Mutational Spectrum and Clinical Features of Patients with LOXHD1 Variants Identified in an 8074 Hearing Loss Patient Cohort


 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Subjects
2.2. Variant Analysis
2.3. Clinical Evaluations
2.4. Haplotype Analysis
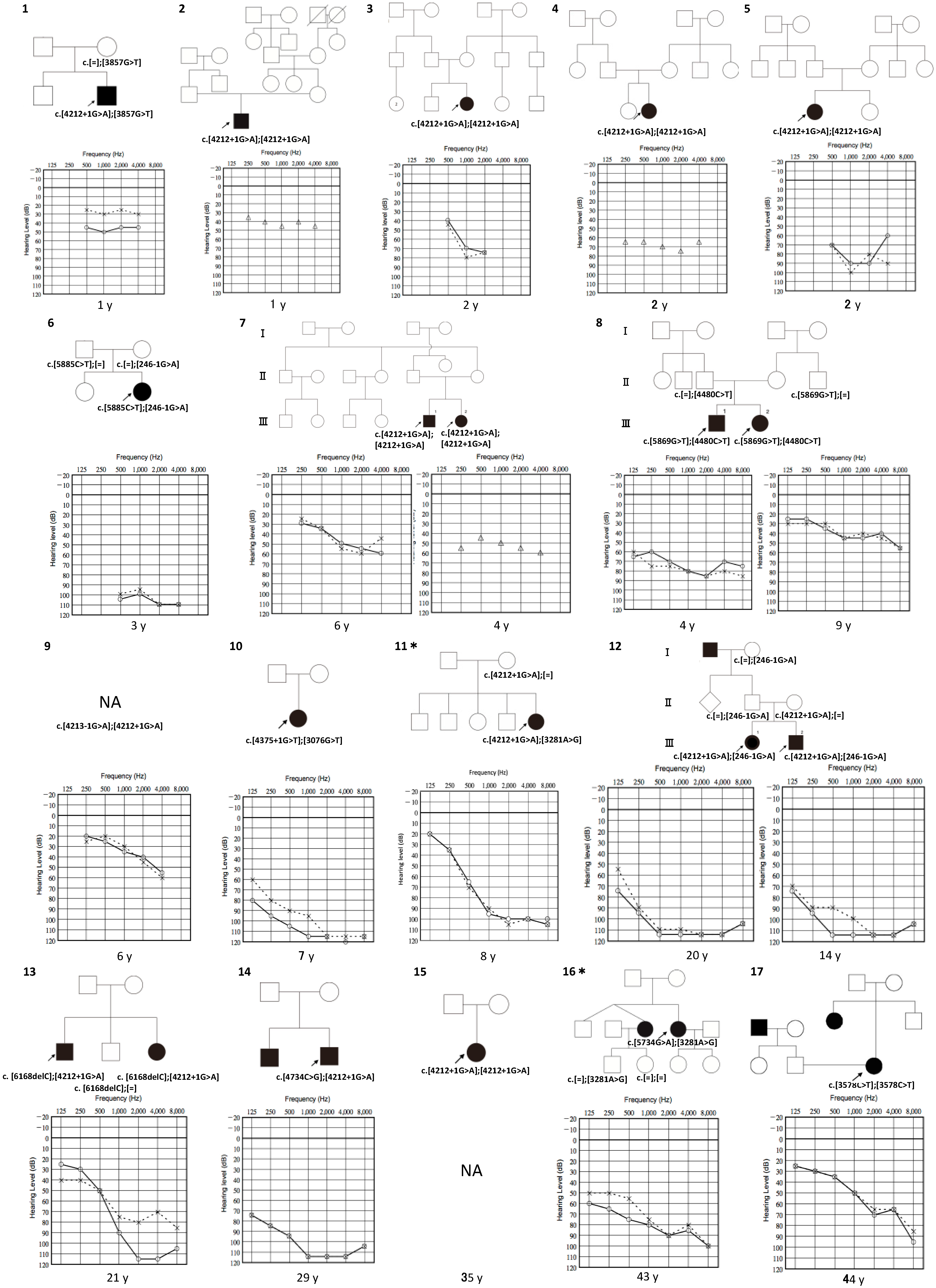
