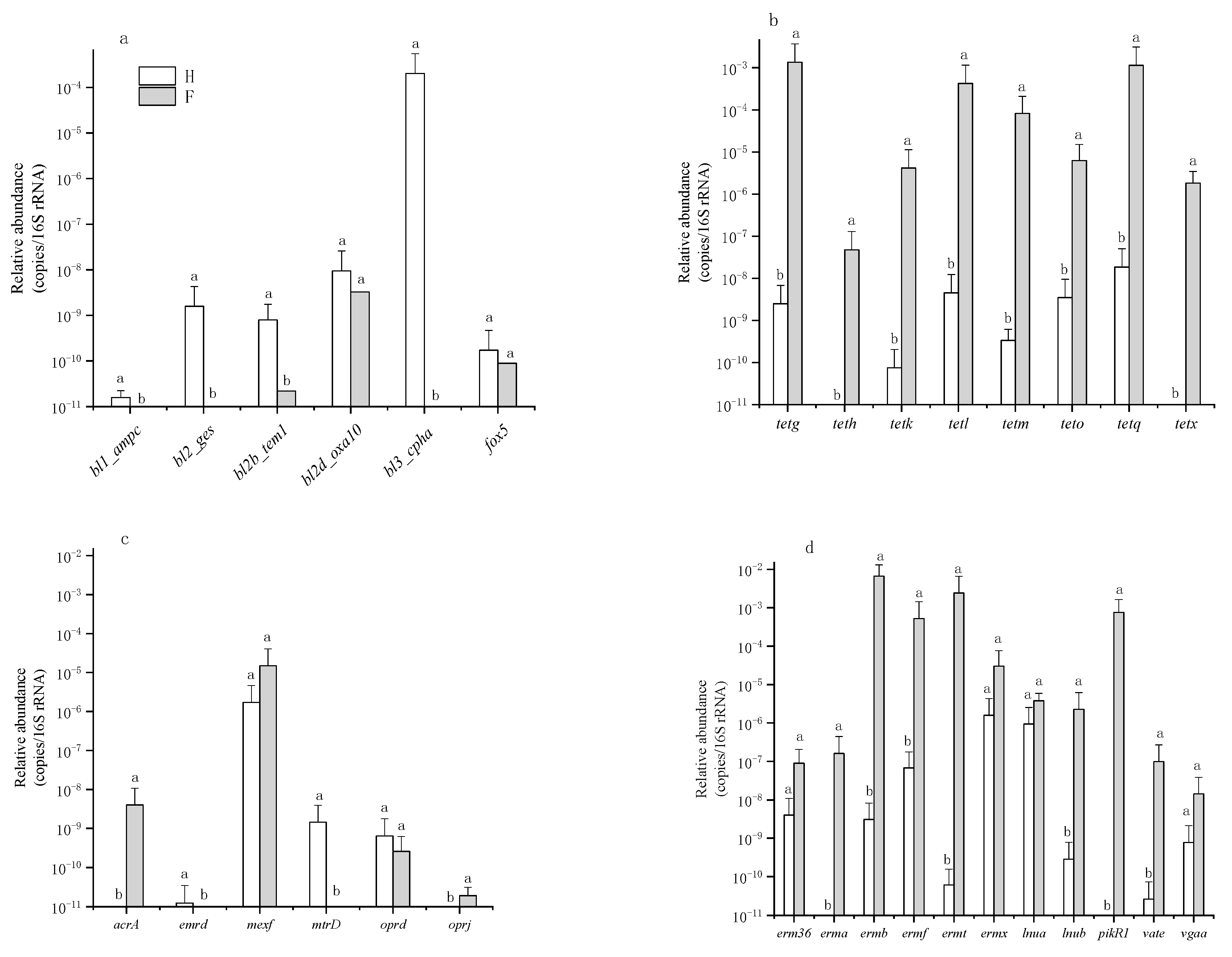
3.1. Occurrence and Abundance of ARGs
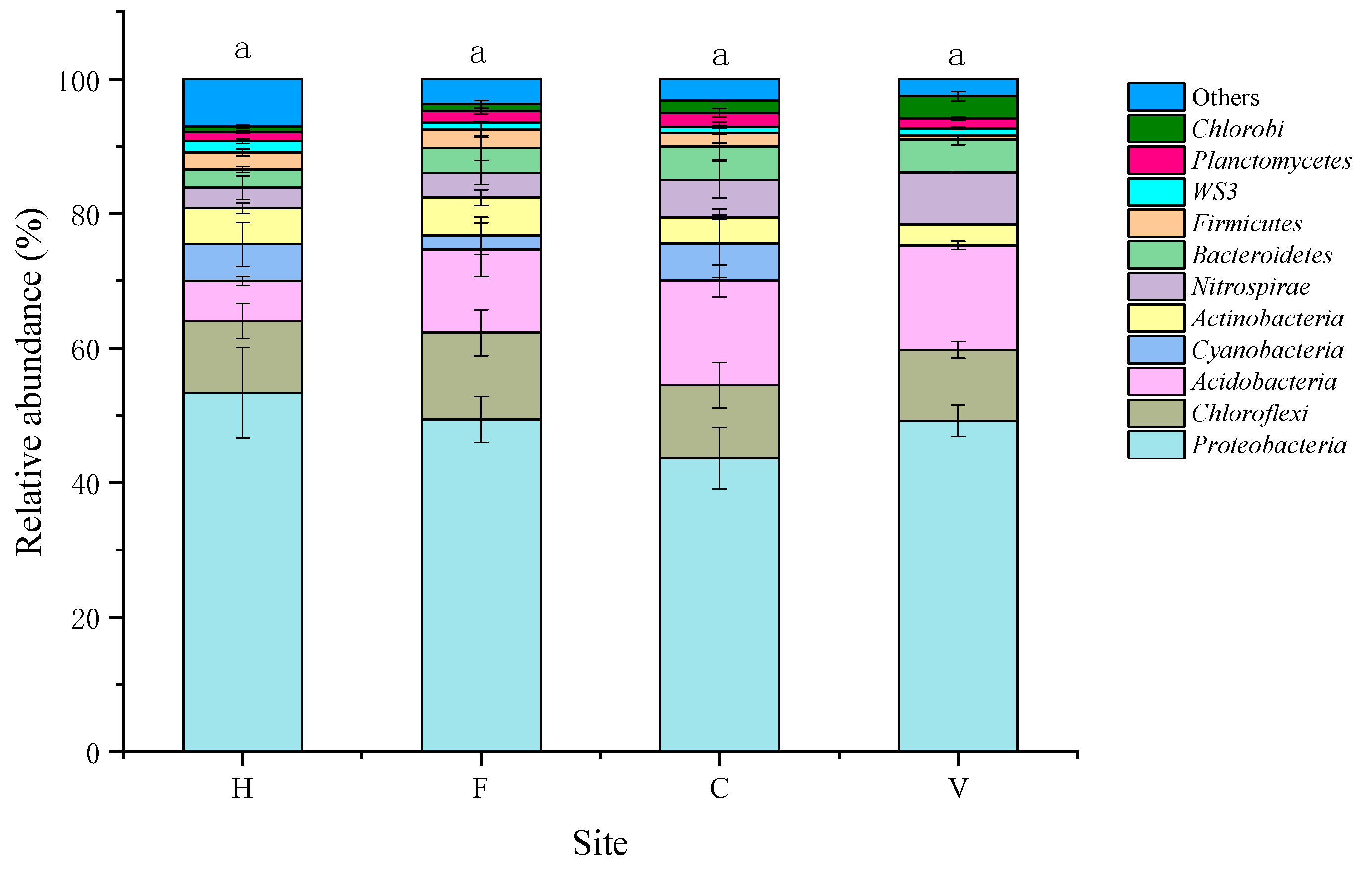
3.2. Characterization of Bacterial Community
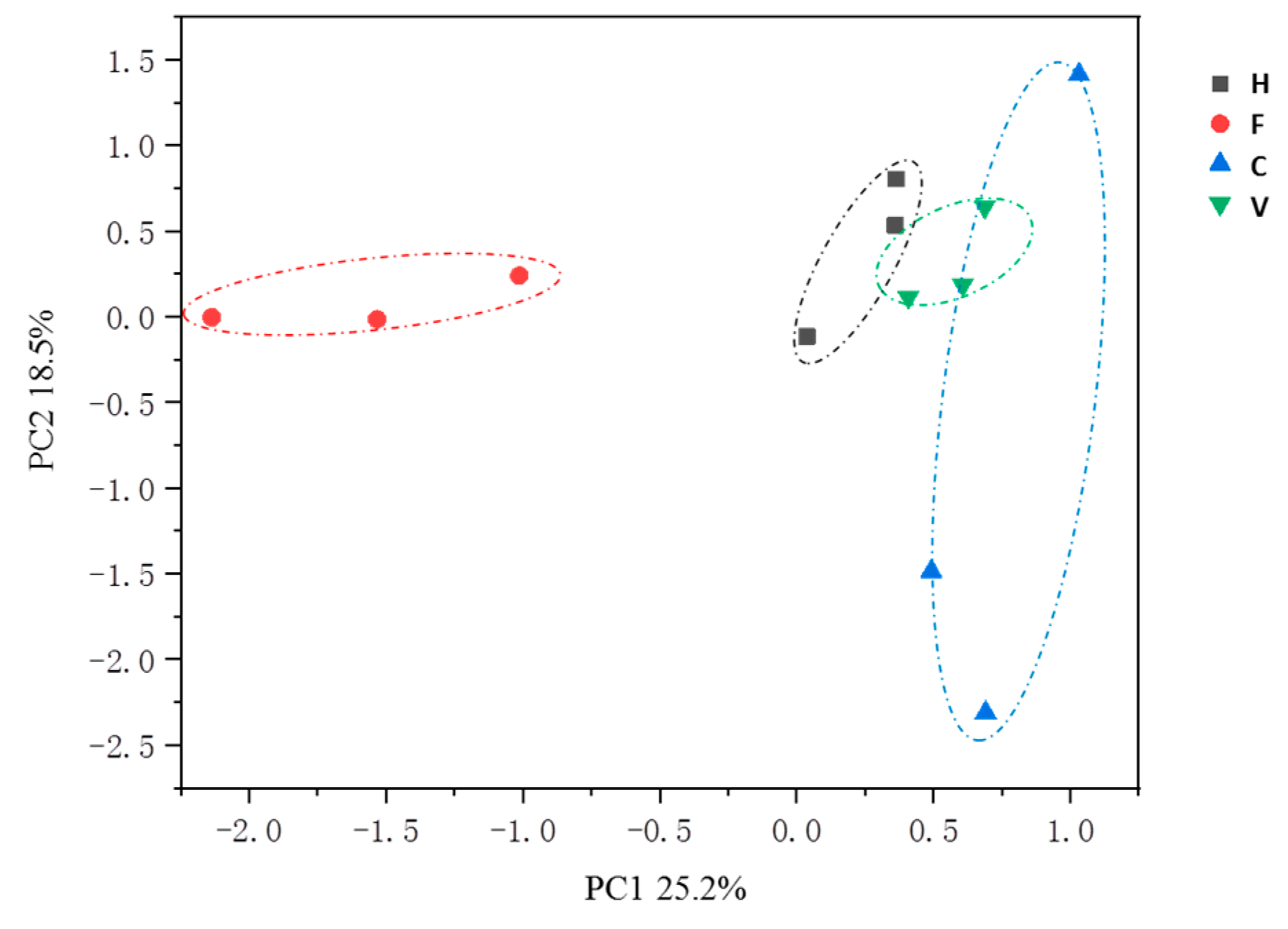
3.3. Correlation of ARGs with Bacterial Community
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Wright, G.D. Antibiotic resistance in the environment: A link to the clinic? Curr. Opin. Microbiol. 2010, 13, 589–594. [Google Scholar] [CrossRef] [PubMed]
- WHO. Global Action Plan on Antimicrobial Resistance; WHO: Geneva, Switzerland, 2015. [Google Scholar]
- Zhang, Q.Q.; Ying, G.G.; Pan, C.G.; Liu, Y.S.; Zhao, J.L. Comprehensive evaluation of antibiotics emission and fate in the river basins of China: Source analysis, multimedia modeling, and linkage to bacterial resistance. Environ. Sci. Technol. 2015, 49, 6772–6782. [Google Scholar] [CrossRef] [PubMed]
- Massive Antibiotic Pollution in China’s Rivers ‘Fueled by Abuse’. Available online: https://www.bignewsnetwork.com/news/234545909/massive-antibiotic-pollution-in-china-rivers-fueled-by-abuse (accessed on 26 February 2019).
