The Use of Pluripotent Stem Cell-Derived Organoids to Study Extracellular Matrix Development during Neural Degeneration

Department of Chemical and Biomedical Engineering, FAMU-FSU College of Engineering, Florida State University, Tallahassee, FL 32310, USA
*
Author to whom correspondence should be addressed.
†
Current address: Waisman Center, University of Wisconsin-Madison, Madison, WI 53706, USA.
‡
These two authors contributed equally to this work.
Cells 2019, 8(3), 242; https://doi.org/10.3390/cells8030242
Submission received: 19 February 2019
/
Revised: 6 March 2019
/
Accepted: 8 March 2019
/
Published: 14 March 2019
(This article belongs to the Special Issue Stem Cell-based Therapy and Disease Modeling)
Abstract
:The mechanism that causes the Alzheimer’s disease (AD) pathologies, including amyloid plaque, neurofibrillary tangles, and neuron death, is not well understood due to the lack of robust study models for human brain. Three-dimensional organoid systems based on human pluripotent stem cells (hPSCs) have shown a promising potential to model neurodegenerative diseases, including AD. These systems, in combination with engineering tools, allow in vitro generation of brain-like tissues that recapitulate complex cell-cell and cell-extracellular matrix (ECM) interactions. Brain ECMs play important roles in neural differentiation, proliferation, neuronal network, and AD progression. In this contribution related to brain ECMs, recent advances in modeling AD pathology and progression based on hPSC-derived neural cells, tissues, and brain organoids were reviewed and summarized. In addition, the roles of ECMs in neural differentiation of hPSCs and the influences of heparan sulfate proteoglycans, chondroitin sulfate proteoglycans, and hyaluronic acid on the progression of neurodegeneration were discussed. The advantages that use stem cell-based organoids to study neural degeneration and to investigate the effects of ECM development on the disease progression were highlighted. The contents of this article are significant for understanding cell-matrix interactions in stem cell microenvironment for treating neural degeneration.
1. Introduction of Alzheimer’s Disease Pathology
1.1. Alzheimer’s Disease Pathology and Progression
Alzheimer’s disease (AD) is a very common, incurable age-associated and economically costly disease characterized by progressive neurodegeneration, which causes deterioration and damage of neurons within the cerebral cortex, loss of memory, and cognitive decline [1]. It is the most common type of dementia and over 30 million people are suffering from this disease all over the world [2]. About 500,000 people die of AD every year and this number increases year by year. Currently, no effective treatments or available drugs can cure AD and the total costs in 2010 were around $172 billion in the United States [2], which makes it a heavy economic burden not only on the dementia patients but also on our global society. Most of clinical cases in AD develop late-onset symptoms (after the age of 65), also called sporadic AD (SAD). And about 2–5% of disease burden is an early-onset type, called familial AD (FAD). FAD is related to genetic mutations, such as mutations in amyloid precursor protein (APP), presenilin-1 (PS1), and presenilin-2 (PS2) [3]. The gene of PS1 encodes the catalytic subunit of γ-secretase that mediates APP cleavage for the generation of amyloid β42 (Aβ42) peptides [4].
AD is identified by three cardinal features in human brain: Aβ plaques, neurofibrillary tangles (NFTs), and neuron loss. Aβ plaques are usually formed because of the increased production or deficient clearance of neuronal Aβ caused by unknown reasons. APP is proteolyzed to the soluble and nontoxic Aβ monomers by β- and γ-secretases. The monomeric Aβ peptides then aggregate to form toxic Aβ oligomers and further generate the insoluble fibrils, which ultimately form the plaques (Figure 1) [5]. Some extracellular matrix (ECM) components, e.g., heparan sulfate proteoglycans (HSPG), are considered to promote extracellular formation of amyloid plaques [6]. In addition, a healthy body has the mechanism to remove Aβ peptides and balance their generation and clearance. For instance, matrix metalloproteinase 3 (MMP3), a collagen IV proteinase, could degrade Aβ components [7,8]. Also, a defective clearance of Aβ and an unbalance between Aβ generation and clearance have been demonstrated in SAD [9]. Thus, the peculiar generation and flawed removal of Aβ peptides cause Aβ accumulation. The more accumulations of Aβ peptides occur, the more senile plaques are formed in the human brain.
The NFTs in AD are composed of paired helical filaments consisting of hyperphosphorylated tau (p-tau), a protein associated with the microtubules [10]. The normal form of tau is soluble, whereas it depicts strong ability to assemble and become insoluble under physiological conditions. The over phosphorylation of tau contributes to its disassembly from microtubules and its aggregation to form insoluble fibers, that is, the NFTs. Protein kinases, such as mitogen-activated protein kinase (MAPK), glycogen synthase kinase-3 (GSK-3), and Jun N-terminal kinase/stress-activated protein kinases (JNK/SAPK), could upregulate phosphorylation of tau. Many studies support the notion that Aβ may interplay with tau-derived NFT formation, while critical questions about Aβ-induced tau pathology in AD are still unanswered. Overall, the perturbation of tau kinases and tau phosphatases leads to the abnormal hyperphosphorylation of tau, which results in NFT generation and toxicity in neurons.
The Aβ plaque and NFT formation show toxicity to neuronal cells. However, the causes of synaptic dysfunctions and selective loss of vulnerable neurons, particularly the subtypes, such as pyramidal, cholinergic, noradrenergic, and serotonergic neurons, remain elusive [11]. Studies show that Aβ plaques could trigger an inflammatory process by increasing the secretion of pro-inflammatory factors, e.g., IL-6 and TNF-α, which impair microglia and the surrounding neural cells, and finally result in neurodegeneration and neuron loss [12,13]. Based on the Aβ plaque deposition and cortical NFT formation, AD initiates when the connection between entorhinal cortex and hippocampus begins to disappear at the transentorhinal stage [5,14]. Other neuronal cells, such as GABAergic interneurons, show their function loss during the advanced stage of AD, i.e., the isocortical stage.
1.2. Current Challenges and the Demand for a Good AD Model
Although the pathophysiological changes of AD are characterized, it is unclear what factors induce the disease and how selective neuronal loss in AD is caused. Many hypotheses have been proposed for the causes of pathological characteristics [5,9]. These hypotheses include: (1) amyloid hypothesis, which posits that the gene mutations of APP and PS1/2 induce the overexpression or deficient removal of Aβ toxic species, and thus lead to the other two cardinal changes associated with AD (Figure 1); (2) tau hypothesis, which believes that the hyperphosphorylated tau protein disassembles from microtubules and subsequently induces neuronal death (Figure 1); (3) unknown triggering hypothesis, which postulates that some uncertain factors lead to the neurodegeneration, both directly and indirectly, through the formation of plaques and tangles. The last case seems to incorporate both the amyloid and tau hypotheses to interpret the etiology of AD. Although both amyloid and tau hypotheses could explain some of the pathological hallmarks and clinical observations about AD, there is still a controversy over which one occurs first. Thus, it is necessary to establish the disease models of AD, given that it is difficult to obtain patient brain samples.
To date, the modeling of AD for mechanistic and pathological studies mainly relies on non-human animal models and non-neuronal human cells. Non-neuronal cells usually lack the specific cellular structures of neurons and fail to recapitulate the signaling pathway and other physiology of neurons. Therefore, they cannot capture the three pathological changes in AD [15]. The transgenic animal models, usually the mice, have been developed for discovering genetic mutations in FAD [16,17,18]. To some extent, these models could interpret the pathogenicity of Aβ accumulation, plaque formation, and tauopathy. For instance, one study has developed a transgenic AD mouse model with a mutation in APP [17]. The model showed senile plaque formation and disconnection of synapses, but the presence of NFTs and neuronal loss was never found. Although animal models displayed the benefits for understanding AD neuropathology, they fail to reflect human brain anatomy, genetics, and the AD-associated neuron loss. More importantly, pharmacological testing and candidate drug target screening for AD using these animal models have shown no successful development for AD therapeutics till now [19]. Therefore, a more robust and suitable model that could capture all the three cardinal characteristics of AD is demanded. Recently, human adult neural stem cell-derived models through 3D culture in vitro, although limited by the cell source, were found to increase Aβ plaque depositions and tau phosphorylation [20,21]. In another study, Choi et al. reported a 3D thin-layer culture system by embedding human neural stem cells into Matrigel to model AD [22]. The overexpression of FAD mutations in APP and PS1 in embedded cells could induce both extracellular Aβ plaque and phosphorylated tau aggregation in the soma and neurites. These studies accurately paved the way for using human induced pluripotent stem cells (hiPSCs), a more reproducible and scalable cell type, to model AD in vitro.
2. Human Pluripotent Stem Cells for Modeling AD
Human PSCs (hPSCs) include human embryonic stem cells (ESCs) derived from blastocysts and hiPSCs reprogrammed from somatic cells. HPSCs show the properties of unlimited self-renewal and potent differentiation capacity. Theoretically, hPSCs could give rise to the three-germ layers, including ectoderm, endoderm, and mesoderm, and generate every cell type in the body (e.g., neurons, heart, pancreatic, and liver cells). HiPSCs were pioneered by Shinya Yamanaka’s lab in 2006 that the introduction of four reprogramming factors including Oct4, Sox2, c-Myc, and Klf4 (also dubbed Yamanaka factors) could convert adult somatic cells into PSCs [23]. Using hiPSCs to model AD shows great advantages compared to other cell lines or model systems. First, patient-specific cell lines can be easily established just through collecting biopsy from patients (e.g., drops of blood or a piece of skin) and reprogramming the tissue to hiPSCs (Figure 2). Therefore, the ethical controversy using human embryos for human ESCs can be avoided. For AD, the patient-specific hiPSCs loaded with gene mutations can easily recapitulate the mechanism of disease progression, especially for the early-onset type. Thus, the transgenic technology for specific genetic manipulation used in animal models becomes unnecessary. HiPSCs are also clinically advantageous since the use of autologous tissue ideally surpasses the patient’s immune rejection. Moreover, hiPSCs with unlimited self-renewal capacity could help to provide large numbers of patient-specific neuronal cells for in vitro research and clinical applications.
2.1. AD Models Using hPSCs
Generally, three methods are utilized to establish AD pathological phenotype using hPSCs (summarized in Table 1). The first method is chemical induction, in which extrinsic chemicals, such as Aβ42 oligomers and aftin5 (an Aβ42 inducer), are used to treat healthy hPSC-derived neural cells and induce AD-related phenotypes [24,25,26]. The addition of extrinsic chemicals to the normal hPSC-derived neurons could recapitulate some AD events, such as cytotoxicity of neuronal cells. However, the progression and some important AD pathologies, such as extracellular Aβ plaque formation, are difficult to recapitulate. The second method is to use genetic tools, such as lentivirus vectors or CRISPR-Cas9 gene-editing system, to overexpress AD-related genes, such as APP, PS1/2, and apolipoprotein E3/E4 (APOE3/E4), in hPSC-derived cells [27,28,29]. Using human ESC-derived neurons with recombinant human APOE2, APOE3, or APOE4, Huang et al. showed that all three APOE isoforms could upregulate APP and Aβ production but with different efficacy (APOE4 > APOE3 > APOE2) [29]. Park et al. co-cultured neurons and astrocytes derived from hPSCs overexpressing FAD-APP mutations with immortalized SV40 microglia in a microfluidics-based system to study neuro-inflammatory activity in AD [30]. For this approach, overexpression of mutant proteins is needed to induce AD phenotype.
The third method is to use AD patient-specific iPSCs carrying AD phenotype and mutations [42,59]. HiPSCs derived from familial and sporadic AD patients have been differentiated into various types of neuronal cells for studying specific AD pathologies (Table 1). The patients generally had genetic mutations in APP, PS1, or PS2, in the case of FAD, and mutations in APOE3/E4 for SAD. Most of the hPSC-derived AD models used either 2D or embryoid body/neurosphere differentiation protocols to generate forebrain neurons, such as cortical glutamatergic neurons, GABAergic interneurons, or cholinergic neurons. The neuronal cells were characterized through gene and/or neuronal marker expression and tested for action potentials and calcium-handling ability for functional assessments. The AD-related pathology including elevated Aβ42 production and hyperphosphorylated tau was indicated in these models [71,72].
There are two main classes of mutations for familial AD: (1) mutations in APP; and (2) the mutations in PS1 or PS2 [73]. Aβ is a cleavage product of APP, so the dysfunction of APP processing can cause AD (Figure 1). PS1/PS2 mediate the regulated proteolytic events of several proteins including γ-secretase [39], which plays an important role in Aβ generation. Mutations in PS1/PS2 also lead to Aβ plaque accumulation. Human iPSC lines were derived from familial (APP mutation) and sporadic AD patients and differentiated into cortical neurons (expressing TBR1 and SATB2) [37]. In FAD-derived cells, extracellular Aβ42 increased along with the decrease in intracellular Aβ2 compared to control neural cells. For SAD-derived cells, extracellular Aβ42 did not change while intracellular Aβ42 decreased compared to control of neural cells. Astrocytes also accumulated intracellular Aβ, which increased endoplasmic reticulum (ER) stress and reactive oxygen species (ROS). To improve ER stress and inhibit ROS generation, drugs including BSI, DHA, NSC23766, and dibenzoylmethane were evaluated. DHA decreased BiP (an HSP70 molecular chaperone located in the lumen of the ER) protein level, cleaved caspase 4 and peroxiredoxin-4, and decreased ROS generation. Differential response to DHA was observed for different patient-specific hiPSCs, indicating that DHA may be used for a subset of AD patients.
To evaluate if hiPSCs can recapitulate AD phenotype in the patients and understand the relationship between APP processing and tau phosphorylation, hiPSCs were derived from two familial (mutations in APP) and two sporadic AD patients [36]. Neurons from AD patients (two familial and one sporadic) had elevated levels of Aβ40, p-tau, and active glycogen synthase kinase-3β (aGSK-3β). β-secretase inhibitors, but not γ-secretase inhibitors, reduced p-tau and aGSK-3β levels. In AD, synaptic loss is one pathological observation, and the decreased synapsin I is usually observed in the affected human brain. In this study, no synaptic loss was observed and an extended culture period may be required to observe synaptic loss.
Neural progenitor cells (NPCs) derived from hiPSCs from PS1 FAD patients were also characterized [39]. The differentiation from hiPSCs used monolayer-based protocol with dual SMAD inhibition for 14 days. The derived NPCs displayed normal electrophysiology. However, an increased ratio of Aβ42/Aβ40 was observed in the conditioned medium compared to the control cell lines. Genetic profiling identified the genes that were differentially regulated due to PS1 mutations in order to find the molecules that might have a developmental role in FAD pathology. Three targets were found, including NLRP2, ASB2, and NDP. In particular, NDP expression decreased in the hippocampus of the late-onset AD brains. Moreover, synaptic dysfunction was observed in hiPSC-derived cortical neurons with autosomal dominant AD mutations or trisomy of chromosome 21 [58]. The released Aβ peptides from the FAD neurons with APP or PS1 mutations blocked hippocampal long-term potentiation (LTP), while neurons with trisomy 21 inhibited LTP through extracellular tau [58]. In another study, APOE4-expressing neurons showed elevated levels of tau phosphorylation, which was not related to the increased Aβ production, and the cells displayed GABAergic neuron degeneration [66].
2.2. Neural Tissue Patterning of hPSCs
The advancements using hPSCs to model AD started with the generation of neuronal cells or tissues that are directly affected by the disease through neural patterning (Figure 2). The key to success of this utilization is to efficiently generate specific neuronal subtypes or brain-like tissues from hPSCs. Neural tissue patterning is a complex process governed by intrinsic and extrinsic factors including morphogens and cell-ECM interactions. During the development of mammalian brain, early neural progenitors of the neural tube are derived through the inhibition of bone morphogenetic protein (BMP) and activin signaling. The cells are then specified into neuronal subtypes of each brain region along the rostral-caudal axis by tuning Wnt and retinoic acid (RA) signaling pathways, and along the dorsal-ventral axis mainly by the gradient of sonic hedgehog (SHH) proteins. Based on this knowledge, protocols utilizing the morphogens to mimic brain development have been established to produce brain region-specific neuronal subtypes derived from hPSCs (Figure 3). For example, cortical glutamatergic neurons and GABAergic neurons from hPSCs were acquired by the modulation of SHH and fibroblast growth factor (FGF)-2 signals [25,74], while hindbrain/spinal motor neurons were efficiently derived through the activation of SHH, RA, and Wnt signaling pathways [75]. HPSC-derived spherical brain-like tissues, such as cerebral organoids with definable forebrain, midbrain, and hindbrain/spinal cord layers [76], and cortical spheroids with laminated regions [77] were generated by the default neural development without extrinsic factors. Interestingly, recent studies have obtained the whole spectrum of brain region-specific neural progenitors and neuronal subtypes from hiPSCs [78,79], which indicates the remarkable value of hiPSC-based models for the study and treatment of patient-specific neurological diseases. For AD, the Aβ deposition, tau tangles, and neuronal loss ultimately occur in the cerebral cortex, leading to the cortical symptoms related to language, attention, and visuospatial orientation. Therefore, studies modeling AD with hiPSCs often begin with the generation of forebrain cortical neurons. Differentiation to forebrain neuronal fates is based on the “default” induction strategies without exogenous patterning factors after dual-SMAD inhibition in a monolayer culture or embryoid body (EB)-based culture [77,80].
2.3. A Novel Neural Patterning Method: Organoid Technology
The organoid systems derived from EB-based cultures emerge as a mixed cell population in a 3D platform. Relying on the intrinsic ability of self-organization of hiPSCs, and sometimes with the help of suitable exogenous factors (e.g., Matrigel), organoids recapitulate a large number of biological events in vivo [82,83]. Organoids are 3D spatial organization including heterogeneous tissue-specific cells, cell-cell interactions, and cell-ECM interactions, and exhibit certain physiological functions similar to tissues or organs in human body [84]. During organoid formation, ECM is one of the most important patterning factors that regulate and assist the self-organization and differentiation of stem cells within the organoids (Figure 4) [85]. ECMs, either secreted by stem cells or derived from artificial scaffolds, provide physical supports for cell attachment and organization and additional supplementation of signaling cues for cell growth and differentiation [86,87]. This organoid technology bridges a gap in the existing in vitro 3D culture systems by providing a robust approach for tissue patterning of stem cells and is more representative of in vivo situations [88,89,90].