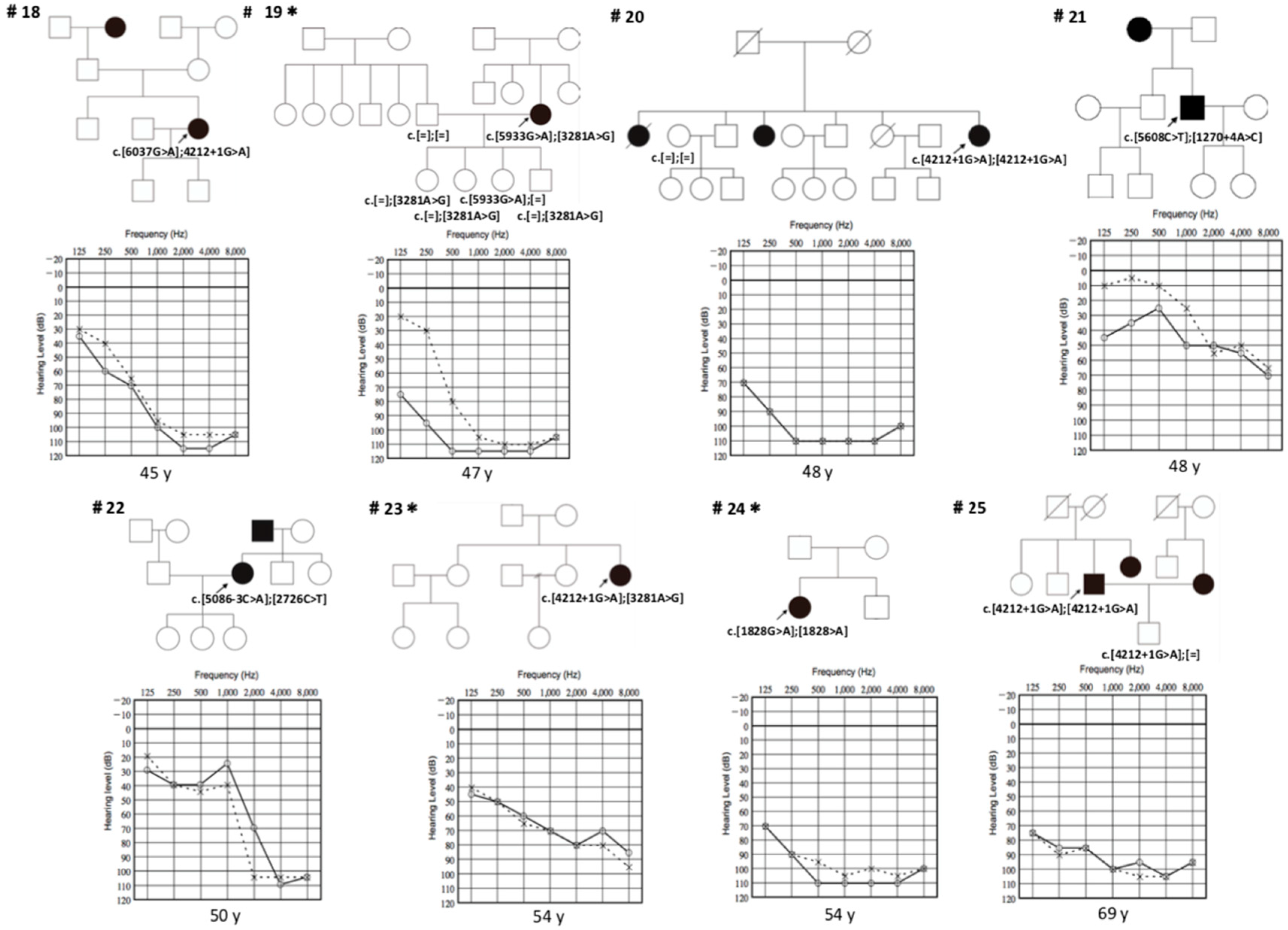
3. Results
3.1. Detected Variations
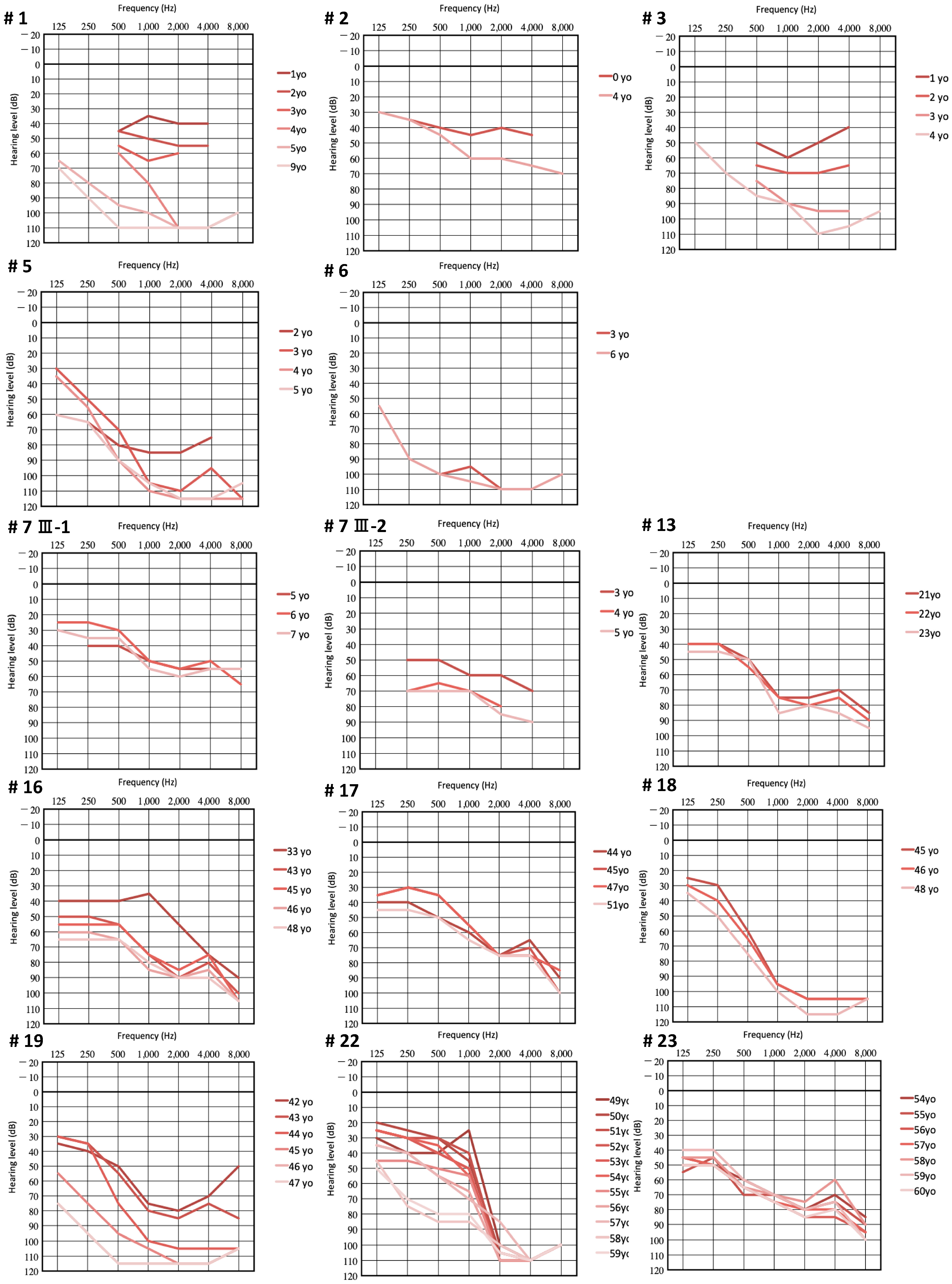
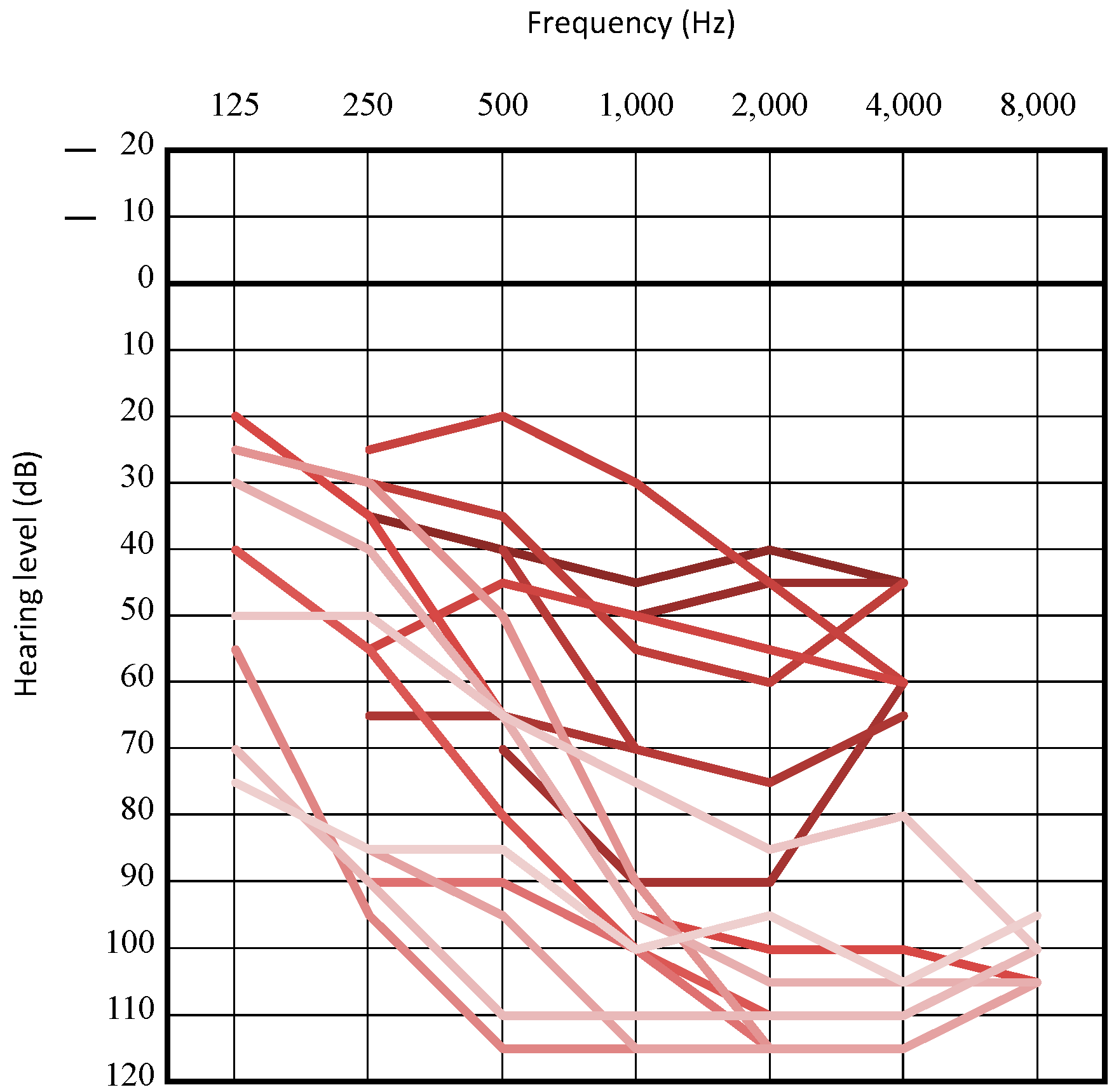
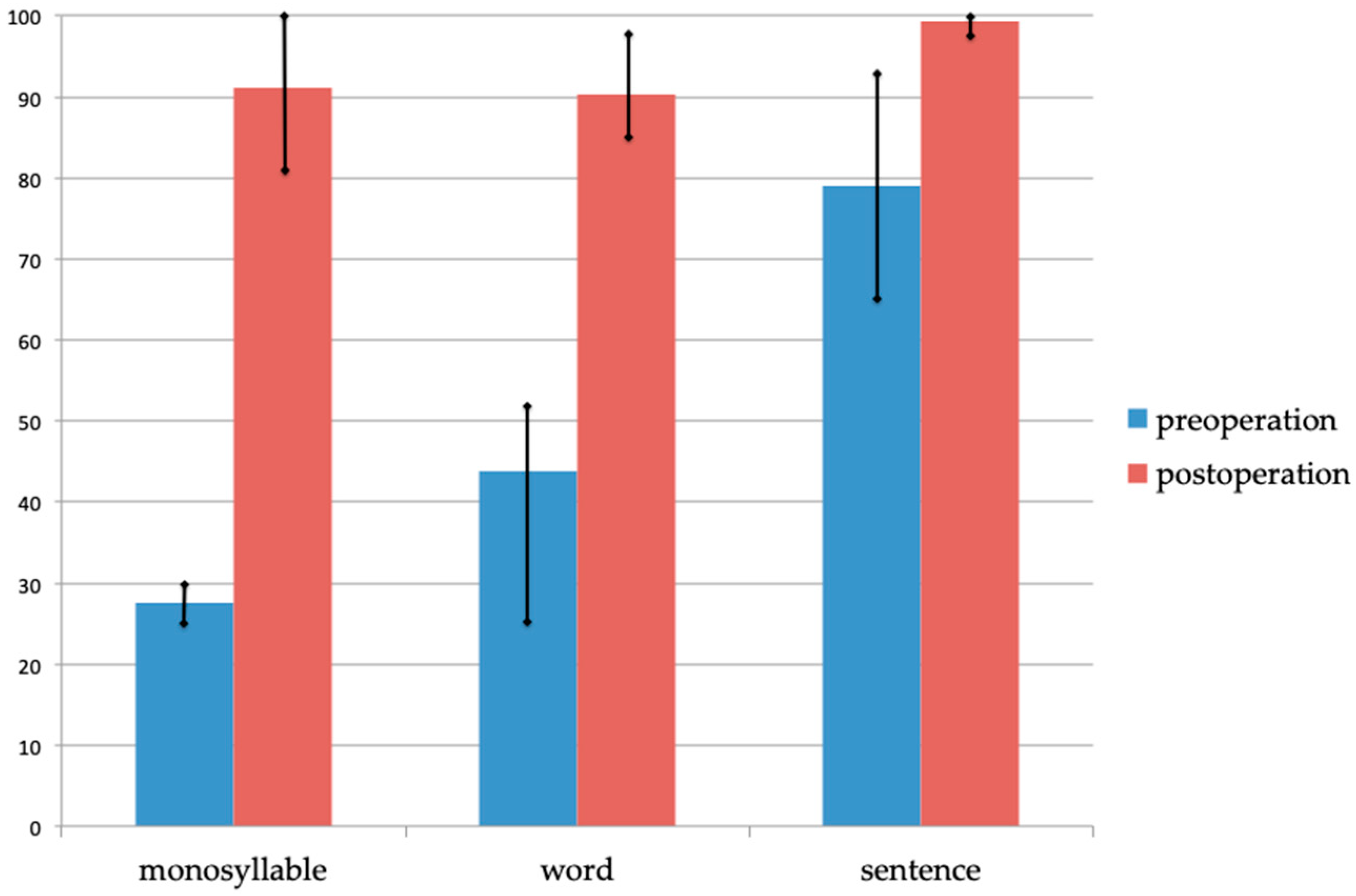
3.2. Clinical Features of Patients with LOXHD1 Variants
3.3. Recurrent Variant
3.4. Intervention
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Data Availability Statement
References
- Morton, C.C.; Nance, W.E. Newborn Hearing Screening—A Silent Revolution. N. Engl. J. Med. 2006, 354, 2151–2164. [Google Scholar] [CrossRef] [PubMed]
- Hereditary Hearing Loss Homepage. Available online: https://hereditaryhearingloss.org (accessed on 5 June 2019).