- Holman, D.B.; Chenier, M.R. Antimicrobial use in swine production and its effect on the swine gut microbiota and antimicrobial resistance. Can. J. Microbiol. 2015, 61, 785–798. [Google Scholar] [CrossRef] [PubMed]
- Van Boeckel, T.P.; Brower, C.; Gilbert, M.; Grenfell, B.T.; Levin, S.A.; Robinson, T.P.; Teillant, A.; Laxminarayan, R. Global trends in antimicrobial use in food animals. Proc. Natl. Acad. Sci. USA 2015, 112, 5649–5654. [Google Scholar] [CrossRef] [PubMed]
- Fang, H.; Wang, H.F.; Cai, L.; Yu, Y.L. Prevalence of antibiotic resistance genes and bacterial pathogens in long-term manured greenhouse soils as revealed by metagenomic survey. Environ. Sci. Technol. 2015, 49, 1095–1104. [Google Scholar] [CrossRef] [PubMed]
- Forsberg, K.J.; Patel, S.; Gibson, M.K.; Lauber, C.L.; Knight, R.; Fierer, N.; Dantas, G. Bacterial phylogeny structures soil resistomes across habitats. Nature 2014, 509, 612. [Google Scholar] [CrossRef] [PubMed]
- Xiao, K.Q.; Li, B.; Ma, L.P.; Bao, P.; Zhou, X.; Zhang, T.; Zhu, Y.G. Metagenomic profiles of antibiotic resistance genes in paddy soils from South China. FEMs Microbiol. Ecol. 2016, 92, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Chang, X.S.; Meyer, M.T.; Liu, X.Y.; Zhao, Q.; Chen, H.; Chen, J.A.; Qiu, Z.Q.; Yang, L.; Cao, J.; Shu, W.Q. Determination of antibiotics in sewage from hospitals, nursery and slaughter house, wastewater treatment plant and source water in Chongqing region of Three Gorge Reservoir in China. Environ. Pollut. 2010, 158, 1444–1450. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, Y.; Veillette, M.; Duchaine, C. Airborne bacteria and antibiotic resistance genes in hospital rooms. Aerobiologia 2010, 26, 185–194. [Google Scholar] [CrossRef]
- Kummerer, K.; Henninger, A. Promoting resistance by the emission of antibiotics from hospitals and households into effluent. Clin. Microbiol. Infect. 2003, 9, 1203–1214. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.W.; Yang, Y.; Liang, X.M.; Yu, K.; Zhang, T.; Li, X.D. Metagenomic profiles of antibiotic resistance genes (args) between human impacted estuary and deep ocean sediments. Environ. Sci. Technol. 2013, 47, 12753–12760. [Google Scholar] [CrossRef] [PubMed]
- Christgen, B.; Yang, Y.; Ahammad, S.Z.; Li, B.; Rodriquez, D.C.; Zhang, T.; Graham, D.W. Metagenomics shows that low-energy anaerobic-aerobic treatment reactors reduce antibiotic resistance gene levels from domestic wastewater. Environ. Sci. Technol. 2015, 49, 2577–2584. [Google Scholar] [CrossRef] [PubMed]
- Garner, E.; Wallace, J.S.; Argoty, G.A.; Wilkinson, C.; Fahrenfeld, N.; Heath, L.S.; Zhang, L.Q.; Arabi, M.; Aga, D.S.; Pruden, A. Metagenomic profiling of historic Colorado Front Range flood impact on distribution of riverine antibiotic resistance genes. Sci. Rep. 2016, 6, 38432. [Google Scholar] [CrossRef] [PubMed]
- Jia, S.Y.; Zhang, X.X.; Miao, Y.; Zhao, Y.T.; Ye, L.; Li, B.; Zhang, T. Fate of antibiotic resistance genes and their associations with bacterial community in livestock breeding wastewater and its receiving river water. Water Res. 2017, 124, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Lau, C.H.F.; Li, B.; Zhang, T.; Tien, Y.C.; Scott, A.; Murray, R.; Sabourin, L.; Lapen, D.R.; Duenk, P.; Topp, E. Impact of pre-application treatment on municipal sludge composition, soil dynamics of antibiotic resistance genes, and abundance of antibiotic-resistance genes on vegetables at harvest. Sci. Total Environ. 2017, 587, 214–222. [Google Scholar] [CrossRef] [PubMed]