The first generation of neural rosettes from human ESCs was derived in 2001 by forming spontaneously differentiated EBs and then plating EBs on coated dishes for neuroepithelial cluster formation [91]. Combining the EB-based rosette patterning and serum-free culture, the so-called SFEBq approach, i.e., serum-free floating culture of EB-like aggregates with quick re-aggregation, was developed and could generate remarkably large rosettes with lumens and apicobasal structures from hPSCs in 2008 [92]. In 2011, the self-organizing optic cups with retinal specifications from hPSCs were derived using the entirely 3D EB-based neural culture [93]. This study indicates that neural tissues with histologically accurate architecture could be patterned only in 3D floating culture of hPSCs. All these studies paved the road for the advent of a new neural patterning technology, that is, organoid. In 2013, Lancaster et al. derived cerebral organoids with a broad brain regionalization to model microcephaly using EB-based culture with Matrigel embedding and agitation [76]. The organoids could reach up to 4 mm in diameter with fluid-filled cavities that resembled ventricles rather than the small neural-tube-like lumens found in rosettes. After that, the organoid-based neural patterning methods of hPSCs have been utilized for mimicking human brain development, modeling neurological disease in vitro, and testing potential drug candidates [81].
Using the SFEBq method, multilayer structures were derived from hiPSCs with lower and upper cortical layer fates in dorsal forebrain [94]. The 3D aggregates can generate inhibitory neurons and display neuronal connectivity (expression of synapsin I and PSD95). Genomics study showed the correlation to the brain development at postconceptional weeks 4–10. Another 3D culture was used to generate laminated cerebral cortex-like structure from hPSCs [77]. The cells were induced for forebrain fate using dual SMAD inhibition for six days, followed by FGF-2/epidermal growth factor treatment for 19 days, and the maturation with brain-derived neurotrophic factor (BDNF) and NT3. At day 18, 85% PAX6+ cells and >80% of FOXG1+ cells were observed. The organoid size increased from 300 µm at two weeks to 4 mm at 2.5 months. The formed human cortical spheroids corresponded to the late mid-fetal periods. The spheroids had a distinct ventricular zone and sub-ventricular zone. The cells also formed superficial cortical layers (expressing BRN2 and SATB2 for layer II-IV, emerged at day 76) and deep cortical layers (expressing CTIP2-layer V and TBR1-layer VI, showed up at day 52). The presence of GFAP+ cells showed the astrogenesis (3–8% cells). Functional characterizations include: (1) calcium imaging for spontaneous calcium spikes; (2) Na+/K+ currents; and (3) the firing of action potentials. Slices of spheroids showed spontaneous synaptic activity, which can be reduced by a glutamate receptor blocker. This study demonstrated the patterning and specification of different neuronal and glial cell types which can be used for large-scale drug screening.
The organoid technology is a promising method to mimic human brain development and could generate brain-like tissues for modeling neurological diseases, including autosomal recessive primary microcephaly [95], Zika virus infection [96], autism spectrum disorder [97], Timothy syndrome [98], brain tumors [99,100], and others (see the review by Amin et al., 2018 [101]). Lots of studies have shown the feasibility of the organoid methods to model AD pathogenesis [24,52,60,61]. Our previous studies found that dynamic culture of cerebral organoids promoted cortical layer structure compared to static culture, and the 3D brain organoids from hiPSCs with PS1 M146V mutation could recapitulate some AD-related phenotype, such as Aβ secretion and neuron death [59,102]. By using hiPSCs with APOE4/E3 mutations, Lin et al. showed that APOE4 forebrain organoids displayed the increased Aβ aggregation and hyperphosphorylated tau [61]. Taken together, 3D hPSC-derived organoids provide structured neural tissues and specific microenvironments that can be used to model AD-associated pathology.
2.4. Effects of ECMs on Neural Patterning of hPSCs
The central nervous system (CNS) is characterized by a functional network of neurons, glia, and their secreted ECMs. ECMs constitute the essential physical structures for neural cells and provide diverse biochemical signals for neurogenesis, neural cell migration and differentiation, and synaptic plasticity. ECMs also play an important role in neural tissue patterning of hPSCs. Studies performed with decellularized ECMs from different PSC derivatives indicated that ECMs guided PSC differentiation into the cell types residing in the tissue from which the ECMs were derived [103,104]. The ECM-stem cell interactions are mainly mediated by integrins, a large family of heterodimeric transmembrane receptors connecting with an intracellular cytoskeleton. Thus, cell migration, organization, and differentiation could be regulated through ECM-integrin-cytoskeleton connections. For instance, the binding between α6β1 integrin and laminin is the key player for neural stem cell adhesion to the vascular structures [105]. ECMs may also control stem cell fate decisions through the modulation of intracellular signaling. For example, our previous study demonstrated that undifferentiated PSC-derived ECMs could upregulate Wnt/β-catenin signaling, while NPC-derived ECMs promoted neural patterning of PSCs through the inhibition of Wnt signaling pathway [106].
The biophysical properties, such as elastic modulus, geometry, and Poisson’s ratio of matrix, also impact growth, proliferation, and neural differentiation of PSCs [107,108]. Stem cells cultured on hydrogels with varied stiffness indicated that substrate elastic modulus can alter critical cellular events, such as ECM assembly, cell motility, and cell spreading [109,110,111]. For neural patterning, a large number of studies showed the important effects of matrix stiffness or elastic modulus (Table 2) [111,112,113,114,115,116,117,118]. Saha et al. first showed that soft substrates supported neuron differentiation from NPCs, while a higher elastic modulus (E ~ 1 to 10 kPa) promoted glial cell generation [111]. Leipzig et al. further demonstrated that substrates with Young’s modulus (E) below 1 kPa favored neuronal differentiation, those in the range of 1 and 3.5 kPa supported astrocytes, and above 7 kPa could enhance oligodendrocyte derivation [112]. By culturing hPSCs on Matrigel-coated polyacrylamide substrates, a very soft matrix (E ~ 0.1 kPa) was found to support early neural differentiation of hPSCs [119]. Normally, cells sense elasticity during the attachment on the substrate through focal adhesions and formation of stress fibers. Their responses to the matrix properties rely on myosin-directed contraction and cell-ECM adhesions, which involve integrins, cadherins, and other adhesion molecules [120]. The Poisson’s ratio is another important biophysical cue that influences stem cell behaviors, as the nuclei of ESCs exhibit a negative Poisson’s ratio in the pluripotent-state [121]. Our previous work found that Poisson’s ratio of matrix could confound the effects of elastic modulus on PSC neural differentiation [108]. In conclusion, ECMs serve as a reservoir of biochemical and biophysical factors that impact stem cell growth, organization, and differentiation.
3. Proteoglycans in the ECMs of AD Brain
The brain ECMs express low amounts of fibrous proteins, such as collagen and laminin, but high amounts of glycosaminoglycans (GAG) compared to other tissue types. The ECMs in the brain include glycoproteins and proteoglycans originated from neurons or glia. Proteoglycans (PGs) are a group of glycoproteins that carry covalently bound sulfated GAG chains which play important roles in cell differentiation, tissue morphogenesis, and phenotypic stabilization via cell-matrix adhesiveness and/or binding to growth regulators. GAGs are composed of repeating disaccharide units attached to a core protein through serine residue and carbohydrate linkage regions. Depending on the disaccharide structures, PGs can be divided into chondroitin/dermatan sulfate, heparin/heparan sulfate (HS), and keratan sulfate side chains. In the CNS, the majority of proteoglycans have either heparin sulfate or chondroitin sulfate side chains. PGs are expressed at different stages of human brain development and are produced non-homogenously by different neural cell types [144].
3.1. Chondroitin Sulfate Proteoglycans (CSPGs) in Brain Development
CSPG family consists of four members: (1) lecticans, a family including aggrecan, versican, neurocan, and brevican; (2) phosphacan; (3) small leucine-rich proteoglycans, e.g., decorin and biglycan; (4) other CSPGs including neuroglycan-C and NG2 [145]. Distribution and expression of these CSPGs vary during human brain development. Aggrecan and versican are distributed in ECMs of various non-neuronal tissues as well as neural tissues, whereas neurocan and brevican appear to show a neural tissue-specific distribution. Versicans are found to express in the post-natal development of brains. Brevican is expressed predominantly by astrocytes, not neurons. NG2, a large transmembrane CSPG, is expressed by oligodendrocyte progenitor cells in CNS. Neurocan, a secretory CSPG, is reported to be synthesized mainly by neuronal cells.
The CSPGs are either secreted as ECMs or inserted to the plasma membrane of the cells. The attachment of CSPGs to the cell membrane is either through a collagenous ligand or by core proteins and cell surface-associated CSPGs such as aggrecan. The concentrations of aggrecan and brevican increase during brain development. The concentration of neurocan also increases with development to reach a peak in the early postnatal period and declines thereafter. The full-length neurocan is the major variant in pre- and early postnatal brains, while it is hardly detectable in the mature brain. Phosphacan and receptor-type protein tyrosine phosphatase are produced by both glial cells and neuronal cells. Variations in sulfate content and localization on the same GAG chains or the same core protein are tissue-specific or cell-specific. The expression of specific GAG biosynthetic and modifying enzymes by individual neural cell types dictates the type of carbohydrate modification of the core proteins expressed in each cell type [146].
3.2. Heparin/HSPG in Brain Development
Heparan sulfate proteoglycans are heavily glycosylated proteins, in which some HS and GAG chains are covalently attached to a core protein, which can be either surface proteins (e.g., syndecans/glypicans) or secreted proteins (e.g., agrin/collagen type XVIII/perlecan) [147]. N-Syndecan (syndecan-3), a transmembrane HSPG, has the capacity to bind to heparin-binding growth factors with a neurite-promoting activity, such as FGF-2, pleiotrophin/HBGAM, and midkine. This matrix-growth factor interaction is found to be the basis for the axonal development of neural cells. Also, N-syndecan works as a receptor molecule for pleiotrophin, which then enhances the phosphorylation of intracellular cytoskeleton-regulating molecules, such as cortactin and fyn-kinase. It was found that the association of cortactin and fyn-kinase with N-syndecan was increased after induction of long-lasting synaptic sensitivity called long-term potentiation.
N-syndecan regulates the neuronal activity-dependent connectivity through the fyn signaling pathway. N-syndecan, together with syndecan-2, also appears to be involved in the maturation of synapses. Syndecan-2 interacts with calcium/calmodulin-dependent serine protein kinase, a PDZ family protein that induces the formation of mature dendritic spines, and supports the development of postsynaptic specialization [148]. Heparin is one of the glycosaminoglycans that bind to different proteins and thereby promote various neural functions, such as preventing blood clotting, neuronal communication, and brain development.
3.3. Impacts of CSPG on AD Pathology
The motor and sensory areas of the cortical brain, which show cytoskeletal changes during AD development, were found to express prominent levels of CSPGs. These results suggested the importance of CSPGs on AD development, e.g., synaptic loss. Numerous molecular events occur in the post-synaptic density of an excitatory synapse in response to a train of pre-synaptic action potentials. One of these factors is the presence of peri-synaptic ECMs, a lattice of chondroitin sulfate-bearing proteoglycans, termed lecticans (e.g., aggrecan, brevican, neurocan, and versican), bound to hyaluronic acid near their N-terminus and tenascin-R near their C-terminus. Alterations in brain ECMs occur early in AD progression. The synaptic loss is observed prior to neuronal cell death, and the loss of synapses in the outer molecular layer of the hippocampal dentate gyrus is more highly correlated with cognitive impairment than other classical AD pathology including NFTs and senile plaques. ECMs can respond to network activity by incorporating secreted molecules, by shedding extracellular domains of transmembrane molecules, or by freeing products of activity-dependent proteolytic cleavage as signaling messengers.
Lecticans, also known as hyalectans, have been studied for their influence on the plasticity of synapses because of the elevated expression in AD patients. Lecticans are found to contribute to diminished synaptic plasticity. Involvement of CSPGs in hippocampal synaptic plasticity was confirmed using the enzyme that specifically digests chondroitin sulfates, chondroitinase ABC (ChABC) [149]. Injection of ChABC in young AD mice elevated lectican levels and resulted in a reversal of contextual fear memory deficits and restoration to normal long-term potentiation. ChABC treatment results in the removal of CS chains from lectican core proteins (and other CS bearing PGs). These studies demonstrated that removal of CS chains stimulated neural plasticity. Increasing evidence has demonstrated that CS-bearing ECM molecules increase with age and are associated with AD. CS-bearing PGs bind to Aβ, but it remains unknown whether Aβ binds to brain-derived lecticans. Injection of ChABC increased synaptic density surrounding plaques and reduced Aβ burden in the s/m of the hippocampus [150].
3.4. Impacts of Heparin/HSPGs on AD Pathology
HSPGs are found to grow concomitantly with amyloid filament formation. HSPGs have a negative charge and thereby concentrate on the ligands and protect them from proteolysis while making insoluble complexes [151]. Heparin and HSPGs regulate the activity of a number of proteases involved in AD. HSPGs promote Aβ aggregation, stabilize Aβ fibrils, and inhibit Aβ degradation. Perlecan and agrin are found to be the important HSPGs that are involved in the pathogenesis and localized with Aβ deposits in AD brain. Perlecan accelerates Aβ fibril formation and maintains the Aβ fibril stability by using Perlecan’s HS-GAG chains [152]. Agrin also binds to Aβ and accelerates fibril formation and protects fibrillar Aβ from proteolysis (Figure 5) [153]. HSPGs bind Aβ with high affinity and promote intracellular Aβ uptake in multiple cell types and thereby increase the cytotoxicity [154].
HSPGs are also the target of Aβ-induced oxidative stress production. Studies propose that minimal chain length is a prerequisite for efficient fibril polymerization and deposition. Fragmentation of HS using heparinase III was found to inhibit the polymerization and deposition of amyloid peptides [155]. Heparinase is a glucuronidase that specifically cleaves heparan sulfate, thereby preventing Aβ-HSPG interactions. The formed HS oligosaccharides then bind amyloid monomers to prevent them from polymerizing and assembling into larger aggregates. Treatment of cells with heparinase III also reduced the HSPG-based Aβ-induced ROS production.
HSPGs modulate the Aβ-associated neuroinflammation and Aβ clearance from the human brain. Several in vitro studies indicated the interaction of Aβ with GAGs including HS and heparin, a HS analogue with a higher sulfation degree [156]. All those studies describe that the N-terminus of Aβ is a HS-binding motif and this sequence supports the interaction between Aβ and HSPGs [157]. Studies also suggest that the Aβ peptides compete with FGF-2 for HS binding site. The interaction depends on several factors, such as the sulfation pattern, chain length of the GAGs, and the pH. The higher sulfation in heparin boosted the affinity and the degree of Aβ-HS complex formation was higher compared to Aβ-HSPG complex. Lower binding of heparin fragments shorter than 6-sugar units to Aβ also suggested the requirement of GAG chain length for the interaction [158]. It is also proposed that the Aβ-HS interaction is mutually protective because HS is protected from heparanase degradation and Aβ is protected from protease degradation.
Cellular degradation of HSPGs using heparanase (an endo-β-glucuronidase that specifically cleaves HS side chains of HSPGs) decreased the cytotoxicity of Aβ peptides on the cells [154]. Aβ clearance from the cells is performed in different ways, including degradation by proteolytic enzymes or receptor-mediated Aβ transport across the blood-brain barrier, in which the main receptor is low-density lipoprotein receptor-related protein-1 (LRP-1) [159,160]. The complex interactions of Aβ precursor protein ApoE, LRP-1, and HSPG facilitate Aβ internalization. Recent studies found that HSPGs and LRP-1 mediate Aβ internalization in a seemingly cooperative manner, in which HSPGs regulate Aβ binding to cell surface through LRP-1 [159]. These findings suggest that cell surface HS mediates Aβ internalization and toxicity. Tau binding and internalization also depend on HSPGs, which are critical mediators of tau fibril internalization. Tau binding to HSPGs is required for transcellular propagation [161]. Further understanding of the interactions between ECM molecules and Aβ peptides and their effects on neuronal differentiation should help to develop new treatment methods for AD.
3.5. Heparin-Based Therapy for Neural Degeneration
Aβ-related activation and the followed neuroinflammation are dependent on the region 1–11 of the peptide [162]. Related studies of the molecules that have the ability to pharmacologically target this region indicate the possible therapeutic effects of heparin [163]. In vitro treatment of neurons with heparin reduced Aβ-associated cytotoxicity, revealing the capability of heparin on AD treatment. Heparin, which has been widely used as an anticoagulant in clinical use, has the capability to bind to the region 12–17 of Aβ (HHQK) [157], near to the contact and complement activation residues. The heparin-binding regions of these proteins are characterized by clusters of arginines and lysines. These clusters form centers of highly positive charge density that electrostatically interact with the acidic groups of heparin.
The binding of heparin to the regions of Aβ occurs as electrostatic interactions between cationic sites of Aβ and anionic sulfate residues of heparin. This causes steric changes in the Aβ and influences the biological function, which in turn reduces the inflammation of neural cells by inhibiting Aβ complement and contact systems and thereby reduces the neurodegeneration. Complement and contact systems are related inflammatory cascades and are dependent on the reciprocal activation. These systems share a major inhibitor factor (C1 INH) with each other (Figure 6), which is downregulated in AD brain. Inhibition of complement activation by heparin might also be due to the potentiation of C1-1NH. C1-1NH attaches to the enzymes and works as an inhibitor of spontaneous activation of the complementary system. The addition of low molecular weight heparin reduced plaques and Aβ accumulation in a mouse AD model [164]. Association between heparin and the Aβ peptides was found to be pH-dependent. Heparin-binding to the peptides reduced the concentration of the peptides and the expression of kinnogen.