- Yokota, Y.; Moteki, H.; Nishio, S.Y.; Yamaguchi, T.; Wakui, K.; Kobayashi, Y.; Ohyama, K.; Miyazaki, H.; Matsuoka, R.; Abe, S.; et al. Frequency and clinical features of hearing loss caused by STRC deletions. Sci. Rep. 2019, 9, 4408. [Google Scholar] [CrossRef] [PubMed]
- Müller, U.; Grillet, N. The genetics of progressive hearing loss: A link between hearing impairment and dysfunction of mechanosensory hair cells. Future Neurol. 2010, 5, 9–12. [Google Scholar] [CrossRef] [PubMed]
- Grillet, N.; Schwander, M.; Hildebrand, M.S.; Sczaniecka, A.; Kolatkar, A.; Velasco, J.; Webster, J.A.; Kahrizi, K.; Najmabadi, H.; Kimberling, W.J.; et al. Mutations in LOXHD1, an Evolutionarily Conserved Stereociliary Protein, Disrupt Hair Cell Function in Mice and Cause Progressive Hearing Loss in Humans. Am. J. Hum. Genet. 2009, 85, 328–337. [Google Scholar] [CrossRef] [PubMed]
- Nishio, S.Y.; Usami, S.I. Deafness Gene Variations in a 1120 Nonsyndromic Hearing Loss Cohort: Molecular Epidemiology and Deafness Mutation Spectrum of Patients in Japan. Ann. Otol. Rhinol. Laryngol. 2015, 124, 49S–60S. [Google Scholar] [CrossRef] [PubMed]
- Kitano, T.; Miyagawa, M.; Nishio, S.Y.; Moteki, H.; Oda, K.; Ohyama, K.; Miyazaki, H.; Hidaka, H.; Nakamura, K.I.; Murata, T.; et al. POU4F3 mutation screening in Japanese hearing loss patients: Massively parallel DNA sequencing-based analysis identified novel variants associated with autosomal dominant hearing loss. PLoS ONE 2017, 12, e0177636. [Google Scholar] [CrossRef] [PubMed]
- Nishio, S.; Moteki, H.; Usami, S. Simple and efficient germline copy number variant visualization method for the Ion AmpliSeqTM custom panel. Mol. Genet. Genomic Med. 2018, 6, 678–686. [Google Scholar] [CrossRef]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Mazzoli, M.; Van Camp, G.; Newton, V.; Giarbini, N.; Declau, F.; Parving, A. Recommendations for the Description of Genetic and Audiological Data for Families with Nonsyndromic Hereditary Hearing Impairment. Audiol. Med. 2003, 1, 148–150. [Google Scholar] [CrossRef]
- Human Genetic Variation Database. Available online: http://www.genome.med.kyoto-u.ac.jp/ SnpDB/index.html (accessed on 19 June 2019).
- 3.5KJPN—Integrative Japanese Genome Variation Database. Available online: https:// jmorp.megabank.tohoku.ac.jp/201905/ (accessed on 19 June 2019).
- Wesdorp, M.; Schreur, V.; Beynon, A.J.; Oostrik, J.; van de Kamp, J.M.; Elting, M.W.; van den Boogaard, M.J.H.; Feenstra, I.; Admiraal, R.J.C.; Kunst, H.P.M.; et al. Further audiovestibular characterization of DFNB77, caused by deleterious variants in LOXHD1, and investigation into the involvement of Fuchs corneal dystrophy. Clin. Genet. 2018, 94, 221–231. [Google Scholar] [CrossRef] [PubMed]
- Sakuma, N.; Moteki, H.; Takahashi, M.; Nishio, S.Y.; Arai, Y.; Yamashita, Y.; Oridate, N.; Usami, S.I. An effective screening strategy for deafness in combination with a next-generation sequencing platform: A consecutive analysis. J. Hum. Genet. 2016, 61, 253–261. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zeng, H.; Li, X.; Guo, W.; Han, Z.; Li, P.; Zhao, X.; Wang, X.; Xia, L.; Ma, S. Identification of novel common mutations among patients with non-syndromic hearing loss with high-throughput gene capture technology. Zhonguha Yi Xue Yi Chuan Xue Za Zhi 2016, 33, 758–761. [Google Scholar] [CrossRef]
- Minami, S.B.; Mutai, H.; Namba, K.; Sakamoto, H.; Matsunaga, T. Clinical characteristics of a Japanese family with hearing loss accompanied by compound heterozygous mutations in LOXHD1. Auris Nasus Larynx 2016, 43, 309–613. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Horta, O.; Duman, D.; Foster II, J.; Sırmacı, A.; Gonzalez, M.; Mahdieh, N.; Fotouhi, N.; Bonyadi, M.; Cengiz, F.B.; Menendez, I.; et al. Whole-Exome Sequencing Efficiently Detects Rare Mutations in Autosomal Recessive Nonsyndromic Hearing Loss. PLoS ONE 2012, 7, e50628. [Google Scholar] [CrossRef] [PubMed]
- Sloan-Heggen, C.M.; Bierer, A.O.; Shearer, A.E.; Kolbe, D.L.; Nishimura, C.J.; Frees, K.L.; Ephraim, S.S.; Shibata, S.B.; Booth, K.T.; Campbell, C.A.; et al. Comprehensive genetic testing in the clinical evaluation of 1119 patients with hearing loss. Hum. Genet. 2016, 135, 441–450. [Google Scholar] [CrossRef] [PubMed]
- Eppsteiner, R.W.; Shearer, A.E.; Hildebrand, M.S.; Deluca, A.P.; Ji, H.; Dunn, C.C.; Black-ziegelbein, E.A.; Casavant, T.L.; Braun, T.A.; Scheetz, T.E.; et al. Mutation: The Spiral Ganglion Hypothesis. Hear. Res. 2013, 292, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Mori, K.; Moteki, H.; Kobayashi, Y.; Azaiez, H.; Booth, K.T.; Nishio, S.Y.; Sato, H.; Smith, R.J.H.; Usami, S.I. Mutations in LOXHD1 Gene Cause Various Types and Severities of Hearing Loss. Ann. Otol. Rhinol. Laryngol. 2015, 124 (Suppl. 1), 135S–141S. [Google Scholar] [CrossRef]
