Portable Analyzer for On-Site Determination of Dissolved Organic Carbon—Development and Field Testing

Abstract
:1. Introduction
2. Materials and Methods
2.1. Development and Setup of a Portable Prototype
2.2. DOC Decomposition Experiments
2.3. Optimization of the Electrolyte Composition
2.4. Method Validation
2.5. Field Testing
2.6. Chemical Analysis
2.6.1. Total Oxidizing Agents
2.6.2. Hydrogen Peroxide
2.6.3. Further Parameter
3. Results
3.1. Development and Technical Optimization of the Portable Prototype
3.1.1. Influence of the Degree of Crystallinity of the BDD Anode
3.1.2. Selection of the Cathode Material
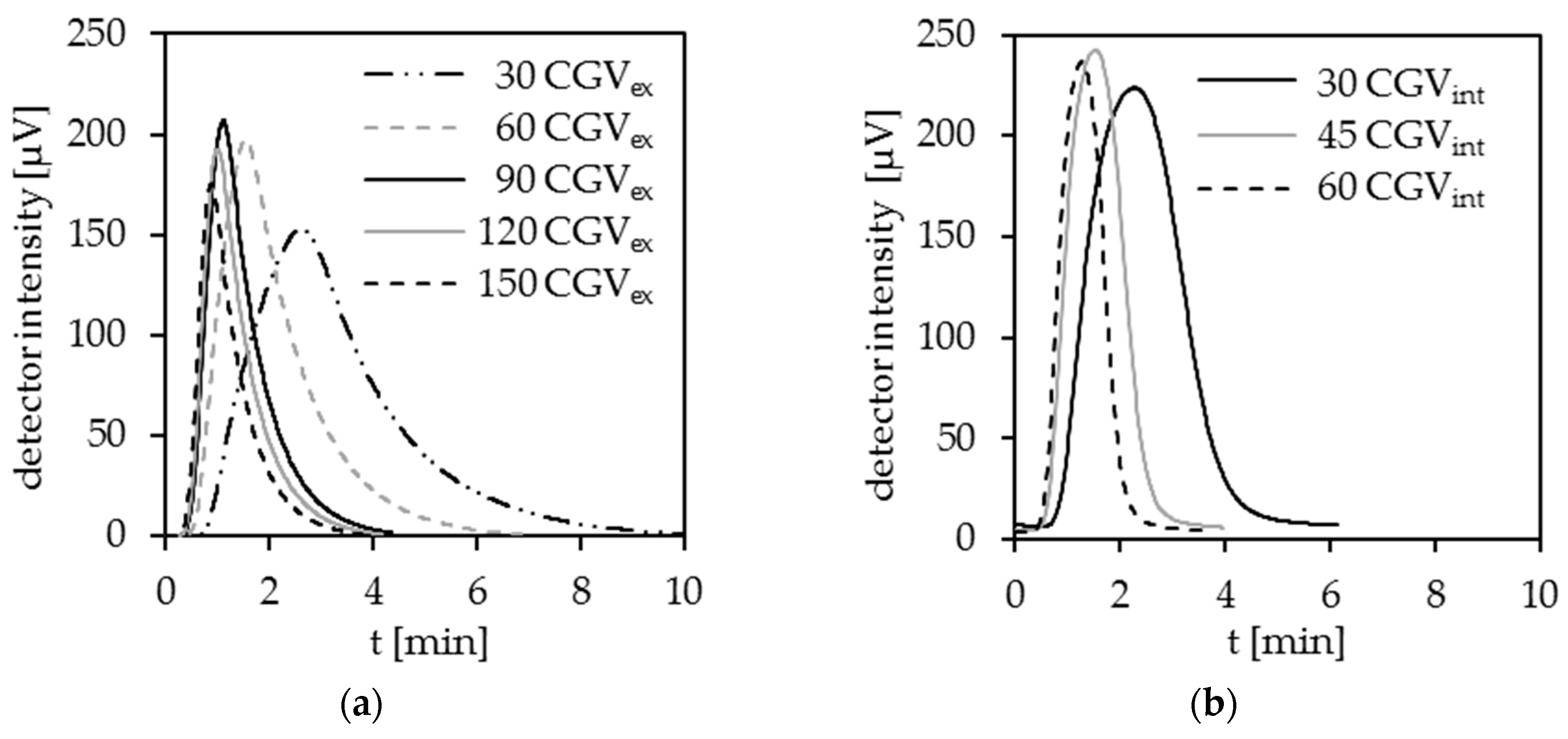
3.1.3. Transport of the Generated CO2
Influence of External Carrier Gas
Influence of Internal Carrier Gas
Combined Use of Internal and External Carrier Gas
3.2. Optimization of the Electrolyte Composition
3.3. Validation and Field Testing of the Prototype
4. Conclusions
- no burning technology needed
- no catalyst or ultrapure carrier gases needed
- short analysis time of under 5 min
- on-site measurement installation in under 5 min
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Visco, G.; Campanella, L.; Nobili, V. Organic carbons and TOC in waters: An overview of the international norm for its measurements. Microchem. J. 2005, 79, 185–191. [Google Scholar] [CrossRef]
- Lin, H.; Wu, J.; Zhang, H. Degradation of bisphenol A in aqueous solution by a novel electro/Fe3+/peroxydisulfate process. Sep. Purif. Technol. 2013, 117, 18–23. [Google Scholar] [CrossRef]
- Urbansky, E.T. Total organic carbon analyzers as tools for measuring carbonaceous matter in natural waters. J. Environ. Monit. 2001, 3, 102–112. [Google Scholar] [CrossRef] [PubMed]
- Avagyan, A.; Runkle, B.R.K.; Kutzbach, L. Application of high-resolution spectral absorbance measurements to determine dissolved organic carbon concentration in remote areas. J. Hydrol. 2014, 517, 435–446. [Google Scholar] [CrossRef]
- Vogl, J.; Heumann, K.G. Development of an ICP−IDMS method for dissolved organic carbon determinations and its application to chromatographic fractions of heavy metal complexes with humic substances. Anal. Chem. 1998, 70, 2038–2043. [Google Scholar] [CrossRef]
- Tepuš, B.; Simonič, M. Uncertainty of the result of TOC determination in water samples. Accredit. Qual. Assur. 2007, 12, 357–364. [Google Scholar] [CrossRef]
- Schmalz, V.; Börnick, H.; Worch, E. Technische Universität Dresden. Verfahren und Vorrichtung zur Bestimmung des Gesamten Organisch Gebundenen Kohlenstoffs in Einer Wässrigen Probe. Patent DE 10 2008 010 581 A1, 12 February 2008. [Google Scholar]
- Comninellis, C. Electrocatalysis in the electrochemical conversion/combustion of organic pollutants for waste water treatment. Electrochim. Acta 1994, 39, 1857–1862. [Google Scholar] [CrossRef]