- Su, J.Q.; An, X.L.; Li, B.; Chen, Q.L.; Gillings, M.R.; Chen, H.; Zhang, T.; Zhu, Y.G. Metagenomics of urban sewage identifies an extensively shared antibiotic resistome in China. Microbiome 2017, 5, 84. [Google Scholar] [CrossRef] [PubMed]
- Shiraki, K.; Yamada, H.; Yoshida, Y.; Ohno, A.; Watanabe, T.; Watanabe, T.; Watanabe, H.; Watanabe, H.; Yamaguchi, M.; Tokuoka, F.; et al. Improved photocatalytic air cleaner with decomposition of aldehyde and aerosol-associated influenza virus infectivity in indoor air. Aerosol Air Qual. Res. 2017, 17, 2901–2912. [Google Scholar] [CrossRef]
- Lin, H.H.; Ezzati, M.; Murray, M. Tobacco smoke, indoor air pollution and tuberculosis: A systematic review and meta-analysis. PLoS Med. 2007, 4, 173–189. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Gao, B.F.; Jin, Y.H.; Xu, N.; Guo, T.P. Acupuncture for common cold: A systematic review and meta-analyze protocol. Medicine 2018, 97, 10. [Google Scholar] [CrossRef] [PubMed]
- Fisman, D.N. Of Time and the river: How our understanding of legionellosis has changed since 1976. J. Infect. Dis. 2018, 217, 171–173. [Google Scholar] [CrossRef] [PubMed]
- Poole, J.A.; Romberger, D.J. Immunological and inflammatory responses to organic dust in agriculture. Curr. Opin. Allergy Clin. Immunol. 2012, 12, 126–132. [Google Scholar] [CrossRef] [PubMed]
- Mendell, M.J. Commentary: Air conditioning as a risk for increased use of health services. Int. J. Epidemiol. 2004, 33, 1123–1126. [Google Scholar] [CrossRef] [PubMed]
- Graudenz, G.S.; Oliveira, C.H.; Tribess, A.; Mendes, C.; Latorre, M.R.D.O.; Kalil, J. Association of air-conditioning with respiratory symptoms in office workers in tropical climate. Indoor Air 2005, 15, 62–66. [Google Scholar] [CrossRef] [PubMed]
- Su, J.Q.; Xia, Y.; Yao, H.Y.; Li, Y.Y.; An, X.L.; Singh, B.K.; Zhang, T.; Zhu, Y.G. Metagenomic assembly unravel microbial response to redox fluctuation in acid sulfate soil. Soil Biol. Biochem. 2017, 105, 244–252. [Google Scholar] [CrossRef]
- Zhou, J.Z.; Bruns, M.A.; Tiedje, J.M. DNA recovery from soils of diverse composition. Appl. Environ. Microbiol. 1996, 62, 316–322. [Google Scholar] [PubMed]
- Long, X.E.; Huang, Y.; Chi, H.F.; Li, Y.Y.; Ahmad, N.; Yao, H.Y. Nitrous oxide flux, ammonia oxidizer and denitrifier abundance and activity across three different landfill cover soils in Ningbo, China. J. Clean Prod. 2018, 170, 288–297. [Google Scholar] [CrossRef]
- Turner, S.; Pryer, K.M.; Miao, V.P.; Palmer, J.D. Investigating deep phylogenetic relationships among cyanobacteria and plastids by small subunit rRNA sequence analysis. J. Eukaryot. Microbiol. 1999, 46, 327–338. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Pena, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef] [PubMed]
- Su, J.Q.; Wei, B.; Ou-Yang, W.Y.; Huang, F.Y.; Zhao, Y.; Xu, H.J.; Zhu, Y.G. Antibiotic resistome and its association with bacterial communities during sewage sludge composting. Environ. Sci. Technol. 2015, 49, 7356–7363. [Google Scholar] [CrossRef] [PubMed]
- An, X.L.; Chen, Q.L.; Zhu, D.; Zhu, Y.G.; Gillings, M.R.; Su, J.Q. Impact of wastewater treatment on the prevalence of integrons and the genetic diversity of integron gene cassettes. Appl. Environ. Microbiol. 2018, 84, e02766-17. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.H.; Qiao, M.; Su, J.Q.; Chen, Z.; Zhou, X.; Zhu, Y.G. High throughput profiling of antibiotic resistance genes in urban park soils with reclaimed water irrigation. Environ. Sci. Technol. 2014, 48, 9079–9085. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, W.Y.; Huang, F.Y.; Zhao, Y.; Li, H.; Su, J.Q. Increased levels of antibiotic resistance in urban stream of Jiulongjiang River, China. Appl. Microbiol. Biotechnol. 2015, 99, 5697–5707. [Google Scholar] [CrossRef] [PubMed]