- Seco, C.Z.; Wesdorp, M.; Feenstra, I.; Pfundt, R.; Hehir-Kwa, J.Y.; Lelieveld, S.H.; Castelein, S.; Gilissen, C.; De Wijs, I.J.; Admiraal, R.J.C.; et al. The diagnostic yield of whole-exome sequencing targeting a gene panel for hearing impairment in the Netherlands. Eur. J. Hum. Genet. 2017, 25, 308–314. [Google Scholar] [CrossRef]
- Atik, T.; Onay, H.; Aykut, A.; Bademci, G.; Kirazli, T.; Tekin, M.; Ozkinay, F. Comprehensive analysis of deafness genes in families with autosomal recessive nonsyndromic hearing loss. PLoS ONE 2015, 10, e0142154. [Google Scholar] [CrossRef] [PubMed]
- Posey, J.E.; Harel, T.; Liu, P.; Rosenfeld, J.A.; James, R.A.; Coban Akdemir, Z.H.; Walkiewicz, M.; Bi, W.; Xiao, R.; Ding, Y.; et al. Resolution of Disease Phenotypes Resulting from Multilocus Genomic Variation. N. Engl. J. Med. 2016, 376, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Abouelhoda, M.; Sobahy, T.; El-Kalioby, M.; Patel, N.; Shamseldin, H.; Monies, D.; Al-Tassan, N.; Ramzan, K.; Imtiaz, F.; Shaheen, R.; et al. Clinical genomics can facilitate countrywide estimation of autosomal recessive disease burden. Genet. Med. 2016, 18, 1244–1249. [Google Scholar] [CrossRef] [PubMed]
- Vozzi, D.; Morgan, A.; Vuckovic, D.; D’Eustacchio, A.; Abdulhadi, K.; Rubinato, E.; Badii, R.; Gasparini, P.; Girotto, G. Hereditary hearing loss: A 96 gene targeted sequencing protocol reveals novel alleles in a series of Italian and Qatari patients. Gene 2014, 542, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.; Sun, F.; Zhang, J.; Tang, Y.; Qiu, J.; Wang, Z.; Zhang, L. Genetic Etiology Study of Ten Chinese Families with Nonsyndromic Hearing Loss. Neural Plast. 2018, 2018, 4920980. [Google Scholar] [CrossRef] [PubMed]
- Morgan, A.; Lenarduzzi, S.; Cappellani, S.; Pecile, V.; Morgutti, M.; Orzan, E.; Ghiselli, S.; Ambrosetti, U.; Brumat, M.; Gajendrarao, P.; et al. Genomic Studies in a Large Cohort of Hearing Impaired Italian Patients Revealed Several New Alleles, a Rare Case of Uniparental Disomy (UPD) and the Importance to Search for Copy Number Variations. Front. Genet. 2018, 9, 681. [Google Scholar] [CrossRef] [PubMed]
- Lebeko, K.; Sloan-Heggen, C.M.; Noubiap, J.J.N.; Dandara, C.; Kolbe, D.L.; Ephraim, S.S.; Booth, K.T.; Azaiez, H.; Santos-Cortez, R.L.P.; Leal, S.M.; et al. Targeted genomic enrichment and massively parallel sequencing identifies novel nonsyndromic hearing impairment pathogenic variants in Cameroonian families. Clin. Genet. 2016, 90, 288–290. [Google Scholar] [CrossRef] [PubMed]
- Cabanillas, R.; Diñeiro, M.; Cifuentes, G.A.; Castillo, D.; Pruneda, P.C.; Álvarez, R.; Sánchez-Durán, N.; Capín, R.; Plasencia, A.; Viejo-Díaz, M.; et al. Comprehensive genomic diagnosis of non-syndromic and syndromic hereditary hearing loss in Spanish patients. BMC Med. Genomics 2018, 11, 58. [Google Scholar] [CrossRef]
- Plevova, P.; Paprskarova, M.; Tvrda, P.; Turska, P.; Slavkovsky, R.; Mrazkova, E. STRC deletion is a frequent cause of slight to moderate congenital hearing impairment in the Czech Republic. Otol. Neurotol. 2017, 38, e393–e400. [Google Scholar] [CrossRef]
- Edvardson, S.; Jalas, C.; Shaag, A.; Zenvirt, S.; Landau, C.; Lerer, I.; Elpeleg, O. A deleterious mutation in the LOXHD1 gene causes autosomal recessive hearing loss in Ashkenazi Jews. Am. J. Med. Genet. Part. A 2011, 155, 1170–1172. [Google Scholar] [CrossRef]
- Danial-Farran, N.; Brownstein, Z.; Gulsuner, S.; Tammer, L.; Khayat, M.; Aleme, O.; Chervinsky, E.; Zoubi, O.A.; Walsh, T.; Ast, G.; et al. Genetics of hearing loss in the Arab population of Northern Israel. Eur. J. Hum. Genet. 2018, 26, 1840–1847. [Google Scholar] [CrossRef]
- Shen, N.; Wang, T.; Li, D.; Liu, A.; Lu, Y. Whole-exome sequencing identifies a novel missense variant within LOXHD1 causing rare hearing loss in a Chinese family. BMC Med. Genet. 2019, 20, 30. [Google Scholar] [CrossRef] [PubMed]
- Riazuddin, S.A.; Parker, D.S.; McGlumphy, E.J.; Oh, E.C.; Iliff, B.W.; Schmedt, T.; Jurkunas, U.; Schleif, R.; Katsanis, N.; Gottsch, J.D. Mutations in LOXHD1, a recessive-deafness locus, cause dominant late-onset Fuchs corneal dystrophy. Am. J. Hum. Genet. 2012, 90, 533–539. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Allele Frequency Information | ANNOVAR dbNSFP ver 3.5 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Base Change | AA Change | Pathogenecity | ExAC03 | gnomAD | SIFT | PP2 HVAR | LRT | MutTaster | MutAssessor | CADD | Reference |
| c.246 − 1G > A | Pathogenic (PVS1,PM2,PM3,PP1,PP3) | - | - | - | - | - | D (0.81) | - | 26.7 | This Study | |