Biologically active beta-site amyloid precursor protein cleaving enzyme 1 (BACE1) interacts directly with HS/heparin and has a high relative affinity for these polysaccharides. It has been reported that heparin inhibits BACE1 activity in vitro and thereby regulates the production of Aβ peptides [163,165]. It is also suggested that Aβ is a metalloprotein that expresses both high and low metal affinity sites. The AD patients are characterized by elevated levels of copper and zinc in the hippocampal region. These metals have been found to accelerate the accumulation of Aβ and promote the ROS release to the cells. Cu2+ acts as a bridge between anionic groups on HSPGs and cationic groups of the NFTs. It was also found that free carboxyl groups are required for HSPG binding to NFTs. Heparin can bind to the extracellular superoxide dismutase, a homotetrameric Cu2+ and Zn2+ containing glycoprotein, and regulate the activity of these metals which reduce accumulation of Aβ peptides (Figure 6).
4. Studying ECM Effects in hiPSC-Derived Forebrain Organoids
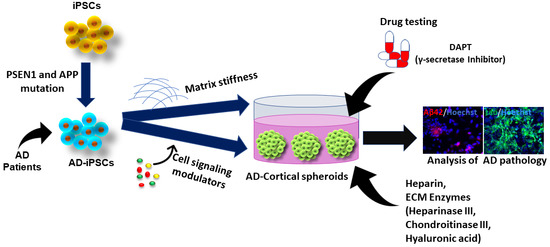
Combining the knowledge of forebrain organoids and the ECM development in AD pathology, our studies used heparin (competes for Aβ affinity with HSPG), heparinase III (digests HSPGs), chondroitinase (digests CSPGs), and hyaluronic acid (HA) to treat the cortical and hippocampal forebrain spheroids/organoids exposed to Aβ42 oligomers [166]. Briefly, cortical spheroids were generated using dual SMAD inhibition followed with FGF-2 and cyclopamine (an Shh inhibitor). In the case of hippocampal differentiation, the hiPSCs were treated with dual SMAD inhibitors, cyclopamine, and IWP4 (a Wnt inhibitor), followed by treatment with CHIR99021 (a Wnt activator) and BDNF. Exogenous addition of Aβ42 oligomers was found to reduce the expression of β-tubulin III, suggesting the associated neuronal cell death. The treatments of ECM-related molecules with Aβ42 oligomers promoted β-tubulin III expression, indicating the protective effect of these ECM related molecules. Further analysis on the forebrain and hippocampal markers revealed that ECM enzymes were capable of rescuing TBR1+ cells in cortical forebrain spheroids and PROX1+ neurons in the hippocampal spheroids in the presence of Aβ42 oligomers. These results suggest the necessity of further evaluation of various brain ECMs on forebrain spheroid patterning and cell survival and the related neurological disorders to derive better therapeutic interventions. In addition, heparin-conjugated HA hydrogels were synthesized and characterized [107]. A comparative study with heparin-HA hydrogels with varying modulus reveals that the lower modulus (300–400 Pa) hydrogel supports the forebrain fate whereas the higher modulus (1000–1300 Pa) hydrogel supports hindbrain fate of hiPSCs. This patterning effects may be regulated through modulation of the Hippo/YAP signaling.
5. Conclusions and Perspectives
Recently, the findings of adult neurogenesis in humans have sharply declined, which raises questions about animal models to study human central nervous system [167]. The emerging hPSC technology and organoid methods represent promising opportunities to investigate the development of human brain and neurological diseases. These stem cell-based organoid systems have recapitulated some biological events, such as temporal and spatial organization of brain tissues, neuronal-glial cell interactions, and cell-matrix interactions. However, these models need to show promise to reveal new features of normal and pathological phenomena of neurogenesis. One aspect of the interests is that the patient-derived hPSCs with mutations, such as APP or PS1/PS2, could be used to study the roles of specific genes in FAD development and progression. Another aspect is that organoids provide a more advanced in vitro tool to investigate complex niche interactions, such as how HSPGs are involved in Aβ pathogenesis. While the organoid models have great advantages, they are also challenged by the low homogeneity and reproducibility. The brain organoids may be different from each other not only in size but also structure, which makes it difficult to control the quality and predict the readouts. Future improvements, such as novel biomaterials combined with new culture system design (e.g., microfluidics), may be utilized to better control neural patterning in a more accurate and predictable way.
Author Contributions
Y.Y. wrote the first half of the manuscript, summarized tables, and prepared some figures. J.B. wrote the second half of the manuscript and prepared some figures. M.M. helped literature review. Y.L. conceived the projects, wrote, and revised the manuscript.
Funding
This work is supported by FSU start up fund, FSU Bridge Grant, NSF BRIGE Award (No. 1342192), NSF Career Award (No. 1652992 to Y.L.), and by NIH R03NS102640 (Y.L.).
Acknowledgments
We would like to thank Liqing Song for her help with checking references. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Stelzmann, R.A.; Norman Schnitzlein, H.; Reed Murtagh, F. An English translation of Alzheimer’s 1907 paper, “Über eine eigenartige Erkankung der Hirnrinde”. Clin. Anat. 1995, 8, 429–431. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Dementia: A Public Health Priority; World Health Organization: Genève, Switzerland, 2012. [Google Scholar]
- Holtzman, D.M.; Morris, J.C.; Goate, A.M. Alzheimer’s disease: The challenge of the second century. Sci. Transl. Med. 2011, 3, 77sr71. [Google Scholar] [CrossRef] [PubMed]
- Bekris, L.M.; Yu, C.-E.; Bird, T.D.; Tsuang, D.W. Genetics of Alzheimer disease. J. Geriatr. Psychiatry Neurol. 2010, 23, 213–227. [Google Scholar] [CrossRef] [PubMed]
- Palmer, A.M. Neuroprotective therapeutics for Alzheimer’s disease: Progress and prospects. Trends Pharmacol. Sci. 2011, 32, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Van Horssen, J.; Wilhelmus, M.M.; Heljasvaara, R.; Pihlajaniemi, T.; Wesseling, P.; de Waal, R.M.; Verbeek, M.M. Collagen XVIII: A Novel Heparan Sulfate Proteoglycan Associated with Vascular Amyloid Depositions and Senile Plaques in Alzheimer’s Disease Brains. Brain Pathol. 2002, 12, 456–462. [Google Scholar] [CrossRef] [PubMed]
- Meighan, S.E.; Meighan, P.C.; Choudhury, P.; Davis, C.J.; Olson, M.L.; Zornes, P.A.; Wright, J.W.; Harding, J.W. Effects of extracellular matrix-degrading proteases matrix metalloproteinases 3 and 9 on spatial learning and synaptic plasticity. J. Neurochem. 2006, 96, 1227–1241. [Google Scholar] [CrossRef] [Green Version]
- Mlekusch, R.; Humpel, C. Matrix metalloproteinases-2 and-3 are reduced in cerebrospinal fluid with low beta-amyloid1–42 levels. Neurosci. Lett. 2009, 466, 135–138. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.-C.; Jiang, Z.-F. Accumulated amyloid-β peptide and hyperphosphorylated tau protein: Relationship and links in Alzheimer’s disease. J. Alzheimer Dis. 2009, 16, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Köpke, E.; Tung, Y.C.; Shaikh, S.; Alonso, A.D.; Iqbal, K.; Grundke-Iqbal, I. Microtubule-associated protein tau. Abnormal phosphorylation of a non-paired helical filament pool in Alzheimer disease. J. Biol. Chem. 1993, 268, 24374–24384. [Google Scholar] [PubMed]
- Palmer, A.M. Pharmacotherapy for Alzheimer’s disease: Progress and prospects. Trends Pharmacol. Sci. 2002, 23, 426–433. [Google Scholar] [CrossRef]
- Prokop, S.; Miller, K.R.; Heppner, F.L. Microglia actions in Alzheimer’s disease. Acta Neuropathol. 2013, 126, 461–477. [Google Scholar] [CrossRef]
- Heppner, F.L.; Ransohoff, R.M.; Becher, B. Immune attack: The role of inflammation in Alzheimer disease. Nat. Rev. Neurosci. 2015, 16, 358. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Rüb, U.; Schultz, C.; Tredici, K.D. Vulnerability of cortical neurons to Alzheimer’s and Parkinson’s diseases. J. Alzheimer Dis. 2006, 9, 35–44. [Google Scholar] [CrossRef]
- Xu, X.-H.; Zhong, Z. Disease modeling and drug screening for neurological diseases using human induced pluripotent stem cells. Acta Pharmacol. Sin. 2013, 34, 755. [Google Scholar] [CrossRef]
- Duff, K.; Eckman, C.; Zehr, C.; Yu, X.; Prada, C.-M.; Perez-Tur, J.; Hutton, M.; Buee, L.; Harigaya, Y.; Yager, D. Increased amyloid-β42 (43) in brains of mice expressing mutant presenilin 1. Nature 1996, 383, 710. [Google Scholar] [CrossRef]
- Hsiao, K.; Chapman, P.; Nilsen, S.; Eckman, C.; Harigaya, Y.; Younkin, S.; Yang, F.; Cole, G. Correlative memory deficits, Aβ elevation, and amyloid plaques in transgenic mice. Science 1996, 274, 99–103. [Google Scholar] [CrossRef]
- Sturchler-Pierrat, C.; Abramowski, D.; Duke, M.; Wiederhold, K.-H.; Mistl, C.; Rothacher, S.; Ledermann, B.; Bürki, K.; Frey, P.; Paganetti, P.A. Two amyloid precursor protein transgenic mouse models with Alzheimer disease-like pathology. Proc. Natl. Acad. Sci. USA 1997, 94, 13287–13292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franco, R.; Cedazo-Minguez, A. Successful therapies for Alzheimer’s disease: Why so many in animal models and none in humans? Front. Pharmacol. 2014, 5, 146. [Google Scholar] [CrossRef]
- Nam, H.; Lee, K.-H.; Nam, D.-H.; Joo, K.M. Adult human neural stem cell therapeutics: Current developmental status and prospect. World J. Stem Cells 2015, 7, 126. [Google Scholar] [CrossRef] [PubMed]
- Jakel, R.J.; Schneider, B.L.; Svendsen, C.N. Using human neural stem cells to model neurological disease. Nat. Rev. Genet. 2004, 5, 136. [Google Scholar] [CrossRef]
- Choi, S.H.; Kim, Y.H.; Hebisch, M.; Sliwinski, C.; Lee, S.; D’Avanzo, C.; Chen, H.; Hooli, B.; Asselin, C.; Muffat, J.; et al. A three-dimensional human neural cell culture model of Alzheimer’s disease. Nature 2014, 515, 274–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narazaki, G.; Uosaki, H.; Teranishi, M.; Okita, K.; Kim, B.; Matsuoka, S.; Yamanaka, S.; Yamashita, J.K. Directed and systematic differentiation of cardiovascular cells from mouse induced pluripotent stem cells. Circulation 2008, 118, 498–506. [Google Scholar] [CrossRef] [PubMed]
- Pavoni, S.; Jarray, R.; Nassor, F.; Guyot, A.C.; Cottin, S.; Rontard, J.; Mikol, J.; Mabondzo, A.; Deslys, J.P.; Yates, F. Small-molecule induction of Abeta-42 peptide production in human cerebral organoids to model Alzheimer’s disease associated phenotypes. PLoS ONE 2018, 13, e0209150. [Google Scholar] [CrossRef] [PubMed]
- Vazin, T.; Ball, K.A.; Lu, H.; Park, H.; Ataeijannati, Y.; Head-Gordon, T.; Poo, M.M.; Schaffer, D.V. Efficient derivation of cortical glutamatergic neurons from human pluripotent stem cells: A model system to study neurotoxicity in Alzheimer’s disease. Neurobiol. Dis. 2014, 62, 62–72. [Google Scholar] [CrossRef]
- Yan, Y.; Bejoy, J.; Xia, J.; Guan, J.; Zhou, Y.; Li, Y. Neural patterning of human induced pluripotent stem cells in 3-D cultures for studying biomolecule-directed differential cellular responses. Acta Biomater. 2016, 42, 114–126. [Google Scholar] [CrossRef] [PubMed]