- Looft, T.; Johnson, T.A.; Allen, H.K.; Bayles, D.O.; Alt, D.P.; Stedtfeld, R.D.; Sul, W.J.; Stedtfeld, T.M.; Chai, B.L.; Cole, J.R.; et al. In-feed antibiotic effects on the swine intestinal microbiome. Proc. Natl. Acad. Sci. USA 2012, 109, 1691–1696. [Google Scholar] [CrossRef] [PubMed]
- Qiao, M.; Ying, G.G.; Singer, A.C.; Zhu, Y.G. Review of antibiotic resistance in China and its environment. Environ. Int. 2018, 110, 160–172. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.X.; Feng, J.; Yin, X.L.; Liu, J.; Fu, W.J.; Berendonk, T.U.; Zhang, T.; Li, X.Y.; Li, B. Antibiotic resistome in landfill leachate from different cities of China deciphered by metagenomic analysis. Water Res. 2018, 134, 126–139. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.Y.; Chapman, S.J.; Nicol, G.W.; Yao, H.Y. Nitrification and nitrifiers in acidic soils. Soil Biol. Biochem. 2018, 116, 290–301. [Google Scholar] [CrossRef]
- McEachran, A.D.; Blackwell, B.R.; Hanson, J.D.; Wooten, K.J.; Mayer, G.D.; Cox, S.B.; Smith, P.N. Antibiotics, bacteria, and antibiotic resistance genes: Aerial transport from cattle feed yards via particulate matter. Environ. Health Perspect. 2015, 123, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Cao, C.; Jiang, W.J.; Wang, B.Y.; Fang, J.H.; Lang, J.D.; Tian, G.; Jiang, J.K.; Zhu, T.F. Inhalable microorganisms in Beijing’s PM2.5 and PM10 pollutants during a severe smog event. Environ. Sci. Technol. 2014, 48, 1499–1507. [Google Scholar] [CrossRef] [PubMed]
- Behzad, H.; Gojobori, T.; Mineta, K. Challenges and opportunities of airborne metagenomics. Genome Biol. Evol. 2015, 7, 1216–1226. [Google Scholar] [CrossRef] [PubMed]
- Miletto, M.; Lindow, S.E. Relative and contextual contribution of different sources to the composition and abundance of indoor air bacteria in residences. Microbiome 2015, 3, 61. [Google Scholar] [CrossRef] [PubMed]
- Prussin, A.J.; Marr, L.C. Sources of airborne microorganisms in the built environment. Microbiome 2015, 3, 78. [Google Scholar] [CrossRef] [PubMed]
- Adams, R.I.; Miletto, M.; Lindow, S.E.; Taylor, J.W.; Bruns, T.D. Airborne bacterial communities in residences: Similarities and differences with fungi. PLoS ONE 2014, 9, e91283. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.L.; Zhao, F.Z.; Zhang, X.X.; Li, K.; Li, C.R.; Ye, L.; Li, M. Metagenomic profiling of ARGs in airborne particulate matters during a severe smog event. Sci. Total Environ. 2018, 615, 1332–1340. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.J.; Sun, J.Y.; Zhang, X.Y.; Zhang, Y.M.; Zhang, L.; Che, H.C.; Ma, Q.L.; Yu, X.M.; Yue, Y.; Zhang, Y.W. Characterization of submicron aerosols and effect on visibility during a severe haze-fog episode in Yangtze River Delta, China. Atmos. Environ. 2015, 120, 307–316. [Google Scholar] [CrossRef]
- Zhu, Y.G.; Johnson, T.A.; Su, J.Q.; Qiao, M.; Guo, G.X.; Stedtfeld, R.D.; Hashsham, S.A.; Tiedje, J.M. Diverse and abundant antibiotic resistance genes in Chinese swine farms. Proc. Natl. Acad. Sci. USA 2013, 110, 3435–3440. [Google Scholar] [CrossRef] [PubMed]