| c.1270 + 4A > C | Likely Pathogenic (PVS1, PM2) | - | - | - | - | - | - | - | - | This Study | |
| c.1828G > A | p.E610K | Uncertain Significance (PM3, PP3, BS1) | 4.602e−05 | 5.3e−05 | D (0.614) | D (0.85) | D (0.629) | D (0.548) | M (0.743) | 25 | [14] * |
| c.2726C > T | p.T909M | Uncertain Significance (PM2, BP4) | - | 6.785e−06 | D (0.443) | B (0.214) | N (0.383) | D (0.81) | L (0.263) | 10.25 | This Study |
| c.3076G > T | p.V1026F | Uncertain Significance (PM2, PM3, PP3, PP5) | - | - | D (0.721) | D (0.754) | D (0.537) | D (0.81) | H (0.932) | 15.91 | [15] |
| c.3281A > G | p.D1094G | Uncertain significance (PP3, PM7, PP3, PP5, BS1) | 0 | 5.883e−05 | D (0.555) | D (0.916) | D (0.843) | D (0.81) | H (0.973) | 15.22 | [16] * |
| c.3578C > T | p.A1193V | Uncertain Significance (PM2, PP3) | - | - | D (0.682) | D (0.818) | D (0.523) | D (0.81) | H (0.970) | 33 | This Study |
| c.3857G > T | p.G1286V | Likely Pathogenic (PM2, PM3, PP1, PP3) | - | - | D (0.784) | D (0.971) | D (0.743) | D (0.81) | H (0.967) | 26.1 | This Study |
| c.4212 + 1G > A | Pathogenic (PVS1, PM2, PM7, PP3, PP5) | - | 2.65e−05 | - | - | - | D (0.81) | - | 24.2 | [17] | |
| c.4213 − 1G > A | Pathogenic (PVS1, PM2, PM3) | - | 6.6e−06 | - | - | - | D (0.81) | - | 26 | This Study | |
| c.4375 + 1G > T | Likely Pathogenic (PVS1, PM2, PM3, PP5) | - | - | - | - | - | D (0.81) | - | 26.6 | [16] | |
| c.4480C > T | p.E1494X | Pathogenic (PVS1, PM2, PM3, PP1, PP3, PP5) | 0.0006 | 0.0006 | - | - | D (0.843) | A (0.81) | - | 38 | [18,19,20,21] |
| c.4734C > G | p.Y1578X | Pathogenic (PVS1, PM2, PM3) | - | - | - | - | D (0.559) | A (0.81) | - | 35 | This Study |
| c.5086 − 3C > A | Pathogenic (PVS1, PM2, PP3) | - | 6.623e−26 | - | - | - | - | - | This Study | ||
| c.5608C > T | p.R1870W | Uncertain Significance (PM2, PP3) | 4.499e−05 | 3.939e−05 | D (0.784) | D (0.916) | - | D (0.48) | M (0.924) | 34 | This Study |
| c.5734G > A | p.D1912N | Uncertain Significance (PM2, PM3, PP3,) | - | - | D (0.912) | D (0.916) | - | D (0.588) | M (0.632) | 28.7 | This Study |
| c.5869G > T | p.E1957X | Pathogenic (PVS1, PM2, PM3, PP1, PP5) | - | - | - | - | - | A (0.81) | 54 | [21] | |
| c.5885C > T | p.1962M | Uncertain Significance (PM2, PP1, PP3, PP5) | 4.44e−05 | 6.544e−06 | D (0.784) | D (0.916) | - | D (0.52) | L (0.44) | 34 | [22] |
| c.5933G > A | p.G1978D | Likely Pathogenic (PM2, PM3, PP1, PP3) | 4.531e−05 | 6.565e−06 | D (0.614) | D (0.679) | - | D (0.588) | L (0.52) | 34 | This Study |
| c.6037G > A | p.G2013R | Uncertain Significance (PM2, PM3, PP3) | - | - | D (0.682) | D (0.971) | - | D (0.81) | L (0.52) | 33 | This Study |
| c.6168delC | p.C2057Vfs * 42 | Pathogenic (PVS1, PM2, PM3) | - | - | - | - | - | - | - | - | This Study |
| Family No. | Base Change | Amino Acid Change | Onset | Sex | Tinnitus | Verigo | Vestibular Examination (R/L) | Progression | Severity | Configuration |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | c.[4212 + 1G > A]; [3857G > T] | p.[?]; [G1286V] | 0 | m | - | - | Normal/Normal | + | Mild | Flat |
| 2 | c.[4212 + 1G > A]; [4212 + 1G > A] | p.[?]; [?] | 0 | m | - | - | NA | - | Moderate | Flat |
| 3 | c.[4212 + 1G > A]; [4212 + 1G > A] | p.[?]; [?] | 0 | f | - | - | NA | + | Moderate | Precipitous |
| 4 | c.[4212 + 1G > A]; [4212 + 1G > A] | p.[?]; [?] | 0 | f | - | - | NA | - | Moderate | Flat |
| 5 | c.[4212 + 1G > A]; [4212 + 1G > A] | p.[?]; [?] | 0 | f | - | - | NA | - | Severe | Flat |
| 6 | c.[5885C > T]; [246 − 1G > A] | p.[1962M]; [?] | 0 | f | NA | NA | Normal/Normal | + | Profound | Flat |
| 7 III-1 | c.[4212 + 1G > A]; [4212 + 1G > A] | p.[?]; [?] | 0 | m | NA | - | NA | - | Moderate | Flat |
| III-2 | c.[4212 + 1G > A]; [4212 + 1G > A] | p.[?]; [?] | 0 | f | NA | - | NA | - | Moderate | Flat |
| 8 III-1 | c.[5869G > T]; [4480C > T] | p.[E1957X]; [E1494X] | 0 | f | - | - | NA | + | Severe | Flat |
| III-2 | c.[5869G > T]; [4480C > T] | p.[E1957X]; [E1494X] | 7 | m | - | - | NA | + | Moderate | Flat |
| 9 | c.[4213 − 1G > A]; [4212 + 1G > A] | p.[?]; [?] | 5 | m | - | - | NA | - | Mild | Sloping |
| 10 | c.[4375 + 1G > T]; [3076G > T] | p.[?]; [V1026F] | 7 | f | - | - | NA | - | Profound | Sloping |
| 11 | c.[4212 + 1G > A]; [3281A > G] | p.[?]: [D1094G] | 0 | f | - | - | NA | + | Severe | Sloping |
| 12 III-1 | c.[4212 + 1G > A]; [246-1G>A] | p.[?]; [?] | 2 | f | - | - | NA | + | Profound | Flat |
| III-2 | c.[4212 + 1G>A]; [ 246 − 1G > A] | p.[?]; [?] | 0 | m | - | - | NA | + | Profound | Flat |
| 13 | c.[6168delC]; [4212 + 1G > A] | p.[C2057Vfs * 42]; [?] | 3 | m | + | - | Normal/Normal | + | Severe | Precipitous |
| 14 | c.[4734C > G]; [4212 + 1G > A] | p.[Y1578X]; [?] | 0 | m | - | - | NA | + | Profound | Sloping |
| 15 | c.[4212 + 1G > A]; [4212 + 1G > A] | p.[?]; [?] | 4 | f | NA | NA | NA | - | NA | NA |