- Chen, X.; Chen, G.; Gao, F.; Yue, P.L. High-performance Ti/BDD electrodes for pollutant oxidation. Environ. Sci. Technol. 2003, 37, 5021–5026. [Google Scholar] [CrossRef] [PubMed]
- Boye, B.; Brillas, E.; Marselli, B.; Michaud, P.-A.; Comninellis, C.; Farnia, G.; Sandonà, G. Electrochemical incineration of chloromethylphenoxy herbicides in acid medium by anodic oxidation with boron-doped diamond electrode. Electrochim. Acta 2006, 51, 2872–2880. [Google Scholar] [CrossRef]
- Iniesta, J.; Michaud, P.A.; Panizza, M.; Comninellis, C. Electrochemical oxidation of 3-methylpyridine at boron-doped diamond electrode: Application to electroorganic synthesis and wastewater treatment. Electrochem. Comm. 2001, 3, 346–351. [Google Scholar] [CrossRef]
- Panizza, M.; Michaud, P.A.; Cerisola, G.; Comninellis, C. Anodic oxidation of 2-naphthol at boron-doped diamond electrodes. J. Electroanal. Chem. 2001, 507, 206–214. [Google Scholar] [CrossRef]
- Guinea, E.; Arias, C.; Cabot, P.L.; Garrido, J.A.; Rodríguez, R.M.; Centellas, F.; Brillas, E. Mineralization of salicylic acid in acidic aqueous medium by electrochemical advanced oxidation processes using platinum and boron-doped diamond as anode and cathodically generated hydrogen peroxide. Water Res. 2008, 42, 499–511. [Google Scholar] [CrossRef] [PubMed]
- El-Ghenymy, A.; Garrido, J.A.; Centellas, F.; Arias, C.; Cabot, P.L.; Rodríguez, R.M.; Brillas, E. Electro-Fenton and photoelectro-Fenton degradation of sulfanilic acid using a boron-doped diamond anode and an air diffusion cathode. J. Phys. Chem. A 2012, 116, 3404–3412. [Google Scholar] [CrossRef] [PubMed]
- Glorian, H.; Schmalz, V.; Kürbis, S.; Börnick, H.; Worch, E.; Dittmar, T. Electrochemical decomposition of dissolved organic carbon using boron-doped diamond technology as basic element of a portable DOC analyzer. J. Electroanal. Chem. 2017, 801, 43–48. [Google Scholar] [CrossRef]
- DIN Deutsches Institut für Normung, e.V.; Arbeitsausschuss Chemische Terminologie(AChT) D. I. für Normung, DIN EN 1484-1997: Anleitungen zur Bestimmung des Gesamten Organischen Kohlenstoffs (TOC) und des Gelösten Organischen Kohlenstoffs (DOC); Beuth Verlag GmbH: Berlin, Germany, 1997. [Google Scholar]
- Lipczynska-Kochany, E.; Sprah, G.; Harms, S. Influence of some groundwater and surface waters constituents on the degradation of 4-chlorophenol by the Fenton reaction. Chemosphere 1995, 30, 9–20. [Google Scholar] [CrossRef]
- Kerwick, M.I.; Reddy, S.M.; Chamberlain, A.H.L.; Holt, D.M. Electrochemical disinfection, an environmentally acceptable method of drinking water disinfection? Electrochim. Acta 2005, 50, 5270–5277. [Google Scholar] [CrossRef]
- Michaud, P.-A.; Mahé, E.; Haenni, W.; Perret, A.; Comninellis, C. Preparation of peroxodisulfuric acid using boron-doped diamond thin film electrodes. Electrochem. Solid-State Lett. 2000, 3, 77–79. [Google Scholar] [CrossRef]