- Koch, P.; Tamboli, I.Y.; Mertens, J.; Wunderlich, P.; Ladewig, J.; Stuber, K.; Esselmann, H.; Wiltfang, J.; Brustle, O.; Walter, J. Presenilin-1 L166P mutant human pluripotent stem cell-derived neurons exhibit partial loss of gamma-secretase activity in endogenous amyloid-beta generation. Am. J. Pathol. 2012, 180, 2404–2416. [Google Scholar] [CrossRef]
- Woodruff, G.; Young, J.E.; Martinez, F.J.; Buen, F.; Gore, A.; Kinaga, J.; Li, Z.; Yuan, S.H.; Zhang, K.; Goldstein, L.S. The presenilin-1 DeltaE9 mutation results in reduced gamma-secretase activity, but not total loss of PS1 function, in isogenic human stem cells. Cell Rep. 2013, 5, 974–985. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.A.; Zhou, B.; Wernig, M.; Sudhof, T.C. ApoE2, ApoE3, and ApoE4 Differentially Stimulate APP Transcription and Abeta Secretion. Cell 2017, 168, 427–441.e421. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Wetzel, I.; Marriott, I.; Dreau, D.; D’Avanzo, C.; Kim, D.Y.; Tanzi, R.E.; Cho, H. A 3D human triculture system modeling neurodegeneration and neuroinflammation in Alzheimer’s disease. Nat. Neurosci. 2018, 21, 941–951. [Google Scholar] [CrossRef] [PubMed]
- Wicklund, L.; Leao, R.N.; Stromberg, A.M.; Mousavi, M.; Hovatta, O.; Nordberg, A.; Marutle, A. Beta-amyloid 1-42 oligomers impair function of human embryonic stem cell-derived forebrain cholinergic neurons. PLoS ONE 2010, 5, e15600. [Google Scholar] [CrossRef]
- Yahata, N.; Asai, M.; Kitaoka, S.; Takahashi, K.; Asaka, I.; Hioki, H.; Kaneko, T.; Maruyama, K.; Saido, T.C.; Nakahata, T.; et al. Anti-Abeta drug screening platform using human iPS cell-derived neurons for the treatment of Alzheimer’s disease. PLoS ONE 2011, 6, e25788. [Google Scholar] [CrossRef]
- Mertens, J.; Stuber, K.; Poppe, D.; Doerr, J.; Ladewig, J.; Brustle, O.; Koch, P. Embryonic stem cell-based modeling of tau pathology in human neurons. Am. J. Pathol. 2013, 182, 1769–1779. [Google Scholar] [CrossRef] [PubMed]
- Yagi, T.; Ito, D.; Okada, Y.; Akamatsu, W.; Nihei, Y.; Yoshizaki, T.; Yamanaka, S.; Okano, H.; Suzuki, N. Modeling familial Alzheimer’s disease with induced pluripotent stem cells. Hum. Mol. Genet. 2011, 20, 4530–4539. [Google Scholar] [CrossRef]
- Shi, Y.C.; Kirwan, P.; Smith, J.; MacLean, G.; Orkin, S.H.; Livesey, F.J. A Human Stem Cell Model of Early Alzheimer’s Disease Pathology in Down Syndrome. Sci. Transl. Med. 2012, 4, 124ra29. [Google Scholar] [CrossRef]
- Israel, M.A.; Yuan, S.H.; Bardy, C.; Reyna, S.M.; Mu, Y.; Herrera, C.; Hefferan, M.P.; Van Gorp, S.; Nazor, K.L.; Boscolo, F.S.; et al. Probing sporadic and familial Alzheimer’s disease using induced pluripotent stem cells. Nature 2012, 482, 216–220. [Google Scholar] [CrossRef] [Green Version]
- Kondo, T.; Asai, M.; Tsukita, K.; Kutoku, Y.; Ohsawa, Y.; Sunada, Y.; Imamura, K.; Egawa, N.; Yahata, N.; Okita, K.; et al. Modeling Alzheimer’s disease with iPSCs reveals stress phenotypes associated with intracellular Abeta and differential drug responsiveness. Cell Stem Cell 2013, 12, 487–496. [Google Scholar] [CrossRef] [PubMed]
- Mertens, J.; Stuber, K.; Wunderlich, P.; Ladewig, J.; Kesavan, J.C.; Vandenberghe, R.; Vandenbulcke, M.; van Damme, P.; Walter, J.; Brustle, O.; et al. APP processing in human pluripotent stem cell-derived neurons is resistant to NSAID-based gamma-secretase modulation. Stem Cell Rep. 2013, 1, 491–498. [Google Scholar] [CrossRef] [PubMed]
- Sproul, A.A.; Jacob, S.; Pre, D.; Kim, S.H.; Nestor, M.W.; Navarro-Sobrino, M.; Santa-Maria, I.; Zimmer, M.; Aubry, S.; Steele, J.W.; et al. Characterization and molecular profiling of PSEN1 familial Alzheimer’s disease iPSC-derived neural progenitors. PLoS ONE 2014, 9, e84547. [Google Scholar] [CrossRef]
- Mahairaki, V.; Ryu, J.; Peters, A.; Chang, Q.; Li, T.; Park, T.S.; Burridge, P.W.; Talbot, C.C., Jr.; Asnaghi, L.; Martin, L.J.; et al. Induced pluripotent stem cells from familial Alzheimer’s disease patients differentiate into mature neurons with amyloidogenic properties. Stem Cells Dev. 2014, 23, 2996–3010. [Google Scholar] [CrossRef]
- Duan, L.S.; Bhattacharyya, B.J.; Belmadani, A.; Pan, L.L.; Miller, R.J.; Kessler, J.A. Stem cell derived basal forebrain cholinergic neurons from Alzheimer’s disease patients are more susceptible to cell death. Mol. Neurodegen. 2014, 9, 3. [Google Scholar] [CrossRef]
- Muratore, C.R.; Rice, H.C.; Srikanth, P.; Callahan, D.G.; Shin, T.; Benjamin, L.N.; Walsh, D.M.; Selkoe, D.J.; Young-Pearse, T.L. The familial Alzheimer’s disease APPV717I mutation alters APP processing and Tau expression in iPSC-derived neurons. Human. Mol. Genet. 2014, 23, 3523–3536. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Waltz, S.; Woodruff, G.; Ouyang, J.; Israel, M.A.; Herrera, C.; Sarsoza, F.; Tanzi, R.E.; Koo, E.H.; Ringman, J.M.; et al. Effect of potent gamma-secretase modulator in human neurons derived from multiple presenilin 1-induced pluripotent stem cell mutant carriers. JAMA Neurol. 2014, 71, 1481–1489. [Google Scholar] [CrossRef]
- Lee, J.K.; Jin, H.K.; Park, M.H.; Kim, B.R.; Lee, P.H.; Nakauchi, H.; Carter, J.E.; He, X.; Schuchman, E.H.; Bae, J.S. Acid sphingomyelinase modulates the autophagic process by controlling lysosomal biogenesis in Alzheimer’s disease. J. Exp. Med. 2014, 211, 1551–1570. [Google Scholar] [CrossRef] [Green Version]
- Young, J.E.; Boulanger-Weill, J.; Williams, D.A.; Woodruff, G.; Buen, F.; Revilla, A.C.; Herrera, C.; Israel, M.A.; Yuan, S.H.; Edland, S.D.; et al. Elucidating molecular phenotypes caused by the SORL1 Alzheimer’s disease genetic risk factor using human induced pluripotent stem cells. Cell Stem Cell 2015, 16, 373–385. [Google Scholar] [CrossRef] [PubMed]
- Moore, S.; Evans, L.D.; Andersson, T.; Portelius, E.; Smith, J.; Dias, T.B.; Saurat, N.; McGlade, A.; Kirwan, P.; Blennow, K.; et al. APP metabolism regulates tau proteostasis in human cerebral cortex neurons. Cell Rep. 2015, 11, 689–696. [Google Scholar] [CrossRef] [PubMed]
- Hossini, A.M.; Megges, M.; Prigione, A.; Lichtner, B.; Toliat, M.R.; Wruck, W.; Schroter, F.; Nuernberg, P.; Kroll, H.; Makrantonaki, E.; et al. Induced pluripotent stem cell-derived neuronal cells from a sporadic Alzheimer’s disease donor as a model for investigating AD-associated gene regulatory networks. BMC Genom. 2015, 16, 84. [Google Scholar]
- Armijo, E.; Gonzalez, C.; Shahnawaz, M.; Flores, A.; Davis, B.; Soto, C. Increased susceptibility to Abeta toxicity in neuronal cultures derived from familial Alzheimer’s disease (PSEN1-A246E) induced pluripotent stem cells. Neurosci. Lett. 2017, 639, 74–81. [Google Scholar] [CrossRef]
- Balez, R.; Steiner, N.; Engel, M.; Munoz, S.S.; Lum, J.S.; Wu, Y.; Wang, D.; Vallotton, P.; Sachdev, P.; O’Connor, M.; et al. Neuroprotective effects of apigenin against inflammation, neuronal excitability and apoptosis in an induced pluripotent stem cell model of Alzheimer’s disease. Sci. Rep. 2016, 6, 31450. [Google Scholar] [CrossRef] [Green Version]
- Liao, M.-C.; Muratore, C.R.; Gierahn, T.M.; Sullivan, S.E.; Srikanth, P.; De Jager, P.L.; Love, J.C.; Young-Pearse, T.L. Single-Cell Detection of Secreted Aβ and sAPPα from Human IPSC-Derived Neurons and Astrocytes. J. Neurosci. 2016, 36, 1730–1746. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.K.; Velazquez Sanchez, C.; Chen, M.; Morin, P.J.; Wells, J.M.; Hanlon, E.B.; Xia, W. Three Dimensional Human Neuro-Spheroid Model of Alzheimer’s Disease Based on Differentiated Induced Pluripotent Stem Cells. PLoS ONE 2016, 11, e0163072. [Google Scholar] [CrossRef]
- Raja, W.K.; Mungenast, A.E.; Lin, Y.T.; Ko, T.; Abdurrob, F.; Seo, J.; Tsai, L.H. Self-Organizing 3D Human Neural Tissue Derived from Induced Pluripotent Stem Cells Recapitulate Alzheimer’s Disease Phenotypes. PLoS ONE 2016, 11, e0161969. [Google Scholar] [CrossRef] [PubMed]
- Jones, V.C.; Atkinson-Dell, R.; Verkhratsky, A.; Mohamet, L. Aberrant iPSC-derived human astrocytes in Alzheimer’s disease. Cell Death Dis. 2017, 8, e2696. [Google Scholar] [CrossRef] [PubMed]
- Muratore, C.R.; Zhou, C.; Liao, M.; Fernandez, M.A.; Taylor, W.M.; Lagomarsino, V.N.; Pearse, R.V., 2nd; Rice, H.C.; Negri, J.M.; He, A.; et al. Cell-type Dependent Alzheimer’s Disease Phenotypes: Probing the Biology of Selective Neuronal Vulnerability. Stem Cell Rep. 2017, 9, 1868–1884. [Google Scholar] [CrossRef] [PubMed]
- Kondo, T.; Imamura, K.; Funayama, M.; Tsukita, K.; Miyake, M.; Ohta, A.; Woltjen, K.; Nakagawa, M.; Asada, T.; Arai, T.; et al. iPSC-Based Compound Screening and In Vitro Trials Identify a Synergistic Anti-amyloid beta Combination for Alzheimer’s Disease. Cell Rep. 2017, 21, 2304–2312. [Google Scholar] [CrossRef]
- Ochalek, A.; Mihalik, B.; Avci, H.X.; Chandrasekaran, A.; Teglasi, A.; Bock, I.; Giudice, M.L.; Tancos, Z.; Molnar, K.; Laszlo, L.; et al. Neurons derived from sporadic Alzheimer’s disease iPSCs reveal elevated TAU hyperphosphorylation, increased amyloid levels, and GSK3B activation. Alzheimers Res. Ther. 2017, 9, 90. [Google Scholar] [CrossRef]
- Oksanen, M.; Petersen, A.J.; Naumenko, N.; Puttonen, K.; Lehtonen, S.; Gubert Olive, M.; Shakirzyanova, A.; Leskela, S.; Sarajarvi, T.; Viitanen, M.; et al. PSEN1 Mutant iPSC-Derived Model Reveals Severe Astrocyte Pathology in Alzheimer’s Disease. Stem Cell Rep. 2017, 9, 1885–1897. [Google Scholar] [CrossRef]
- Hu, N.W.; Corbett, G.T.; Moore, S.; Klyubin, I.; O’Malley, T.T.; Walsh, D.M.; Livesey, F.J.; Rowan, M.J. Extracellular Forms of Abeta and Tau from iPSC Models of Alzheimer’s Disease Disrupt Synaptic Plasticity. Cell Rep. 2018, 23, 1932–1938. [Google Scholar] [CrossRef]
- Yan, Y.; Song, L.; Bejoy, J.; Zhao, J.; Kanekiyo, T.; Bu, G.; Zhou, Y.; Li, Y. Modelling neurodegenerative microenvironment using cortical organoids derived from human stem cells. Tissue Eng. Part A 2018, 24, 1125–1137. [Google Scholar] [CrossRef]
- Gonzalez, C.; Armijo, E.; Bravo-Alegria, J.; Becerra-Calixto, A.; Mays, C.E.; Soto, C. Modeling amyloid beta and tau pathology in human cerebral organoids. Mol. Psychiatry 2018, 23, 2363–2374. [Google Scholar] [CrossRef]
- Lin, Y.T.; Seo, J.; Gao, F.; Feldman, H.M.; Wen, H.L.; Penney, J.; Cam, H.P.; Gjoneska, E.; Raja, W.K.; Cheng, J.; et al. APOE4 Causes Widespread Molecular and Cellular Alterations Associated with Alzheimer’s Disease Phenotypes in Human iPSC-Derived Brain Cell Types. Neuron 2018, 98, 1141–1154. [Google Scholar] [CrossRef]
- Chen, M.; Lee, H.K.; Moo, L.; Hanlon, E.; Stein, T.; Xia, W. Common proteomic profiles of induced pluripotent stem cell-derived three-dimensional neurons and brain tissue from Alzheimer patients. J. Proteom. 2018, 182, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Birnbaum, J.H.; Wanner, D.; Gietl, A.F.; Saake, A.; Kundig, T.M.; Hock, C.; Nitsch, R.M.; Tackenberg, C. Oxidative stress and altered mitochondrial protein expression in the absence of amyloid-beta and tau pathology in iPSC-derived neurons from sporadic Alzheimer’s disease patients. Stem Cell Res. 2018, 27, 121–130. [Google Scholar] [CrossRef]