| 16 | c.[5734G > A]; [3281A > G] | p.[D1912N]; [D1094G] | 30 | f | + | - | NA | + | Severe | Sloping |
| 17 | c.[3578C > T]; [3578C > T] | p.[A1193V]; [A1193V] | 0 | f | + | - | NA | - | Moderate | Precipitous |
| 18 | c.[6037G > A]; [4212 + 1G > A] | p.[G2013R]; [?] | 5 | f | + | - | Normal/Normal | + | Profound | Sloping |
| 19 | c.[5933G > A]; [3281A > G] | p.[G1978D]; [D1094G] | 32 | f | + | - | NA | + | Profound | Precipitous |
| 20 | c.[4212 + 1G > A]; [4212 + 1G > A] | p.[?]; [?] | 0 | f | + | - | NA | - | Profound | Flat |
| 21 | c.[5608C > T]; [1270 + 4A > C] | p.[R1870W]; [?] | 36 | m | NA | NA | NA | + | Mild | Precipitous |
| 22 | c.[5086 − 3C > A]; [2726C > T] | p.[?]; [T909M] | 30 | f | + | - | Normal/Normal | + | Profound | Precipitous |
| 23 | c.[4212 + 1G > A]; [3281A > G] | p.[?]: [D1094G] | 4 | f | NA | NA | Normal/Normal | + | Severe | Sloping |
| 24 | c.[1828G > A]; [1828G > A] | p.[E610K]; [E610K] | 2 | f | NA | NA | NA | + | Profound | Flat |
| 25 | c.[4212 + 1G > A]; [4212 + 1G > A] | p.[?]; [?] | 5 | m | - | - | NA | - | Profound | Flat |
| Nucleotide Change | Amino Acid Change | HL Onset | Severity of HL | Progression | Population | Reference |
|---|---|---|---|---|---|---|
| c.[71delT]; [71delT] | p.[L24Rfs * 74]; [L24Rfs * 74] | Congenital or Prelingual | Severe or Profound | NA | Turkish | [23] |
| c.[442A > T]; [4217C > T] | p.[K148X]; [A1406V] | NA | NA | NA | American | [24] |
| c.[486_487delCTinsGG]; [486_487delCTinsGG] | p.[?]; [?] | NA | NA | NA | Saudi Arabian | [25] |
| c.[894T > G]; [6353G > A] | p.[Y298X]; [G2118E] | Congenital | Mild-moderate | NA | American | [19] |
| c.[1588C > T]; [1588C > T] | p.[E530X]; [E530X] | Childhood | Severe–profound | Progressive | Qatar | [26] |
| c.[1603C > T]; [1938G > A] | p.[R535X]; [K646K] | Childhood | Mild–moderate | NA | American | [19] |
| c.[1618dupA]; [1730T > G] | p.[T540Afs * 24]; [L635P] | Congenital | Severe | Stable–Progressive | Dutch | [14,22] |
| c.[1751C > T]; [5815G > A] | p.[T584]; [D1939N] | 35–40 y.o. | Severe | Progressive | Chinese | [27] |
| c.[1730T > G]; [5869G > A]; [5944C > T] * | p.[L577R]; [E1957K]; [R1982X] | Congenital | Severe–profound | NA | American | [19] |
| c.[1828G > T]; [2641G > A] | p.[E610X]; [G881R] | 2–4 y.o. | Mild | Stable | Dutch | [14] |
| c.[1843C > T]; [3281A > G] | p.[R615W]; [D1094G] | NA | NA | NA | Chinese | [16] |
| c.[1904T > C]; [4678T > C] | p.[L635P]; [C1560R] | 2–3 y.o. | Mild | Stable–Progressive | Dutch | [14] |
| c.[1938G > A]; [4936C > T] | p.[K646K]; [C1560R] | Childhood | Mild–moderate | NA | American | [19] |
| c.[2008C > T]; [2008C > T] | p.[R670X]; [R670X] | Childhood | Moderate–profound | Progressive | Iranian | [5] |
| c.[2696G > C]; [3596T > C] | p.[R899P]; [L1199P] | NA | NA | NA | American | [19] |
| c.[2696G > C]; [3834G > C] | p.[R899P]; [W1278C] | 5 y.o. | Moderate | Stable | Dutch | [14] |
| c.[2696G > C]; [5934C > T] | p.[R899P]; [T1978G] | Congenital | Mild | NA | Dutch | [14] |
| c.[2825_2827delAGA]; [4217C > A] | p.[?]; [A1406E] | Childhood | Mild–Moderate | NA | American | [19] |
| c.[2863G > T]; [2863G > T] | p.[E955X]; [E955X] | NA | NA | NA | Turkey | [18] |
| c.[3061C > T]; [5885C > T] | p.[R1021X]; [T1962M] | 1–10 y.o. | Severe | Progressive | Netherlands | [14,22] |
| c.[3061 + 1G > A]; [6353G > A] | p.[?]; [G2118E] | Congenital | Moderate | NA | Netherlands | [14,22] |
| c.[3071A > G]; [3071A > G] | p.[Y1024C]; [Y1024C] | Early-onset | Severe–profound | NA | Italy | [28] |
| c.[3076G > T]; [4375 + 1G > T] | p.[V1026F]; [?] | 3 y.o. | Profound | Non-progressive | Japanese | [15] |
| c.[3169C > T]; [6353G > A] | p.[V1026F]; [G2118E] | Congenital | Severe | Stable | Dutch | [14] |
| c.[3371G >A]; [3979T > A] | p.[A1124H]; [P1327I] | NA | NA | NA | Cameroon | [29] |
| c.[3571A > G]; [3571A > G] | p.[T1191A]; [T1191A] | Congenital | Severe-profound | NA | Spanish | [30] |
| c.[3748 + 1G > C]; [6353G > A] | p[?]; [G2118E] | Congenital | Moderate–severe | Stable–Progressive | Dutch | [14] |
| c.[4099G > T]; [6162_6164delCCT] | p.[E1367X]; [F2055Nfs * 157] | Congenital | Severe–profound | NA | American | [19] |
| c.[4212 + 1G > A]; [5674G > T] | p.[?]; [V1892F] | Congenital–7 y.o. | Mild–Severe | Progressive | Japanese | [17] |
| c.[4480C > T]; [4480C > T] | p.R1494X[R1494X] | NA | NA | NA | Turkey | [18] |
| c.[4480C > T; 4526G > A]; [4480C > T; 4526G > A] | p.[R1494X;G1509E]; [R1494X;G1509E] | Congenital | Mild–moderate | NA | American | [19] |
| c.[4480C > T]; [4526G > A] | p.[R1494X]; [G1509E] | 40 y.o. | Severe–profound | Progressive | American | [20] |
| c.[4480C > T]; [5869G > T] | p.[R1494X]; [E1957X] | NA | Moderate–severe | Non-progressive | Japanese | [21] |