- Palmas, S.; Polcaro, A.M.; Vacca, A.; Mascia, M.; Ferrara, F. Influence of the operating conditions on the electrochemical disinfection process of natural waters at BDD electrodes. J. Appl. Electrochem. 2007, 37, 1357–1365. [Google Scholar] [CrossRef]
- Davis, J.; Baygents, J.C.; Farrell, J. Understanding persulfate production at boron doped diamond film anodes. Electrochim. Acta 2014, 150, 68–74. [Google Scholar] [CrossRef]
- Neta, P.; Huie, R.E.; Ross, A.B. Rate constants for reactions of inorganic radicals in aqueous solution. J. Phys. Chem. Ref. Data 1988, 17, 1027–1284. [Google Scholar] [CrossRef]
- Mascia, M.; Vacca, A.; Palmas, S.; Polcaro, A.M. Kinetics of the electrochemical oxidation of organic compounds at BDD anodes: Modelling of surface reactions. J. Appl. Electrochem. 2006, 37, 71–76. [Google Scholar] [CrossRef]
- Lutze, H. Sulfate Radical Based Oxidation in Water Treatment. Ph.D. Thesis, Universität Duisburg-Essen, Duisburg, Germany, 2013. Available online: https://duepublico.uni-duisburg-essen.de/servlets/DerivateServlet/Derivate-35021/Lutze_Diss.pdf (accessed on 13 February 2016).
- Serrano, K.; Michaud, P.A.; Comninellis, C.; Savall, A. Electrochemical preparation of peroxodisulfuric acid using boron doped diamond thin film electrodes. Electrochim. Acta 2002, 48, 431–436. [Google Scholar] [CrossRef] [Green Version]
- DIN Deutsches Institut für Normung, e.V.; Arbeitsausschuss Chemische Terminologie(AChT). DIN 32645 1994-05: Chemische Analytik; Nachweis-, Erfassungs- und Bestimmungsgrenze; Ermittlung unter Wiederholbedingungen, Begriffe, Verfahren, Auswertung; Beuth Verlag GmbH: Berlin, Germany, 1994. [Google Scholar]
- Liang, C.; Huang, C.-F.; Mohanty, N.; Kurakalva, R.M. A rapid spectrophotometric determination of persulfate anion in ISCO. Chemosphere 2008, 73, 1540–1543. [Google Scholar] [CrossRef] [PubMed]
- Bennedsen, L.R.; Søgaard, E.G.; Muff, J. Development of a spectrophotometric method for on-site analysis of peroxygens during in-situ chemical oxidation applications. Water Sci. Technol. 2014, 70, 1656–1662. [Google Scholar] [CrossRef] [PubMed]
- Klassen, N.V.; Marchington, D.; McGowan, H.C.E. H2O2 Determination by the I3-Method and by KMnO4 Titration. Anal. Chem. 1994, 66, 2921–2925. [Google Scholar] [CrossRef]
- Sellers, R.M. Spectrophotometric determination of hydrogen peroxide using potassium titanium(IV) oxalate. Analyst 1980, 105, 950–954. [Google Scholar] [CrossRef]
- Brandhuber, P.; Korshin, G.V. Methods for the Detection of Residual Concentations of Hydrogen Peroxide in Advanced Oxidation Processes; WateReuse Foundation: Alexandria, VA, USA, 2009. [Google Scholar]
- Fryda, M.; Schäfer, L.; Herrmann, D.; Gandini, D.; Perret, A.; Comninellis, C.; Klages, C.-P.; Heanni, W. Properties of diamond electrodes for wastewater treatment. New Diam. Front. Carbon Technol. 1999, 9, 229–240. [Google Scholar]