- Young, J.E.; Fong, L.K.; Frankowski, H.; Petsko, G.A.; Small, S.A.; Goldstein, L.S.B. Stabilizing the Retromer Complex in a Human Stem Cell Model of Alzheimer’s Disease Reduces TAU Phosphorylation Independently of Amyloid Precursor Protein. Stem Cell Rep. 2018, 10, 1046–1058. [Google Scholar] [CrossRef] [PubMed]
- Ovchinnikov, D.A.; Korn, O.; Virshup, I.; Wells, C.A.; Wolvetang, E.J. The Impact of APP on Alzheimer-like Pathogenesis and Gene Expression in Down Syndrome iPSC-Derived Neurons. Stem Cell Rep. 2018, 11, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Najm, R.; Xu, Q.; Jeong, D.E.; Walker, D.; Balestra, M.E.; Yoon, S.Y.; Yuan, H.; Li, G.; Miller, Z.A.; et al. Gain of toxic apolipoprotein E4 effects in human iPSC-derived neurons is ameliorated by a small-molecule structure corrector. Nat. Med. 2018, 24, 647–657. [Google Scholar] [CrossRef] [PubMed]
- Xiao, G.; Cui, Y.; Ducy, P.; Karsenty, G.; Franceschi, R.T. Ascorbic acid-dependent activation of the osteocalcin promoter in MC3T3-E1 preosteoblasts: Requirement for collagen matrix synthesis and the presence of an intact OSE2 sequence. Mol. Endocrinol. 1997, 11, 1103–1113. [Google Scholar] [CrossRef]
- Hu, W.; Qiu, B.; Guan, W.; Wang, Q.; Wang, M.; Li, W.; Gao, L.; Shen, L.; Huang, Y.; Xie, G.; et al. Direct Conversion of Normal and Alzheimer’s Disease Human Fibroblasts into Neuronal Cells by Small Molecules. Cell Stem Cell 2015, 17, 204–212. [Google Scholar] [CrossRef] [Green Version]
- Espuny-Camacho, I.; Arranz, A.M.; Fiers, M.; Snellinx, A.; Ando, K.; Munck, S.; Bonnefont, J.; Lambot, L.; Corthout, N.; Omodho, L.; et al. Hallmarks of Alzheimer’s Disease in Stem-Cell-Derived Human Neurons Transplanted into Mouse Brain. Neuron 2017, 93, 1066–1081.e1068. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Ward, M.E.; Chen, R.; Liu, K.; Tracy, T.E.; Chen, X.; Xie, M.; Sohn, P.D.; Ludwig, C.; Meyer-Franke, A.; et al. Scalable Production of iPSC-Derived Human Neurons to Identify Tau-Lowering Compounds by High-Content Screening. Stem Cell Rep. 2017, 9, 1221–1233. [Google Scholar] [CrossRef]
- Harasta, A.E.; Ittner, L.M. Alzheimer’s Disease: Insights from Genetic Mouse Models and Current Advances in Human IPSC-Derived Neurons. Adv. Neurobiol. 2017, 15, 3–29. [Google Scholar]
- Jorfi, M.; D’Avanzo, C.; Tanzi, R.E.; Kim, D.Y.; Irimia, D. Human Neurospheroid Arrays for In Vitro Studies of Alzheimer’s Disease. Sci. Rep. 2018, 8, 2450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldstein, L.S.; Reyna, S.; Woodruff, G. Probing the secrets of Alzheimer’s disease using human-induced pluripotent stem cell technology. Neurotherapeutics 2015, 12, 121–125. [Google Scholar] [CrossRef]
- Vazin, T.; Ashton, R.S.; Conway, A.; Rode, N.A.; Lee, S.M.; Bravo, V.; Healy, K.E.; Kane, R.S.; Schaffer, D.V. The effect of multivalent Sonic hedgehog on differentiation of human embryonic stem cells into dopaminergic and GABAergic neurons. Biomaterials 2014, 35, 941–948. [Google Scholar] [CrossRef]
- Du, Z.-W.; Chen, H.; Liu, H.; Lu, J.; Qian, K.; Huang, C.-L.; Zhong, X.; Fan, F.; Zhang, S.-C. Generation and expansion of highly pure motor neuron progenitors from human pluripotent stem cells. Nat. Commun. 2015, 6, 6626. [Google Scholar] [CrossRef] [PubMed]
- Hattori, N. Cerebral organoids model human brain development and microcephaly. Mov. Disord. 2014, 29, 185. [Google Scholar] [CrossRef] [PubMed]
- Paşca, A.M.; Sloan, S.A.; Clarke, L.E.; Tian, Y.; Makinson, C.D.; Huber, N.; Kim, C.H.; Park, J.-Y.; O’rourke, N.A.; Nguyen, K.D. Functional cortical neurons and astrocytes from human pluripotent stem cells in 3D culture. Nat. Methods 2015, 12, 671–678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, H.; Guo, M.; Martins-Taylor, K.; Wang, X.; Zhang, Z.; Park, J.W.; Zhan, S.; Kronenberg, M.S.; Lichtler, A.; Liu, H.-X. Specification of region-specific neurons including forebrain glutamatergic neurons from human induced pluripotent stem cells. PLoS ONE 2010, 5, e11853. [Google Scholar] [CrossRef] [PubMed]
- Imaizumi, K.; Sone, T.; Ibata, K.; Fujimori, K.; Yuzaki, M.; Akamatsu, W. Controlling the Regional Identity of hPSC-Derived Neurons to Uncover Neuronal Subtype Specificity of Neurological Disease Phenotypes. Stem Cell Rep. 2015, 5, 1010–1022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Y.; Kirwan, P.; Livesey, F.J. Directed differentiation of human pluripotent stem cells to cerebral cortex neurons and neural networks. Nat. Protoc. 2012, 7, 1836. [Google Scholar] [CrossRef]
- Suzuki, I.K.; Vanderhaeghen, P. Is this a brain which I see before me? Modeling human neural development with pluripotent stem cells. Development 2015, 142, 3138–3150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pham, M.T.; Pollock, K.M.; Rose, M.D.; Cary, W.A.; Stewart, H.R.; Zhou, P.; Nolta, J.A.; Waldau, B. Generation of human vascularized brain organoids. Neuroreport 2018, 29, 588–593. [Google Scholar] [CrossRef] [PubMed]
- Mansour, A.A.; Goncalves, J.T.; Bloyd, C.W.; Li, H.; Fernandes, S.; Quang, D.; Johnston, S.; Parylak, S.L.; Jin, X.; Gage, F.H. An in vivo model of functional and vascularized human brain organoids. Nat. Biotechnol. 2018, 36, 432–441. [Google Scholar] [CrossRef] [PubMed]
- Wimmer, R.A.; Leopoldi, A.; Aichinger, M.; Wick, N.; Hantusch, B.; Novatchkova, M.; Taubenschmid, J.; Hammerle, M.; Esk, C.; Bagley, J.A.; et al. Human blood vessel organoids as a model of diabetic vasculopathy. Nature 2019, 565, 505–510. [Google Scholar] [CrossRef] [PubMed]
- Lou, Y.R.; Leung, A.W. Next generation organoids for biomedical research and applications. Biotechnol. Adv. 2018, 36, 132–149. [Google Scholar] [CrossRef]
- Fatehullah, A.; Tan, S.H.; Barker, N. Organoids as an in vitro model of human development and disease. Nat. Cell Biol. 2016, 18, 246. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Xu, C.; Ma, T. In vitro organogenesis from pluripotent stem cells. Organogenesis 2014, 10, 159–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, X.; Mead, B.E.; Safaee, H.; Langer, R.; Karp, J.M.; Levy, O. Engineering stem cell organoids. Cell Stem Cell 2016, 18, 25–38. [Google Scholar] [CrossRef] [PubMed]
- Lancaster, M.A.; Knoblich, J.A. Organogenesis in a dish: Modeling development and disease using organoid technologies. Science 2014, 345, 10. [Google Scholar] [CrossRef]
- Passier, R.; Orlova, V.; Mummery, C. Complex tissue and disease modeling using hiPSCs. Cell Stem Cell 2016, 18, 309–321. [Google Scholar] [CrossRef]
- Zhang, S.-C.; Wernig, M.; Duncan, I.D.; Brüstle, O.; Thomson, J.A. In vitro differentiation of transplantable neural precursors from human embryonic stem cells. Nat. Biotechnol. 2001, 19, 1129. [Google Scholar] [CrossRef]
- Eiraku, M.; Watanabe, K.; Matsuo-Takasaki, M.; Kawada, M.; Yonemura, S.; Matsumura, M.; Wataya, T.; Nishiyama, A.; Muguruma, K.; Sasail, Y. Self-Organized Formation of Polarized Cortical Tissues from ESCs and Its Active Manipulation by Extrinsic Signals. Cell Stem Cell 2008, 3, 519–532. [Google Scholar] [CrossRef] [Green Version]
- Eiraku, M.; Takata, N.; Ishibashi, H.; Kawada, M.; Sakakura, E.; Okuda, S.; Sekiguchi, K.; Adachi, T.; Sasai, Y. Self-organizing optic-cup morphogenesis in three-dimensional culture. Nature 2011, 472, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Mariani, J.; Simonini, M.V.; Palejev, D.; Tomasini, L.; Coppola, G.; Szekely, A.M.; Horvath, T.L.; Vaccarino, F.M. Modeling human cortical development in vitro using induced pluripotent stem cells. Proc. Natl. Acad. Sci. USA 2012, 109, 12770–12775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lancaster, M.A.; Renner, M.; Martin, C.A.; Wenzel, D.; Bicknell, L.S.; Hurles, M.E.; Homfray, T.; Penninger, J.M.; Jackson, A.P.; Knoblich, J.A. Cerebral organoids model human brain development and microcephaly. Nature 2013, 501, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Qian, X.; Nguyen, H.N.; Song, M.M.; Hadiono, C.; Ogden, S.C.; Hammack, C.; Yao, B.; Hamersky, G.R.; Jacob, F.; Zhong, C.; et al. Brain-region-specific organoids using mini-bioreactors for modeling ZIKV exposure. Cell 2016, 165, 1238–1254. [Google Scholar] [CrossRef] [PubMed]
- Mariani, J.; Coppola, G.; Zhang, P.; Abyzov, A.; Provini, L.; Tomasini, L.; Amenduni, M.; Szekely, A.; Palejev, D.; Wilson, M.; et al. FOXG1-dependent dysregulation of GABA/Glutamate neuron differentiation in Autism Spectrum Disorders. Cell 2015, 162, 375–390. [Google Scholar] [CrossRef] [PubMed]
- Birey, F.; Andersen, J.; Makinson, C.D.; Islam, S.; Wei, W.; Huber, N.; Fan, H.C.; Metzler, K.R.C.; Panagiotakos, G.; Thom, N.; et al. Assembly of functionally integrated human forebrain spheroids. Nature 2017, 545, 54–59. [Google Scholar] [CrossRef] [Green Version]
- Bian, S.; Repic, M.; Guo, Z.; Kavirayani, A.; Burkard, T.; Bagley, J.A.; Krauditsch, C.; Knoblich, J.A. Genetically engineered cerebral organoids model brain tumor formation. Nat. Methods 2018, 15, 631–639. [Google Scholar] [CrossRef]
- Ogawa, J.; Pao, G.M.; Shokhirev, M.N.; Verma, I.M. Glioblastoma Model Using Human Cerebral Organoids. Cell Rep. 2018, 23, 1220–1229. [Google Scholar] [CrossRef]
- Amin, N.D.; Pasca, S.P. Building Models of Brain Disorders with Three-Dimensional Organoids. Neuron 2018, 100, 389–405. [Google Scholar] [CrossRef]
- Yan, Y.; Song, L.; Madinya, J.; Ma, T.; Li, Y. Derivation of cortical spheroids from human induced pluripotent stem cells in a suspension bioreactor. Tissue Eng. Part A 2018, 24, 418–431. [Google Scholar] [CrossRef]
- Edri, R.; Gal, I.; Noor, N.; Harel, T.; Fleischer, S.; Adadi, N.; Green, O.; Shabat, D.; Heller, L.; Shapira, A.; et al. Personalized Hydrogels for Engineering Diverse Fully Autologous Tissue Implants. Adv. Mater. 2019, 31, e1803895. [Google Scholar] [CrossRef] [PubMed]
- Sart, S.; Yan, Y.; Li, Y.; Lochner, E.; Zeng, C.; Ma, T.; Li, Y. Crosslinking of extracellular matrix scaffolds derived from pluripotent stem cell aggregates modulates neural differentiation. Acta Biomater. 2016, 30, 222–232. [Google Scholar] [CrossRef] [Green Version]
- Shen, Q.; Wang, Y.; Kokovay, E.; Lin, G.; Chuang, S.-M.; Goderie, S.K.; Roysam, B.; Temple, S. Adult SVZ stem cells lie in a vascular niche: A quantitative analysis of niche cell-cell interactions. Cell Stem Cell 2008, 3, 289–300. [Google Scholar] [CrossRef]
- Yan, Y.; Martin, L.M.; Bosco, D.B.; Bundy, J.L.; Nowakowski, R.S.; Sang, Q.-X.A.; Li, Y. Differential effects of acellular embryonic matrices on pluripotent stem cell expansion and neural differentiation. Biomaterials 2015, 73, 231–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bejoy, J.; Wang, Z.; Bijonowski, B.; Yang, M.; Ma, T.; Sang, Q.X.; Li, Y. Differential effects of heparin and hyaluronic acid on neural patterning of human induced pluripotent stem cells. ACS Biomater. Sci. Eng. 2018, 4, 4354–4366. [Google Scholar] [CrossRef]