| c.[4480C > T; 4526G > A]; [6598delG] | p.[R1494X; G1509E]; [D2200Mfs * 21] | Childhood | Severe-profound | NA | American | [19] |
| c.[4623C >G]; [5545G > A] | p.[Y1541X]; [G1849R] | 2 y.o. | Severe | NA | Czech | [31] |
| c.[4714C >T]; [4714C > T] | p.[R1572X]; [R1572X] | Prelingual | Severe–profound | Non-progressive | Ashkenazi Jews | [32] |
| c.[5894dupG]; [5894dupG] | c.[?]; [?] | Prelingual | Profound | Na | Arab | [33] |
| c.[5948C > T]; [5948C > T] | p.[S1983F]; [S1983F] | Congenital | Severe–profound | Non-progressive | Chinese | [34] |
| Distance from the c.4212 + 1G > A Variation (bp) | Allele Frequency (HapMap-JPT) | Marker | No. 2 | No. 3 | No. 4 | No. 5 | No. 7 | No. 25 |
|---|---|---|---|---|---|---|---|---|
| 980,731 | C 0.30 T 0.70 | rs868409 | C/T | C/T | C/C | C/T | C/T | C/C |
| 926,737 | A 0.38 G 0.62 | rs4890557 | A/G | A/G | G/G | A/G | A/A | G/G |
| 877,703 | A 0.27 G 0.73 | rs17732049 | A/G | G/G | G/G | G/G | A/G | G/G |
| 862,524 | T 0.26 C 0.74 | rs3786397 | C/C | T/C | C/C | T/C | C/C | C/C |
| 860,937 | T 0.35 C 0.65 | rs1552329 | T/C | C/C | C/C | T/C | C/C | C/C |
| 811,321 | G 0.37 A 0.63 | rs3760578 | G/G | A/A | A/A | A/A | A/A | A/A |
| 668,955 | T 0.26 C 0.74 | rs12456289 | C/C | C/C | C/C | C/C | C/C | C/C |
| 626,721 | C 0.26 T 0.74 | rs8097963 | T/T | T/T | T/T | T/T | T/T | T/T |
| 570,604 | A 0.28 T 0.72 | rs8087546 | T/T | T/T | T/T | T/T | T/T | T/T |
| 524,148 | A 0.32 G 0.68 | rs673123 | G/G | A/G | G/G | G/G | G/G | G/G |
| 332,094 | C 0.47 T 0.53 | rs9956574 | C/T | C/T | T/T | C/T | T/T | T/T |
| 233,342 | C 0.38 T 0.62 | rs4890637 | T/T | T/T | T/T | C/T | T/T | T/T |
| 134,221 | T 0.32 C 0.68 | rs16978558 | C/C | C/C | C/C | C/C | C/C | C/C |
| 122,941 | C 0.27 A 0.73 | rs3911131 | C/A | A/A | A/A | A/A | A/A | A/A |
| 24,152 | G 0.27 T 0.73 | rs8084298 | T/T | T/T | T/T | T/T | T/T | T/T |
| 15,561 | A 0.29 G 0.71 | rs426303 | G/G | G/G | G/G | G/G | G/G | G/G |
| 0 | c.4212 + 1 G > A | - | - | - | - | - | - | |
| 108,292 | A 0.23 C 0.77 | rs16939868 | C/C | C/C | C/C | C/C | C/C | C/C |
| 206,155 | C 0.27 G 0.73 | rs4121822 | C/C | C/C | C/G | C/G | C/G | C/C |
| 220,199 | G 0.27 A 0.73 | rs4449041 | A/A | A/A | G/G | A/A | G/G | A/A |
| 282,037 | C 0.28 T 0.72 | rs513775 | T/T | T/T | T/T | C/T | T/T | T/T |
| 290,228 | A 0.32 T 0.68 | rs578451 | T/T | T/T | A/T | A/T | T/T | T/T |
| 296,452 | A 0.26 G 0.74 | rs2576050 | G/G | G/G | G/G | A/G | G/G | G/G |
| 444,615 | A 0.28 G 0.72 | rs2576040 | G/G | G/G | G/G | A/G | G/G | G/G |
| 538,731 | G 0.26 A 0.74 | rs16949034 | A/A | A/A | G/A | A/A | G/A | A/A |
| 615,845 | A 0.27 G 0.73 | rs1434529 | G/G | G/G | G/G | G/G | G/G | G/G |
| 631,424 | C 0.24 G 0.76 | rs1398218 | G/G | G/G | G/G | G/G | G/G | G/G |
| 733,575 | C 0.35 T 0.65 | rs1434506 | C/T | C/C | C/T | C/T | T/T | C/C |
| 805,030 | C 0.33 T 0.67 | rs1893784 | C/T | T/T | C/T | T/T | C/T | T/T |
| 822,756 | C 0.39 T 0.61 | rs4986222 | C/C | C/C | C/T | C/T | C/T | C/C |
| 973,908 | C 0.36 T 0.64 | rs1108062 | T/T | T/T | T/T | T/T | C/T | T/T |
| 1,042,874 | G 0.33 A 0.67 | rs3813071 | A/A | A/A | G/A | A/A | A/A | A/A |
| 1,100,150 | T 0.30 G 0.70 | rs12969708 | G/G | G/G | T/G | G/G | G/G | G/G |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maekawa, K.; Nishio, S.-y.; Abe, S.; Goto, S.-i.; Honkura, Y.; Iwasaki, S.; Kanda, Y.; Kobayashi, Y.; Oka, S.-i.; Okami, M.; et al. Mutational Spectrum and Clinical Features of Patients with LOXHD1 Variants Identified in an 8074 Hearing Loss Patient Cohort. Genes 2019, 10, 735. https://doi.org/10.3390/genes10100735
Maekawa K, Nishio S-y, Abe S, Goto S-i, Honkura Y, Iwasaki S, Kanda Y, Kobayashi Y, Oka S-i, Okami M, et al. Mutational Spectrum and Clinical Features of Patients with LOXHD1 Variants Identified in an 8074 Hearing Loss Patient Cohort. Genes. 2019; 10(10):735. https://doi.org/10.3390/genes10100735
Chicago/Turabian StyleMaekawa, Karuna, Shin-ya Nishio, Satoko Abe, Shin-ichi Goto, Yohei Honkura, Satoshi Iwasaki, Yukihiko Kanda, Yumiko Kobayashi, Shin-ichiro Oka, Mayuri Okami, and et al. 2019. "Mutational Spectrum and Clinical Features of Patients with LOXHD1 Variants Identified in an 8074 Hearing Loss Patient Cohort" Genes 10, no. 10: 735. https://doi.org/10.3390/genes10100735
APA StyleMaekawa, K., Nishio, S.-y., Abe, S., Goto, S.-i., Honkura, Y., Iwasaki, S., Kanda, Y., Kobayashi, Y., Oka, S.-i., Okami, M., Oshikawa, C., Sakuma, N., Sano, H., Shirakura, M., Uehara, N., & Usami, S.-i. (2019). Mutational Spectrum and Clinical Features of Patients with LOXHD1 Variants Identified in an 8074 Hearing Loss Patient Cohort. Genes, 10(10), 735. https://doi.org/10.3390/genes10100735