- Hong, P.Y.; Li, X.Z.; Yang, X.F.; Shinkai, T.; Zhang, Y.H.; Wang, X.L.; Mackie, R.I. Monitoring airborne biotic contaminants in the indoor environment of pig and poultry confinement buildings. Environ. Microbiol. 2012, 14, 1420–1431. [Google Scholar] [CrossRef] [PubMed]
- Gillings, M.R.; Gaze, W.H.; Pruden, A.; Smalla, K.; Tiedje, J.M.; Zhu, Y.G. Using the class 1 integron-integrase gene as a proxy for anthropogenic pollution. ISME J. 2015, 9, 1269–1279. [Google Scholar] [CrossRef] [PubMed]
- D’Costa, V.M.; McGrann, K.M.; Hughes, D.W.; Wright, G.D. Sampling the antibiotic resistome. Science 2006, 311, 374–377. [Google Scholar] [CrossRef] [PubMed]
- Livermore, D.M. Are all beta-lactams created equal? Scand. J. Infect. Dis. 1996, 1, 33–43. [Google Scholar]
- Bush, K.; Bradford, P.A. Beta-lactams and beta-lactamase inhibitors: An overview. Cold Spring Harb. Perspect. Med. 2016, 6, a025247. [Google Scholar]
- Jia, W.; Li, G.; Wang, W. Prevalence and antimicrobial resistance of enterococcus species: A hospital-based study in China. Int. J. Environ. Res. Public Health 2014, 11, 3424–3442. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Wang, X.L.; Lang, X.L.; Wang, Y.C.; Dang, Y.; Zhang, F.X.; Tang, J.; Li, X.Y.; Feng, X. Preparation and application of microarrays for the detection of antibiotic resistance genes in samples isolated from Changchun, China. Mol. Biol. Rep. 2010, 37, 1857–1865. [Google Scholar] [CrossRef] [PubMed]
- Rowe, W.P.M.; Baker-Austin, C.; Verner-Jeffreys, D.W.; Ryan, J.J.; Micallef, C.; Maskell, D.J.; Pearce, G.P. Overexpression of antibiotic resistance genes in hospital effluents over time. J. Antimicrob. Chemother. 2017, 72, 1617–1623. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.L.; Shao, M.F.; Wang, Q.; Wang, L.T.; Fang, W.Y.; Ouyang, F.; Li, J. Airborne microbial communities in the atmospheric environment of urban hospitals in China. J. Hazard. Mater. 2018, 349, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Yang, Y.; Ma, L.P.; Ju, F.; Guo, F.; Tiedje, J.M.; Zhang, T. Metagenomic and network analysis reveal wide distribution and co-occurrence of environmental antibiotic resistance genes. ISME J. 2015, 9, 2490–2502. [Google Scholar] [CrossRef] [PubMed]
- Jang, H.M.; Kim, Y.B.; Choi, S.; Lee, Y.; Shin, S.G.; Unno, T.; Kim, Y.M. Prevalence of antibiotic resistance genes from effluent of coastal aquaculture, South Korea. Environ. Pollut. 2018, 233, 1049–1057. [Google Scholar] [CrossRef] [PubMed]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.; Liao, H.; Yao, H. Prevalence of Antibiotic Resistance Genes in Air-Conditioning Systems in Hospitals, Farms, and Residences. Int. J. Environ. Res. Public Health 2019, 16, 683. https://doi.org/10.3390/ijerph16050683
Li Y, Liao H, Yao H. Prevalence of Antibiotic Resistance Genes in Air-Conditioning Systems in Hospitals, Farms, and Residences. International Journal of Environmental Research and Public Health. 2019; 16(5):683. https://doi.org/10.3390/ijerph16050683
Chicago/Turabian StyleLi, Yaying, Hongkai Liao, and Huaiying Yao. 2019. "Prevalence of Antibiotic Resistance Genes in Air-Conditioning Systems in Hospitals, Farms, and Residences" International Journal of Environmental Research and Public Health 16, no. 5: 683. https://doi.org/10.3390/ijerph16050683
APA StyleLi, Y., Liao, H., & Yao, H. (2019). Prevalence of Antibiotic Resistance Genes in Air-Conditioning Systems in Hospitals, Farms, and Residences. International Journal of Environmental Research and Public Health, 16(5), 683. https://doi.org/10.3390/ijerph16050683