- Yan, Y.; Li, Y.; Song, L.; Zeng, C.; Li, Y. Pluripotent stem cell expansion and neural differentiation in 3-D scaffolds of tunable Poisson’s ratio. Acta Biomater. 2017, 49, 192–203. [Google Scholar] [CrossRef] [PubMed]
- Discher, D.E.; Mooney, D.J.; Zandstra, P.W. Growth factors, matrices, and forces combine and control stem cells. Science 2009, 324, 1673–1677. [Google Scholar] [CrossRef] [PubMed]
- Engler, A.J.; Sen, S.; Sweeney, L.; Discher, D.E. Matrix elasticity directs stem cell lineage specification. Cell 2006, 126, 677–689. [Google Scholar] [CrossRef]
- Saha, K.; Keung, A.J.; Irwin, E.F.; Li, Y.; Little, L.; Schaffer, D.V.; Healy, K.E. Substrate modulus directs neural stem cell behavior. Biophys. J. 2008, 95, 4426–4438. [Google Scholar] [CrossRef]
- Leipzig, N.D.; Shoichet, M.S. The effect of substrate stiffness on adult neural stem cell behavior. Biomaterials 2009, 30, 6867–6878. [Google Scholar] [CrossRef] [PubMed]
- Kothapalli, C.R.; Kamm, R.D. 3D matrix microenvironment for targeted differentiation of embryonic stem cells into neural and glial lineages. Biomaterials 2013, 34, 5995–6007. [Google Scholar] [CrossRef] [PubMed]
- Lantoine, J.; Grevesse, T.; Villers, A.; Delhaye, G.; Mestdagh, C.; Versaevel, M.; Mohammed, D.; Bruyere, C.; Alaimo, L.; Lacour, S.P.; et al. Matrix stiffness modulates formation and activity of neuronal networks of controlled architectures. Biomaterials 2016, 89, 14–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madl, C.M.; LeSavage, B.L.; Dewi, R.E.; Dinh, C.B.; Stowers, R.S.; Khariton, M.; Lampe, K.J.; Nguyen, D.; Chaudhuri, O.; Enejder, A.; et al. Maintenance of neural progenitor cell stemness in 3D hydrogels requires matrix remodelling. Nat. Mater. 2017, 16, 1233–1242. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Li, X.; Janairo, R.R.R.; Kwong, G.; Tsou, A.D.; Chu, J.S.; Wang, A.; Yu, J.; Wang, D.; Li, S. Matrix stiffness modulates the differentiation of neural crest stem cells in vivo. J. Cell. Physiol. 2019, 234, 7569–7578. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.Q.; Gao, X.; Wang, A.; Yang, Y.; Liu, S.; Yu, Z.; Song, G.B.; Zhao, H.C. Substrate stiffness affects neural network activity in an extracellular matrix proteins dependent manner. Colloids Surf. B Biointerfaces 2018, 170, 729–735. [Google Scholar] [CrossRef]
- Srinivasan, A.; Chang, S.Y.; Zhang, S.; Toh, W.S.; Toh, Y.C. Substrate stiffness modulates the multipotency of human neural crest derived ectomesenchymal stem cells via CD44 mediated PDGFR signaling. Biomaterials 2018, 167, 153–167. [Google Scholar] [CrossRef]
- Keung, A.J.; Asuri, P.; Kumar, S.; Schaffer, D.V. Soft microenvironments promote the early neurogenic differentiation but not self-renewal of human pluripotent stem cells. Integr. Biol. 2012, 4, 1049–1058. [Google Scholar] [CrossRef] [Green Version]
- Discher, D.E.; Janmey, P.; Wang, Y.-l. Tissue cells feel and respond to the stiffness of their substrate. Science 2005, 310, 1139–1143. [Google Scholar] [CrossRef]
- Pagliara, S.; Franze, K.; McClain, C.R.; Wylde, G.; Fisher, C.L.; Franklin, R.J.M.; Kabla, A.J.; Keyser, U.F.; Chalut, K.J. Auxetic nuclei in embryonic stem cells exiting pluripotency. Nat. Mater. 2014, 13, 638–644. [Google Scholar] [CrossRef]
- Evans, N.D.; Minelli, C.; Gentleman, E.; LaPointe, V.; Patankar, S.N.; Kallivretaki, M.; Chen, X.; Roberts, C.J.; Stevens, M.M. Substrate stiffness affects early differentiation events in embryonic stem cells. Eur. Cells Mater. 2009, 18, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, A.; Arha, M.; Choudhary, S.; Ashton, R.S.; Bhatia, S.R.; Schaffer, D.V.; Kane, R.S. The influence of hydrogel modulus on the proliferation and differentiation of encapsulated neural stem cells. Biomaterials 2009, 30, 4695–4699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chowdhury, F.; Na, S.; Li, D.; Poh, Y.-C.; Tanaka, T.S.; Wang, F.; Wang, N. Material properties of the cell dictate stress-induced spreading and differentiation in embryonic stem cells. Nat. Mater. 2010, 9, 82. [Google Scholar] [CrossRef] [PubMed]
- Musah, S.; Morin, S.A.; Wrighton, P.J.; Zwick, D.B.; Jin, S.; Kiessling, L.L. Glycosaminoglycan-binding hydrogels enable mechanical control of human pluripotent stem cell self-renewal. ACS Nano 2012, 6, 10168–10177. [Google Scholar] [CrossRef] [PubMed]
- Zoldan, J.; Karagiannis, E.D.; Lee, C.Y.; Anderson, D.G.; Langer, R.; Levenberg, S. The influence of scaffold elasticity on germ layer specification of human embryonic stem cells. Biomaterials 2011, 32, 9612–9621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arshi, A.; Nakashima, Y.; Nakano, H.; Eaimkhong, S.; Evseenko, D.; Reed, J.; Stieg, A.Z.; Gimzewski, J.K.; Nakano, A. Rigid Microenvironments Promote Cardiac Differentiation Of Mouse And Human Embryonic Stem Cells. Circ. Res. 2013, 113, 1. [Google Scholar] [CrossRef]
- Eroshenko, N.; Ramachandran, R.; Yadavalli, V.K.; Rao, R.R. Effect of substrate stiffness on early human embryonic stem cell differentiation. J. Biol. Eng. 2013, 7, 7. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, K.; Lim, V.Y.; Shen, J.Y.; Tan, W.; Rajendran, D.; Luo, S.C.; Gao, S.J.; Wan, A.C.A.; Ying, J.Y. Extracellular Matrix-Mediated Differentiation of Human Embryonic Stem Cells: Differentiation to Insulin-Secreting Beta Cells. Tissue Eng. Part A 2014, 20, 424–433. [Google Scholar] [CrossRef]
- Hazeltine, L.B.; Badur, M.G.; Lian, X.; Das, A.; Han, W.; Palecek, S.P. Temporal impact of substrate mechanics on differentiation of human embryonic stem cells to cardiomyocytes. Acta Biomater. 2014, 10, 604–612. [Google Scholar] [CrossRef]
- Maldonado, M.; Wong, L.Y.; Echeverria, C.; Ico, G.; Low, K.; Fujimoto, T.; Johnson, J.K.; Nam, J. The effects of electrospun substrate-mediated cell colony morphology on the self-renewal of human induced pluripotent stem cells. Biomaterials 2015, 50, 10–19. [Google Scholar] [CrossRef]
- Ribeiro, A.J.; Ang, Y.S.; Fu, J.D.; Rivas, R.N.; Mohamed, T.M.; Higgs, G.C.; Srivastava, D.; Pruitt, B.L. Contractility of single cardiomyocytes differentiated from pluripotent stem cells depends on physiological shape and substrate stiffness. Proc. Natl. Acad. Sci. USA 2015, 112, 12705–12710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mittal, N.; Tasnim, F.; Yue, C.; Qu, Y.; Phan, D.; Choudhury, Y.; Tan, M.-H.; Yu, H. Substrate Stiffness Modulates the Maturation of Human Pluripotent Stem-Cell-Derived Hepatocytes. ACS Biomater. Sci. Eng. 2016, 2, 1649–1657. [Google Scholar] [CrossRef]
- Caiazzo, M.; Okawa, Y.; Ranga, A.; Piersigilli, A.; Tabata, Y.; Lutolf, M.P. Defined three-dimensional microenvironments boost induction of pluripotency. Nat. Mater. 2016, 15, 344–352. [Google Scholar] [CrossRef]
- Przybyla, L.; Lakins, J.N.; Weaver, V.M. Tissue Mechanics Orchestrate Wnt-Dependent Human Embryonic Stem Cell Differentiation. Cell Stem Cell 2016, 19, 462–475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richardson, T.; Barner, S.; Candiello, J.; Kumta, P.N.; Banerjee, I. Capsule stiffness regulates the efficiency of pancreatic differentiation of human embryonic stem cells. Acta Biomater. 2016, 35, 153–165. [Google Scholar] [CrossRef] [Green Version]
- Gjorevski, N.; Sachs, N.; Manfrin, A.; Giger, S.; Bragina, M.E.; Ordonez-Moran, P.; Clevers, H.; Lutolf, M.P. Designer matrices for intestinal stem cell and organoid culture. Nature 2016, 539, 560–564. [Google Scholar] [CrossRef]
- Wang, B.; Tu, X.; Wei, J.; Wang, L.; Chen, Y. Substrate elasticity dependent colony formation and cardiac differentiation of human induced pluripotent stem cells. Biofabrication 2018, 11, 015005. [Google Scholar] [CrossRef]
- Dorsey, T.B.; Kim, D.; Grath, A.; James, D.; Dai, G. Multivalent biomaterial platform to control the distinct arterial venous differentiation of pluripotent stem cells. Biomaterials 2018, 185, 1–12. [Google Scholar] [CrossRef]
- Fu, J.; Chuah, Y.J.; Liu, J.; Tan, S.Y.; Wang, D.-A. Respective Effects of Gelatin-Coated Polydimethylsiloxane (PDMS) Substrates on Self-renewal and Cardiac Differentiation of Induced Pluripotent Stem Cells (iPSCs). ACS Biomater. Sci. Eng. 2018, 4, 4321–4330. [Google Scholar] [CrossRef]
- Hirata, M.; Yamaoka, T. Effect of stem cell niche elasticity/ECM protein on the self-beating cardiomyocyte differentiation of induced pluripotent stem (iPS) cells at different stages. Acta Biomater. 2018, 65, 44–52. [Google Scholar] [CrossRef]
- Goetzke, R.; Franzen, J.; Ostrowska, A.; Vogt, M.; Blaeser, A.; Klein, G.; Rath, B.; Fischer, H.; Zenke, M.; Wagner, W. Does soft really matter? Differentiation of induced pluripotent stem cells into mesenchymal stromal cells is not influenced by soft hydrogels. Biomaterials 2018, 156, 147–158. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.G.; Gil, C.H.; Seo, J.; Park, S.J.; Subbiah, R.; Jung, T.H.; Kim, J.S.; Jeong, Y.H.; Chung, H.M.; Lee, J.H.; et al. Mechanotransduction of human pluripotent stem cells cultivated on tunable cell-derived extracellular matrix. Biomaterials 2018, 150, 100–111. [Google Scholar] [CrossRef] [PubMed]
- Bandtlow, C.E.; Zimmermann, D.R. Proteoglycans in the developing brain: New conceptual insights for old proteins. Physiol. Rev. 2000, 80, 1267–1290. [Google Scholar] [CrossRef] [PubMed]
- Galtrey, C.M.; Fawcett, J.W. The role of chondroitin sulfate proteoglycans in regeneration and plasticity in the central nervous system. Brain Res. Rev. 2007, 54, 1–18. [Google Scholar] [CrossRef]
- Schwartz, N.B.; Domowicz, M. Proteoglycans in brain development. Glycoconj. J. 2004, 21, 329–341. [Google Scholar] [CrossRef] [PubMed]
- Sarrazin, S.; Lamanna, W.C.; Esko, J.D. Heparan sulfate proteoglycans. Cold Spring Harb. Perspect. Biol. 2011, 3, a004952. [Google Scholar] [CrossRef] [PubMed]
- Oohira, A.; Shuo, T.; Tokita, Y.; Nakanishi, K.; Aono, S. Neuroglycan C, a brain-specific part-time proteoglycan, with a particular multidomain structure. Glycoconj. J. 2004, 21, 53–57. [Google Scholar] [CrossRef]
- Senkov, O.; Andjus, P.; Radenovic, L.; Soriano, E.; Dityatev, A. Neural ECM molecules in synaptic plasticity, learning, and memory. Prog. Brain Res. 2014, 214, 53–80. [Google Scholar]
- Howell, M.D.; Bailey, L.A.; Cozart, M.A.; Gannon, B.M.; Gottschall, P.E. Hippocampal administration of chondroitinase ABC increases plaque-adjacent synaptic marker and diminishes amyloid burden in aged APPswe/PS1dE9 mice. Acta Neuropathol. Commun. 2015, 3, 54. [Google Scholar] [CrossRef]