- Lochyński, P.; Dittmar, T.; Worch, E.; Kuczewski, K. Electrochemical oxidation of organic compounds on boron-doped diamond electrodes. Przem. Chem. 2015, 1, 2275–2278. [Google Scholar] [CrossRef]
- DIN Deutsches Institut für Normung e.V.; Arbeitsausschuss Chemische Terminologie(AChT). EN ISO 25178-2:2012: Geometrische Produktspezifikation (GPS)—Berflächenbeschaffenheit: Flächenhaft—Teil 2: Begriffe und Oberflächen-Kenngrößen; Beuth Verlag GmbH: Berlin, Germany, 2012. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Electrolytes | H2SO4 96% (μL L−1) | Na2SO4 (g L−1) | c (SO42−) * (mmol L−1) | pH ** (−) | κ ** (mS cm−1) |
|---|---|---|---|---|---|
| pH 1.4 sulf0 | 1850 | 0 | 33 | 1.38 | 17.6 |
| pH 1.4 sulf1 | 3150 | 8 | 112 | 1.40 | 25.8 |
| pH 1.4 sulf2 | 3600 | 25 | 240 | 1.40 | 43.2 |
| pH 1.4 sulf3 | 4450 | 41 | 368 | 1.40 | 43.2 |
| pH 1.4 HNO3 | 0 | 0 | 0 | 1.44 | 17.4 |
| BDD-SiC-C | REM Picture (Resolution 5 μm) | Layer Thickness (μm) | Crystallinity Grain Size (μm) | Surface Area (As/A) * | kQ (mL A−1 s−1) |
|---|---|---|---|---|---|
| 1 |  | 18 | 2.16 ± 0.28 | 1.219 ± 0.0009 | 0.029 |
| 2 |  | 16 | 1.79 ± 0.33 | 1.159 ± 0.0012 | 0.028 |
| 3 |  | 17 | 0.96 ± 0.16 | 1.049 ± 0.0004 | 0.022 |
| 4 |  | 17 | 0.12 ± 0.05 | 1.019 ± 0.0005 | 0.017 |
| CGV (mL min−1) | Peak Height (μV) | Peak Area (μV min) | Analysis Time (min) | Tailing Factor (−) | |
|---|---|---|---|---|---|
| CGVex | 30 | 168 | 453 | 9.10 | 3.1 |
| 60 | 210 | 359 | 6.43 | 2.9 | |
| 90 | 206 | 233 | 4.27 | 2.8 | |
| 120 | 200 | 199 | 4.03 | 3.2 | |
| 150 | 182 | 167 | 3.88 | 3.3 | |
| CGVint | 30 | 217 | 439 | 6.30 | 1.4 |
| 45 | 237 | 393 | 3.97 | 1.2 | |
| 60 | 233 | 345 | 3.55 | 1.2 | |
| CGVint + ex | 90 (45 + 45) | 252 | 222 | 3.05 | 1.2 |
| Parameter | Value | Unit |
|---|---|---|
| Correlation coefficient | 0.9992 | - |
| Relative standard deviation | 2.3 | % |
| Limit of detection | 0.10 | mg L−1 C |
| Limit of quantification | 0.31 | mg L−1 C |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Glorian, H.; Schmalz, V.; Lochyński, P.; Fremdling, P.; Börnick, H.; Worch, E.; Dittmar, T. Portable Analyzer for On-Site Determination of Dissolved Organic Carbon—Development and Field Testing. Int. J. Environ. Res. Public Health 2018, 15, 2335. https://doi.org/10.3390/ijerph15112335
Glorian H, Schmalz V, Lochyński P, Fremdling P, Börnick H, Worch E, Dittmar T. Portable Analyzer for On-Site Determination of Dissolved Organic Carbon—Development and Field Testing. International Journal of Environmental Research and Public Health. 2018; 15(11):2335. https://doi.org/10.3390/ijerph15112335
Chicago/Turabian StyleGlorian, Heinrich, Viktor Schmalz, Paweł Lochyński, Paul Fremdling, Hilmar Börnick, Eckhard Worch, and Thomas Dittmar. 2018. "Portable Analyzer for On-Site Determination of Dissolved Organic Carbon—Development and Field Testing" International Journal of Environmental Research and Public Health 15, no. 11: 2335. https://doi.org/10.3390/ijerph15112335