- Perry, G.; Siedlak, S.L.; Richey, P.; Kawai, M.; Cras, P.; Kalaria, R.N.; Galloway, P.G.; Scardina, J.M.; Cordell, B.; Greenberg, B.D.; et al. Association of heparan sulfate proteoglycan with the neurofibrillary tangles of Alzheimer’s disease. J. Neurosci. 1991, 11, 3679–3683. [Google Scholar] [CrossRef]
- Castillo, G.M.; Ngo, C.; Cummings, J.; Wight, T.N.; Snow, A.D. Perlecan binds to the beta-amyloid proteins (A beta) of Alzheimer’s disease, accelerates A beta fibril formation, and maintains A beta fibril stability. J. Neurochem. 1997, 69, 2452–2465. [Google Scholar] [CrossRef] [PubMed]
- Cotman, S.L.; Halfter, W.; Cole, G.J. Agrin binds to beta-amyloid (Abeta), accelerates abeta fibril formation, and is localized to Abeta deposits in Alzheimer’s disease brain. Mol. Cell Neurosci. 2000, 15, 183–198. [Google Scholar] [CrossRef] [PubMed]
- Sandwall, E.; O’Callaghan, P.; Zhang, X.; Lindahl, U.; Lannfelt, L.; Li, J.P. Heparan sulfate mediates amyloid-beta internalization and cytotoxicity. Glycobiology 2010, 20, 533–541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.P.; Galvis, M.L.; Gong, F.; Zhang, X.; Zcharia, E.; Metzger, S.; Vlodavsky, I.; Kisilevsky, R.; Lindahl, U. In vivo fragmentation of heparan sulfate by heparanase overexpression renders mice resistant to amyloid protein A amyloidosis. Proc. Natl. Acad. Sci. USA 2005, 102, 6473–6477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leveugle, B.; Scanameo, A.; Ding, W.; Fillit, H. Binding of heparan sulfate glycosaminoglycan to beta-amyloid peptide: inhibition by potentially therapeutic polysulfated compounds. Neuroreport 1994, 5, 1389–1392. [Google Scholar] [PubMed]
- Giulian, D.; Haverkamp, L.J.; Yu, J.; Karshin, W.; Tom, D.; Li, J.; Kazanskaia, A.; Kirkpatrick, J.; Roher, A.E. The HHQK domain of beta-amyloid provides a structural basis for the immunopathology of Alzheimer’s disease. J. Biol. Chem. 1998, 273, 29719–29726. [Google Scholar] [CrossRef]
- Lindahl, B.; Westling, C.; Gimenez-Gallego, G.; Lindahl, U.; Salmivirta, M. Common binding sites for beta-amyloid fibrils and fibroblast growth factor-2 in heparan sulfate from human cerebral cortex. J. Biol. Chem. 1999, 274, 30631–30635. [Google Scholar] [CrossRef]
- Yang, L.; Liu, C.C.; Zheng, H.; Kanekiyo, T.; Atagi, Y.; Jia, L.; Wang, D.; N’Songo, A.; Can, D.; Xu, H.; et al. LRP1 modulates the microglial immune response via regulation of JNK and NF-kappaB signaling pathways. J. Neuroinflamm. 2016, 13, 304. [Google Scholar] [CrossRef]
- Yamazaki, Y.; Kanekiyo, T. Blood-Brain Barrier Dysfunction and the Pathogenesis of Alzheimer’s Disease. Int. J. Mol. Sci. 2017, 18, E1965. [Google Scholar] [CrossRef]
- Holmes, B.B.; DeVos, S.L.; Kfoury, N.; Li, M.; Jacks, R.; Yanamandra, K.; Ouidja, M.O.; Brodsky, F.M.; Marasa, J.; Bagchi, D.P.; et al. Heparan sulfate proteoglycans mediate internalization and propagation of specific proteopathic seeds. Proc. Natl. Acad. Sci. USA 2013, 110, E3138–E3147. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Frederickson, R.C.; Brunden, K.R. Neuroglial-mediated immunoinflammatory responses in Alzheimer’s disease: Complement activation and therapeutic approaches. Neurobiol. Aging 1996, 17, 781–787. [Google Scholar] [CrossRef]
- Bergamaschini, L.; Rossi, E.; Vergani, C.; De Simoni, M.G. Alzheimer’s disease: Another target for heparin therapy. Sci. World J. 2009, 9, 891–908. [Google Scholar] [CrossRef] [PubMed]
- Bergamaschini, L.; Rossi, E.; Storini, C.; Pizzimenti, S.; Distaso, M.; Perego, C.; De Luigi, A.; Vergani, C.; De Simoni, M.G. Peripheral treatment with enoxaparin, a low molecular weight heparin, reduces plaques and beta-amyloid accumulation in a mouse model of Alzheimer’s disease. J. Neurosci. 2004, 24, 4181–4186. [Google Scholar] [CrossRef] [PubMed]
- Scholefield, Z.; Yates, E.A.; Wayne, G.; Amour, A.; McDowell, W.; Turnbull, J.E. Heparan sulfate regulates amyloid precursor protein processing by BACE1, the Alzheimer’s beta-secretase. J. Cell Biol. 2003, 163, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Bejoy, J.; Song, L.; Wang, Z.; Sang, Q.X.; Zhou, Y.; Li, Y. Neuroprotective Activities of Heparin, Heparinase III, and Hyaluronic Acid on the Aβ42-treated Forebrain Spheroids Derived from Human Stem Cells. ACS Biomater. Sci. Eng. 2018, 4, 2922–2933. [Google Scholar] [CrossRef]
- Sorrells, S.F.; Paredes, M.F.; Cebrian-Silla, A.; Sandoval, K.; Qi, D.; Kelley, K.W.; James, D.; Mayer, S.; Chang, J.; Auguste, K.I.; et al. Human hippocampal neurogenesis drops sharply in children to undetectable levels in adults. Nature 2018, 555, 377–381. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
A possible pathology of Alzheimer’s disease (AD). It is postulated that AD may be caused by the deposition of Aβ and tau hyperphosphorylation-derived neurofibrillary tangles (NFTs), both of which could activate the caspase-associated apoptosis. In AD brain the monomeric Aβ peptides aggregate to form toxic Aβ oligomers and further generate the insoluble fibrils, which ultimately form the plaques. The toxic Aβ species could trigger an inflammatory response and increase the level of ROS, which may cause neuron death. On the other hand, toxic Aβ species may be transferred into cells and trigger the apoptosis of neurons. NFTs could either cause neuronal cell death or trigger apoptosis of neurons. Aβ may interplay with tau-derived NFTs formation, while critical questions about Aβ-induced tau pathology in AD are still unanswered. ECM: extracellular matrix; APP: amyloid precursor protein; ROS: reactive oxygen species; SAT3: signal transducer and activator of transcription 3; JNK: c-Jun N-terminal kinase; GSK-3β: glycogen synthase kinase-3β.
Figure 1.
A possible pathology of Alzheimer’s disease (AD). It is postulated that AD may be caused by the deposition of Aβ and tau hyperphosphorylation-derived neurofibrillary tangles (NFTs), both of which could activate the caspase-associated apoptosis. In AD brain the monomeric Aβ peptides aggregate to form toxic Aβ oligomers and further generate the insoluble fibrils, which ultimately form the plaques. The toxic Aβ species could trigger an inflammatory response and increase the level of ROS, which may cause neuron death. On the other hand, toxic Aβ species may be transferred into cells and trigger the apoptosis of neurons. NFTs could either cause neuronal cell death or trigger apoptosis of neurons. Aβ may interplay with tau-derived NFTs formation, while critical questions about Aβ-induced tau pathology in AD are still unanswered. ECM: extracellular matrix; APP: amyloid precursor protein; ROS: reactive oxygen species; SAT3: signal transducer and activator of transcription 3; JNK: c-Jun N-terminal kinase; GSK-3β: glycogen synthase kinase-3β.

Figure 2.
Schematic illustration of the process flow of using hiPSCs for AD studies. The neuronal cells derived from iPSCs cells of AD patients can be used for AD pathological analysis and various therapeutic interventions. AD: Alzheimer’s disease.
Figure 2.
Schematic illustration of the process flow of using hiPSCs for AD studies. The neuronal cells derived from iPSCs cells of AD patients can be used for AD pathological analysis and various therapeutic interventions. AD: Alzheimer’s disease.

Figure 3.
Regional specification in neural differentiation of pluripotent stem cells (PSCs) mimicking in vivo regional patterning, and the use of the derived forebrain organoids for AD modeling. The regional patterning of brain organoids was achieved by cell signaling modulators and the corresponding organoids generated were used for disease modeling. DA: dopaminergic; FGF: fibroblast growth factor; RA: retinoic acid; SHH: Sonic Hedgehog; BDNF: Brain-derived neurotropic growth factor; SDF: Stromal cell-derived factor. Adapted and revised from [81].
Figure 3.
Regional specification in neural differentiation of pluripotent stem cells (PSCs) mimicking in vivo regional patterning, and the use of the derived forebrain organoids for AD modeling. The regional patterning of brain organoids was achieved by cell signaling modulators and the corresponding organoids generated were used for disease modeling. DA: dopaminergic; FGF: fibroblast growth factor; RA: retinoic acid; SHH: Sonic Hedgehog; BDNF: Brain-derived neurotropic growth factor; SDF: Stromal cell-derived factor. Adapted and revised from [81].

Figure 4.
Organoid formation from human pluripotent stem cells. (A) Organoids can be generated from human pluripotent stem cells through the embryonic body (EB)-based procedure with the help of exogenous factors, such as extracellular matrix (ECM). Organoids form 3D ordered structures to mimic the in vivo situation of tissues or cells. (B) Organoids form based on the self-organization and self-assembly of stem cells.
Figure 4.
Organoid formation from human pluripotent stem cells. (A) Organoids can be generated from human pluripotent stem cells through the embryonic body (EB)-based procedure with the help of exogenous factors, such as extracellular matrix (ECM). Organoids form 3D ordered structures to mimic the in vivo situation of tissues or cells. (B) Organoids form based on the self-organization and self-assembly of stem cells.

Figure 5.
A hypothetical model of heparan sulfate proteoglycan (HSPG) (e.g., agrin/perlecan) involvement in Aβ pathogenesis. ECM: extracellular matrix.
Figure 5.
A hypothetical model of heparan sulfate proteoglycan (HSPG) (e.g., agrin/perlecan) involvement in Aβ pathogenesis. ECM: extracellular matrix.

Figure 6.
Possible protective actions of heparin in Alzheimer’s disease (AD) pathology. Heparin-based protection can be realized by: (1) reduction of Aβ generation by an action on amyloid precursor protein (APP); (2) prevention of Aβ aggregation/deposition in senile plaques; (3) reduction of the inflammatory response (complement, kinin system); (4) binding to homotetrameric Cu2+ and Zn2+ containing glycoprotein, and regulating the activity of these metals which reduce accumulation of Aβ peptides. (Red line indicates the actions of heparin on Aβ).
Figure 6.
Possible protective actions of heparin in Alzheimer’s disease (AD) pathology. Heparin-based protection can be realized by: (1) reduction of Aβ generation by an action on amyloid precursor protein (APP); (2) prevention of Aβ aggregation/deposition in senile plaques; (3) reduction of the inflammatory response (complement, kinin system); (4) binding to homotetrameric Cu2+ and Zn2+ containing glycoprotein, and regulating the activity of these metals which reduce accumulation of Aβ peptides. (Red line indicates the actions of heparin on Aβ).


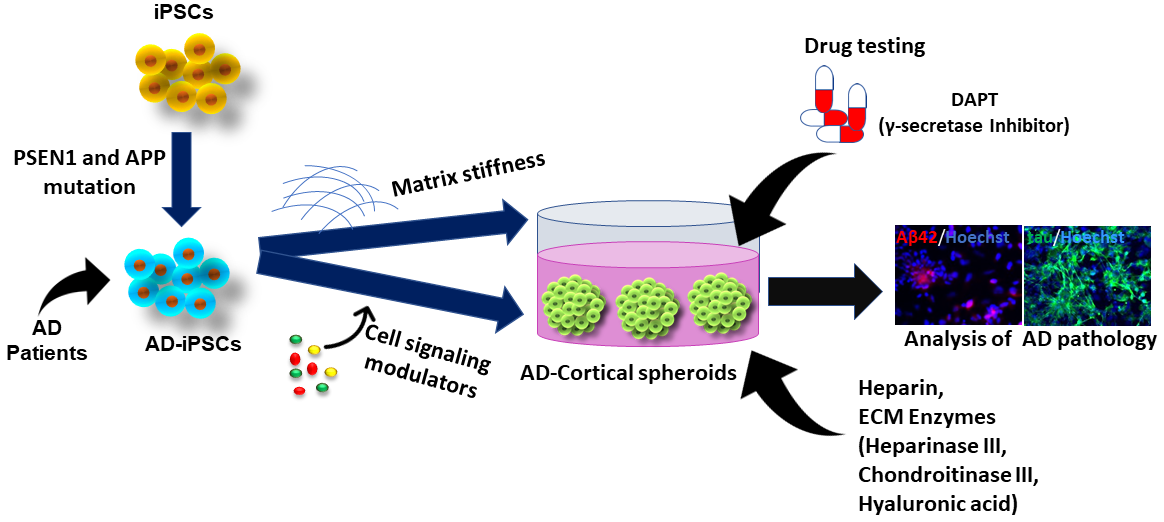{kind=link}
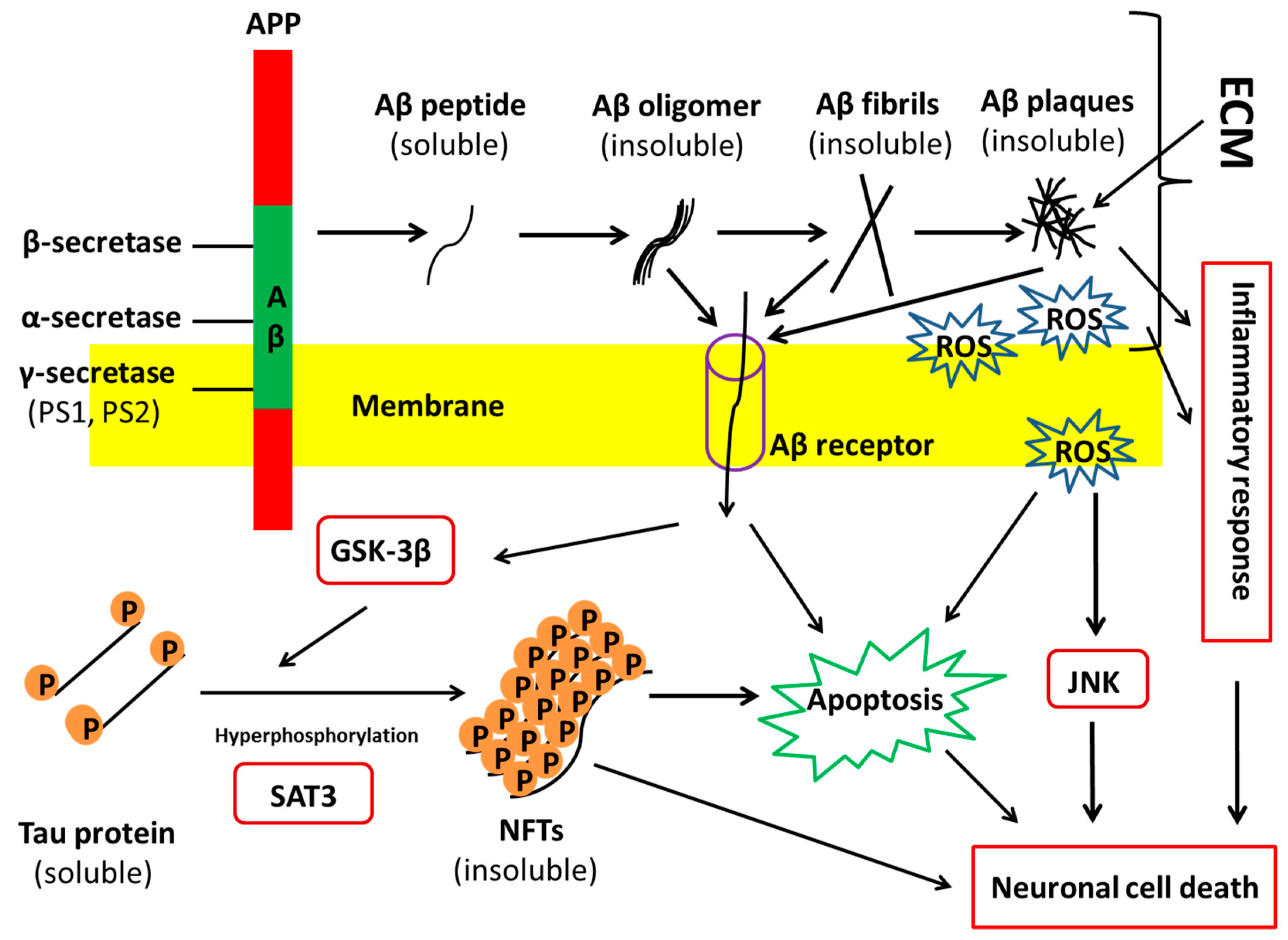{kind=link}
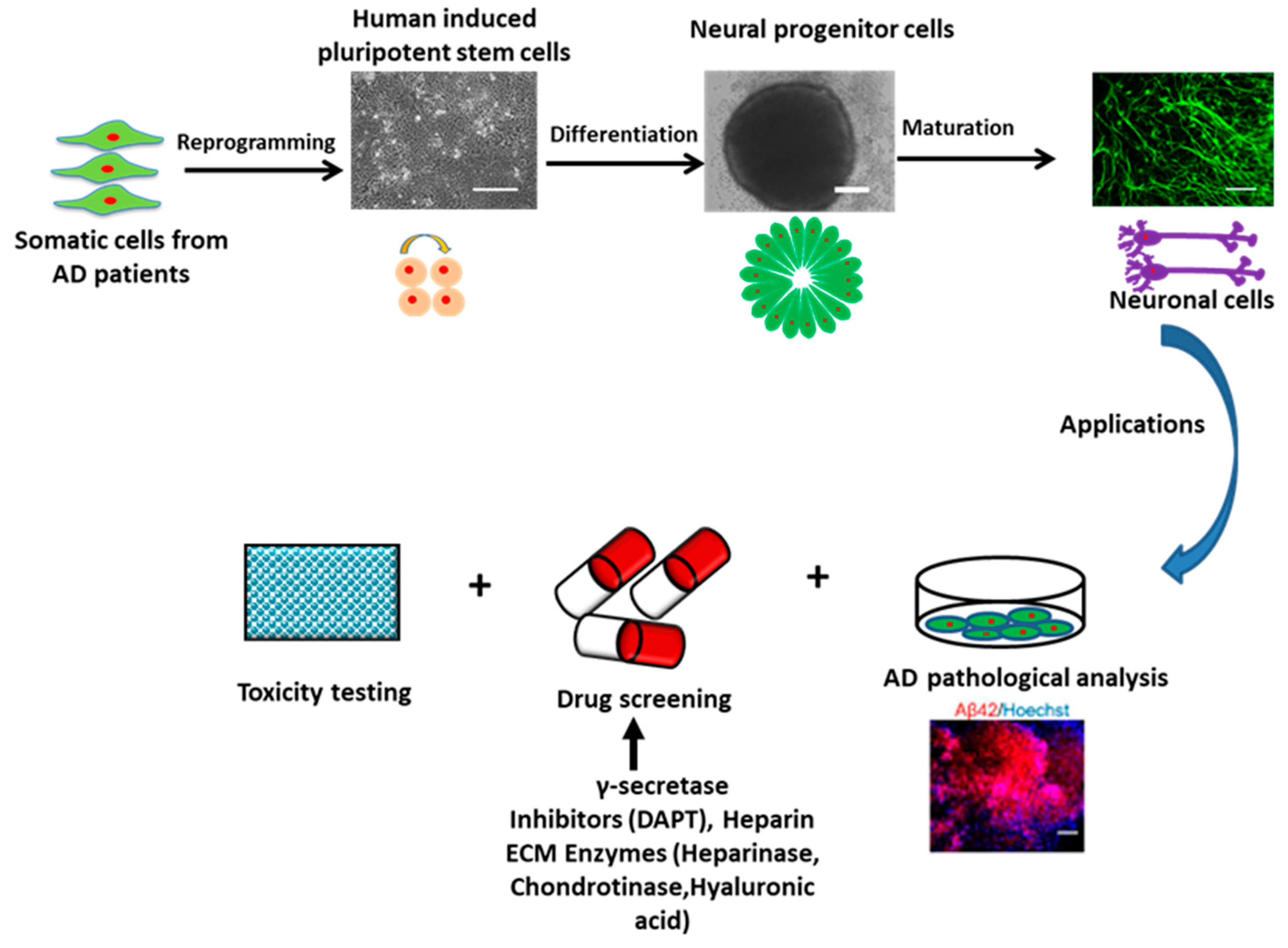{kind=link}
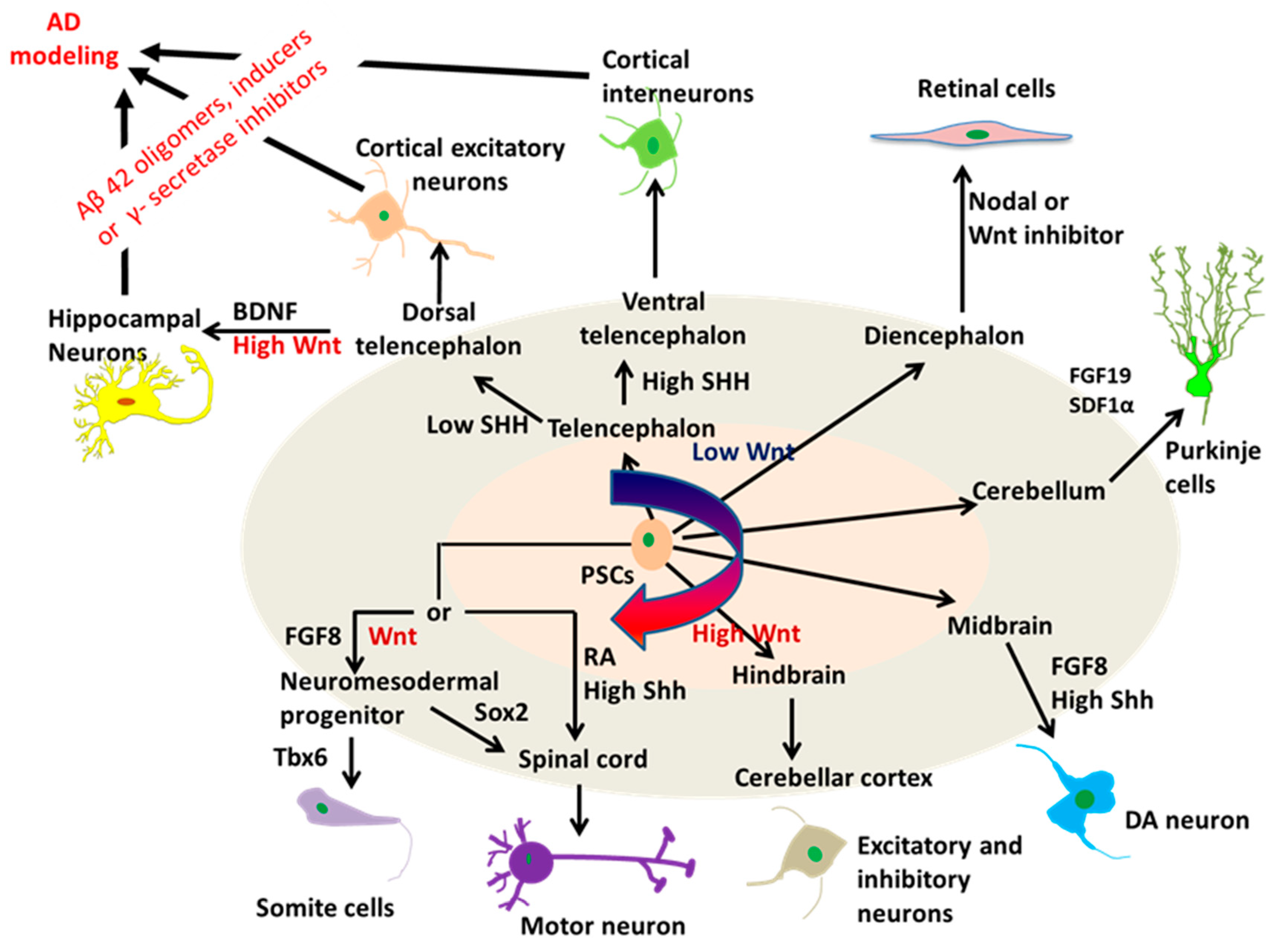{kind=link}
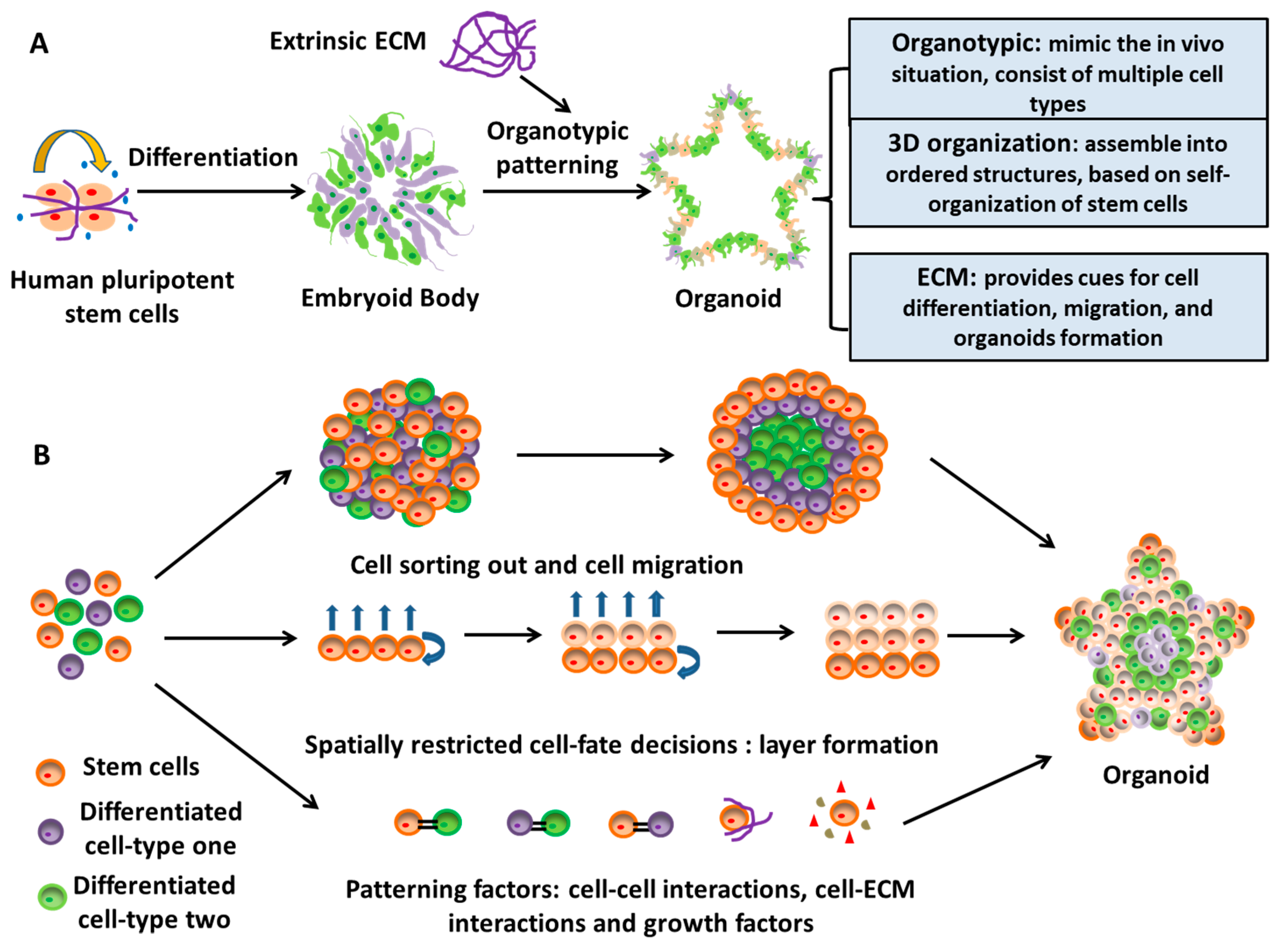{kind=link}
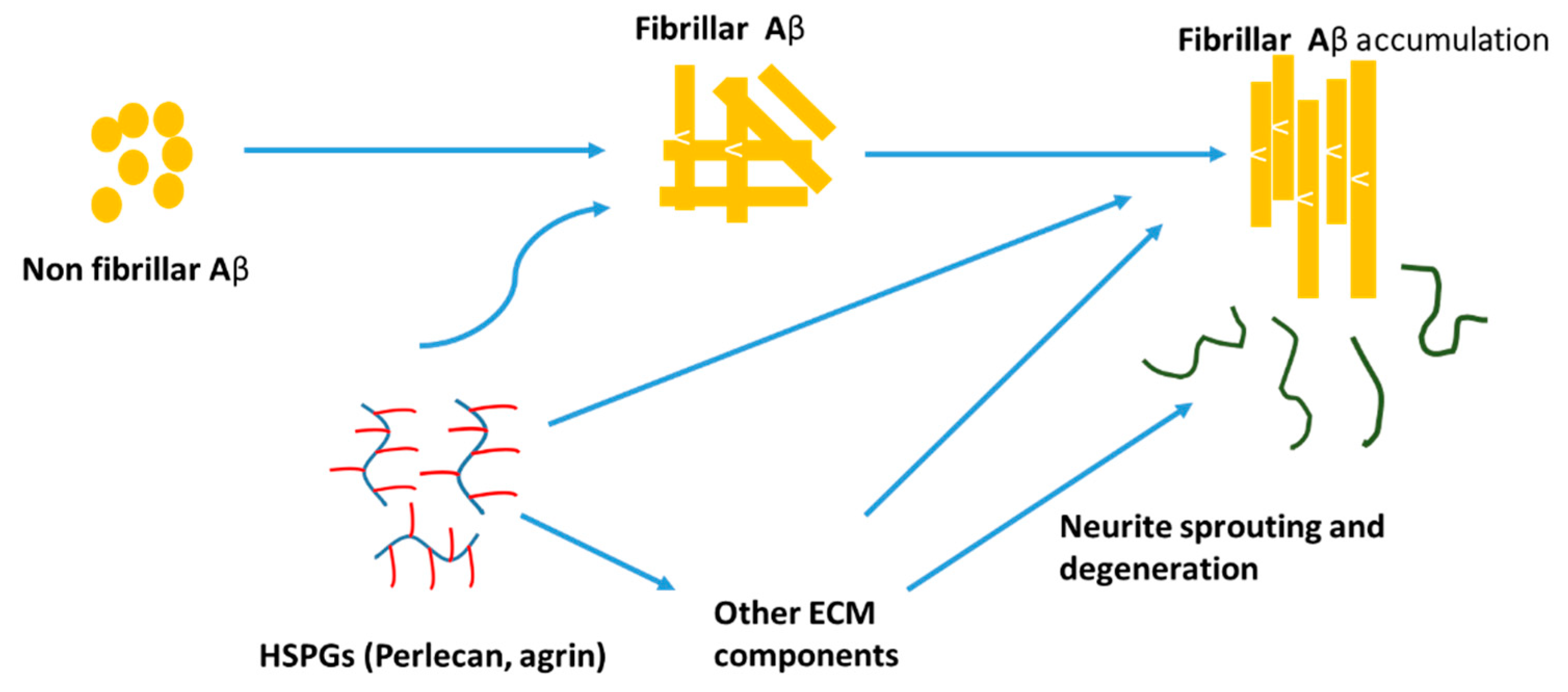{kind=link}
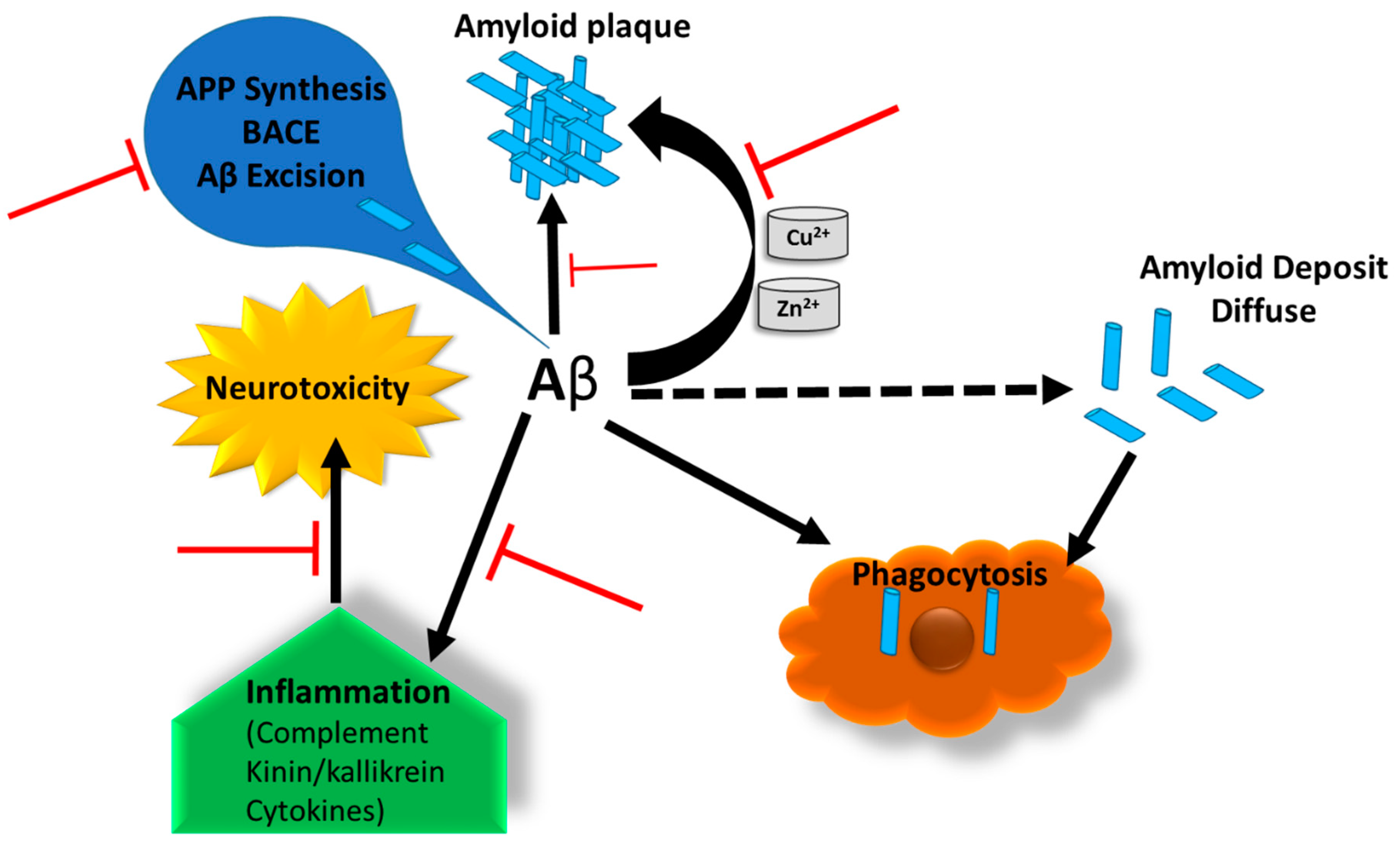{kind=link}
Table 1.
Selected studies of hPSC-based models for Alzheimer’s disease.
| Cell Line | Neural Types | AD Phenotypes | Ref. |
|---|---|---|---|
| Chemicals Induced AD-Phenotypes Using hPSCs | |||
| Human ESC-derived neurons treated with Aβ42 oligomers | 3D neurospheres and 2D, basal forebrain cholinergic neurons expressing ChAT and β-tubulin III | Aβ oligomers suppressed the number of functional neurons | Wicklund et al., 2010 [31] |
| HiPSC-derived neurons treated with β-secretase (BSI) and γ-secretase inhibitor (GSI) and NSAID | 2D, forebrain neurons expressing FOXG1 and TBR1 (62%), CTIP2 (12%), Cux1 (83%) SATB (46%) at day 52 | Differentiated neuronal cells expressed Aβ40 and Aβ42. BSI, GSI, and NSAID partially or fully blocked Aβ production in the hiPSCs-derived neuronal cells | Yahata et al., 2011 [32] |
| Human ESC and hiPSC-derived neurons treated with Aβ42 oligomers | 2D, cortical glutamatergic neurons | Aβ oligomers yielded cell culture age-dependent binding of Aβ and cell death in the glutamatergic populations | Vazin et al., 2014 [25] |
| HiPSC-derived neurons treated with Aβ1-42 oligomers | 3D neurospheres, cortical glutamatergic neurons, and motor neurons | Aβ oligomers caused less cell viability, more caspase expression and higher ROS levels on cortical excitatory neurons population. GSK-3β inhibitor may attenuate Aβ-induced cytotoxicity | Yan et al., 2016 [26] |
| HiPSC-derived neurons induced by Aβ42 inducer (Aftin5) | 3D cortical organoids, neurons expressing NeuN, NCAM, MAP2, and CTIP2 | Increased secretion of Aβ42 and the Aβ42/40 ratio | Pavoni et al., 2018 [24] |
| Overexpression of AD-related gene in hPSCs using genetic modification | |||
| PSEN1 L166P mutant hPSC-derived neurons treated with γ-secretase inhibitor (DAPT) and NSAID | 2D, hPSC-derived neural stem cells (NSCs) expressing Nestin and β-tubulin III | DAPT reduced secretion of both Aβ42 and Aβ40. NSAID reduced secretion of Aβ42. PSEN1 1L166P mutation resulted in elevated Aβ42/40 ratio. | Koch et al., 2012 [27] |
| PSEN1 (∆E9) mutant hiPSCs | 2D, hPSC-derived neural progenitor cells (NPCs) expressing Nestin and tau | The PS1 ∆E9 mutation increases the Aβ42/Aβ40 ratio in human neurons by decreasing Aβ40 | Woodruff et al., 2013 [28] |
| Human ESC-derived neurons model tau pathology | 2D, neurons expressing Nestin, DACH1, SOX2, β-tubulin III, and tau | P-tau impaired the transport of mitochondria and led to axonal degeneration and cell death | Mertens et al., 2013 [33] |
| HPSC-derived neurons co-cultured with ApoE secreted glia | 2D, human neurons generated by forced expression of neurogenin-2 (Ngn2), expressing MAP2 and NeuN | ApoE secreted by glia stimulates neuronal Aβ40 and Aβ42 production with an ApoE4 > ApoE3 > ApoE2 potency rank order | Huang et al., 2017 [29] |
| Human NPCs and hiPSC-derived cells overexpressed APP (K670N/M671L and V717I) mutations | 3D microfluidic platform, tri-culture of neurons, astrocytes, and microglia | Increased Aβ aggregation and p-tau formation, induced microglia recruitment and axonal cleavage. Increased chemokines and cytokines. | Park et al., 2018 [30] |
| AD patient-derived iPSCs | |||
| FAD-hiPSCs with PSEN1/2 mutations | 2D, neurons expressing β-tubulin III (about 80%) and MAP2 | Change in APP processing; increased Aβ42 secretion; responding to γ-secretase inhibitors and modulators. | Yagi et al., 2011 [34] |
| FAD-hiPSCs from a patient with Down’s syndrome (Trisomy 21 defect) | 2D, cortical glutamatergic neurons expressing TBR1, CTIP2, SATB and β-tubulin III | Increased Aβ peptide production, Intracellular and extracellular Aβ42 aggregates. Decreased Aβ40/Aβ42 with γ-secretase inhibitors. Tau hyper-phosphorylation in cell bodies and dendrites. Neuronal cell death. | Shi et al., 2012 [35] |
| FAD-hiPSCs with APP gene duplications and SAD-hiPSCs | 2D, FACS-purified neurons expressing β-tubulin III (>90%) and MAP2 | Neurons from AD patients had higher levels of Aβ40, p-tau, and active glycogen synthase kinase-3β (aGSK-3β). β-secretase inhibitors, not γ-secretase inhibitors, reduced p-tau and aGSK-3b. | Israel et al., 2012 [36] |
| FAD-hiPSCs with APP mutations and SAD-hiPSCs | 2D, cortical neurons expressing β-tubulin III, MAP2, TBR1 and SATB2, and astrocytes expressing GFAP | Intracellular Aβ oligomer formation; reduced extracellular Aβ peptides. | Kondo et al., 2013 [37] |
| FAD-hiPSCs with APP or PSEN1 mutations | 2D, neural stem cells (NSCs) expressing Nestin SOX2, ZO1, β-tubulin III, and MAP2 | Increased the Aβ42/Aβ40 ratio compared to healthy control. With high concentrations of γ-secretase inhibitors (NSAID-based GSMs drugs), Aβ42/Aβ40 ratio was decreased. | Mertens et al., 2013 [38] |
| FAD-hiPSCs with PSEN1 mutations | 2D, NPCs expressing β-tubulin III | Increased the Aβ42/Aβ40 ratio. | Sproul et al., 2014 [39] |
| FAD-hiPSCs with PSEN1 (A246E) mutations | 3D EB-based, neurons expressing Nestin, PAX6, FOXG1, TBR1, STAB2, β-tubulin III, and MAP2 | Increased the Aβ42/Aβ40 ratio, increased expression of FOXG1, mGluR1, and SYT1. | Mahairaki et al., 2014 [40] |
| FAD-hiPSCs with PSEN1 and AG mutations and SAD-hiPSCs with APOE3/E4 mutations | Basal forebrain cholinergic neurons expressing MAP2, ChAT, and VaChT | Elevated Aβ42. With γ-secretase inhibitors, Aβ40 was increased and calcium transient was increased. | Duan et al., 2014 [41] |
| FAD-hiPSCs with APP mutations | 3D EB-based, forebrain neurons expressing MAP2, tau, β-tubulin III, Cux1, TBR1, vGlut1 | Increased Aβ42: Aβ40; Decreased APPsα: APPsβ, γ-secretase inhibitor blocked APPs, β cleavage. Increased total tau and p-tau (Ser262) d100. Aβ antibodies blocked, increased total tau. | Muratore et al., 2014 [42] |
| FAD-hiPSCs with PSEN1 (A246E, H163R or M146L) mutations | 2D, neurons expressing Nestin, PAX6 and SOX1 | Increased the Aβ42/Aβ40 ratio compared to healthy control. Reduced Aβ42 and Aβ38 by γ-secretase inhibitor-GSM4. | Liu et al., 2014 [43] |
| FAD-hiPSCs with PSEN1 mutations | 3D EB based, neurons expressing β-tubulin III | Increased the Aβ42 secretion level. Elevated acid sphingomyelinase (ASM) levels. ASM levels restored by ASM siRNA treatment. | Lee et al., 2014 [44] |
| SAD-hiPSCs with SOR1 variants | 2D, FACS-purified neurons expressing Nestin and MAP2 | Altered induction of SORL1 expression; altered Aβ peptide production. | Young et al., 2015 [45] |
| FAD-hiPSCs with PSEN1 or APP mutations | 2D, cortical excitatory neurons expressing tau | Increased the Aβ42 secretion level. Increased intracellular tau and phosphorylated tau levels. | Moore et al., 2015 [46] |
| SAD-hiPSCs with APP mutations | 2D, neurons expressing Nestin, PAX6 and β-tubulin III | Increased phosphor-tau (p-tau) and active glycogen synthase kinase-3β (aGSK-3β).Reduced p-tau by γ-secretase inhibitor. | Hossini et al., 2015 [47] |
| FAD-hiPSCs with PSEN1 (A246E) mutations and SAD-hiPSC mutations | 2D, neurons expressing Nestin, SOX2, MAP2, and β-tubulin III | Increased Aβ42 for FAD-hiPSCs-derived neurons. | Armijo et al., 2016 [48] |
| FAD-hiPSCs with PSEN1 (P117R)/APOE3/3 mutations and SAD-hiPSCs with APOE3/E4 mutations | 3D neurospheres, neural cells expressing GFAP, and MAP2 | Reduced neurites length and neuronal viability. Elevated levels of nitrite and apoptosis. Hyper-excitable Ca+ signaling phenotype. Protected neurites and cell viability by treatment of apigenin. | Balez et al., 2016 [49] |
| FAD-hiPSCs with APP (V717I) mutations | 3D EB based, forebrain neurons expressing GABA, PVB, and MAP2 | Elevated levels of Aβ and sAPPα. | Liao et al., 2016 [50] |
| SAD-hiPSCs | 3D neuro-spheroid, cortical neurons expressing PAX6, MAP2, NeuN and β-tubulin III | 3D spheroids recapitulated both amyloid β and tau pathology. Reduced Aβ42 and Aβ40 production both in 2D and 3D neurons with BACE1 or γ-secretase inhibitors. | Lee et al., 2016 [51] |
| FAD-human iPSCs with APP or PS1 mutations | 3D brain organoids, neuronal cells expressing SOX2, and MAP2 | 3D organoids recapitulated amyloid β, tau pathology, and endosome abnormalities. Reduced amyloid and tau pathology with β-and γ-secretase inhibitors. | Raja et al., 2016 [52] |
| FAD-hiPSCs with PSEN1 (M146L) mutations and SAD-hiPSCs with APOE4 mutations | 2D differentiation; cortical neurons and astrocytes | Reduced morphological heterogeneity in astrocytes. | Jones et al., 2017 [53] |
| FAD-hiPSCs with APP (V717I) mutations | 3D EB-based differentiation, caudal neurons expressing HOXB4 and rostral neurons expressing TBR1 | Reduced the Aβ42/Aβ40 ratio but increased the Aβ38/Aβ42 ratio for caudal neurons. Higher levels of total and phosphor-tau for rostral neuronal fate. | Muratore et al., 2017 [54] |
| FAD-hiPSCs with PSEN1 (M146L, G384A, H163R or A246E), APP (V717I) mutations and SAD-hiPSCs with APOE4 mutations | 2D, human cortical neurons (iN cells) generated by force expression of neurogenin-2 (Ngn2), iN cells expressing SATB2, MAP2, vGlut1, and TBR2 | iPSC-based screening of pharmaceutical compounds for Aβ phenotypes; anti-Aβ cocktail decreased toxic Aβ levels in neurons derived from patients’ cells. A combination of existing drugs synergistically improved Aβ phenotypes of AD. | Kondo et al., 2017 [55] |
| FAD-hiPSCs with PSEN1 mutations and SAD-hiPSCs with unknown mutations | 2D, cholinergic neurons (VAChT), dopaminergic neurons (TH), GABAergic neurons (GAD2/GAD1), and glutamatergic neurons (vGlut1/2) | Increased levels of extracellular Aβ40 and Aβ42 for FAD and SAD samples. Increased the Aβ42/Aβ40 ratio for FAD sample. Increased levels of p-tau and GSK3β. | Ochalek et al., 2017 [56] |
| FAD-hiPSCs with PSEN1 (∆E9) mutations | 3D EB-based differentiation, astrocytes expressing GFAP and S100β | AD astrocytes increased Aβ42 production, altered cytokine release, dysregulated Ca2+ homeostasis, increased oxidative stress and reduced lactate secretion. | Oksanen et al., 2017 [57] |
| FAD-hiPSC with PSEN1 and APP duplication or hiPSCs from Down’s syndrome (Trisomy 21) | 2D, cortical neurons expressing TBR1, and MAP2 | Synaptic dysfunction (long-term potentiation) caused by PSEN1 and APP duplication secretomes was mediated by Aβ peptides, whereas trisomy 21 neuronal secretomes induced dysfunction through extracellular tau. | Hu et al., 2018 [58] |
| FAD-hiPSCs with PSEN1 (M146V) mutation | 3D cortical organoids, neurons expressing TBR1, SATB2, BRN2, and MAP2 | 3D organoids recapitulated Aβ, tau pathology, and neuronal cell death. Reduced amyloid β with DAPT, heparin and heparinase. | Yan et al., 2018 [59] |
| FAD-hiPSC with PSEN1 (A246E) or hiPSCs from Down’s syndrome (Trisomy 21) | 3D cortical organoids, neurons expressing NeuN, SATB2, TBR1, and MAP2 | Accumulation of Aβ and tau aggregates and induced cellular apoptosis AD organoids. | Gonzalez et al., 2018 [60] |
| SAD-hiPSCs from APOE4/E3 mutations | 3D organoids, neurons, astrocytes, and microglia-like cells | APOE4 organoids displayed increased Aβ aggregation and hyperphosphorylation of tau. | Lin et al., 2018 [61] |
| SAD-hiPSCs from unknown mutations | 3D neuro-spheroid, neurons | AD organoids neuronal dysfunction was similar to AD brain tissue by mass spectrometry-based proteomics analysis. | Chen et al., 2018 [62] |
| SAD-hiPSCs from APOE4/E3 mutations | 2D, neuronal cells expressing MAP2 | Showed aberrant mitochondrial function. Increased levels of ROS and DNA damage. Increased levels of oxidative phosphorylation chain complexes. | Birnbaum et al., 2018 [63] |
| FAD-hiPSCs and SAD-hiPSCs | 2D, FACS-purified neurons | Reduced tau phosphorylation by retromer stabilization. | Young et al., 2018 [64] |
| HiPSCs from a Down’s syndrome patient by controlling APP gene copy number | 2D, cortical neurons | Higher APP gene dosage increased Aβ production, altered the Aβ42/Aβ40 ratio and caused deposition of the pyroglutamate (E3)-containing amyloid aggregates. | Ovchinnikov et al., 2018 [65] |
| SAD-hiPSCs from APOE4/4 or APOE3/3 mutations | 2D, cortical neurons and GABAergic neurons | APOE4 increased Aβ production in human neurons, APOE4-expressing neurons had higher levels of tau phosphorylation. | Wang et al., 2018 [66] |
| FAD-hiPSCs with APP duplication mutants | 2D, FACS-purified neurons | Neuronal cholesteryl esters (CE) regulated the proteasome-dependent degradation of p-tau, CE-mediated Aβ secretion by a cholesterol-binding down in APP, A CYP46A1-CE-tau axis was identified as an early pathway. | van der Kant et al., 2019 [67] |
Note: PSCs, pluripotent stem cells; ESCs, embryonic stem cells; iPSCs, induced pluripotent stem cells; AD, Alzheimer’s disease; Aβ, β-amyloid peptide; 2D, two-dimensional; 3D, three-dimensional; BSI/BACE1, β-secretase inhibitor; GSI, γ-secretase inhibitor; NSAID, nonsteroidal anti-inflammatory drug; ROS, reactive oxygen species; GSK-3β, glycogen synthase kinase 3 beta; NSCs, neural stem cells, NPCs, neural progenitor cells; PSEN1/2, presenlin1/2; APP, amyloid precursor protein; FAD, familial AD; SAD, sporadic AD; EB, embryoid body; FACS, fluorescence-activated cell sorting; ChAT: Choline Acetyltransferase. Other Useful studies: Hu et al., 2015 Cell Stem Cell [68], Human chemical-induced neuronal cells (hciNs) from FAD patient fibroblasts with APP (V717I) or PSEN1 (I167del or A434T or S169del) mutations, increased extracellular Aβ42 level and the Aβ42/Aβ40 ratio. Espuny-Camacho et al., 2017 Neuron [69], Chimeric model of AD generated using hPSCs-derived neurons (hPSC-neurons grafted into AD mice), major degeneration and loss of human neurons in chimeric AD mice, absence of tangle pathology in degenerating human neurons in vivo. Wang et al., 2017 Stem Cell Reports [70], Neurogenin 2 (NGN2)-induced glumatergic neurons (iN cells) from hiPSCs, iN cells are used to identify tau-lowering compounds in LOPAC (Library of Pharmacologically Active Compounds), and identified adrenergic receptors agonists as a class of compounds that reduce endogenous human tau.
Table 2.
Effects of matrix modulus on pluripotent stem cell fate decisions.
| Cell Source | Range of Modulus and Substrates | Effect on Morphology, Proliferation, and Differentiation | Reference |
|---|---|---|---|
| Neural progenitor cells | 0.1 kPa–10 kPa; PA gels based vmIPNs | Soft gel (100–500 Pa) favored neurons, harder gel (1–10 kPa) promoted glial cells. | Saha et al., 2009 [111] |
| Neural progenitor cells | 1–20 kPa; MAC substrates | <1 kPa favored neuronal differentiation; <3.5 kPa supported astrocyte, >7kPa favored oligodendrocyte. | Leipzig et al., 2009 [112] |
| Mouse ESCs | 41–2700 kPa; collagen coated PDMS surface | Increasing substrate stiffness from 41–2700 kPa promoted cell spreading, proliferation, mesendodermal and osteogenic differentiation. | Evans et al., 2009 [122] |
| Rat neural stem cells | 180–20,000 Pa; 3D alginate hydrogel scaffolds | The rate of proliferation of neural stem cells decreased with an increase in the modulus of the hydrogels. Lower stiffness enhanced neural differentiation. | Banerjee et al., 2009 [123] |
| Mouse ESCs | 0.6 kPa; PA gel substrates | Soft substrate supported self-renewal | Chowdhury et al., 2010 [124] |
| Human ESCs and iPSCs | 0.7–10 kPa; GAG-binding hydrogel | The stiff (10 kPa) hydrogel maintained cell proliferation and pluripotency. | Musah et al., 2012 [125] |
| Human ESCs | 0.05–7 MPa, 3D PLLA, PLGA, PCL or PEGDA scaffold coated with matrigel | 50 to 100 kPa supported ectoderm differentiation; 100 to 1000 kPa supported endoderm differentiation; 1.5 to 6 MPa supported mesoderm differentiation. | Zoldan et al., 2011 [126] |
| Human ESCs and iPSCs | 0.1–75 kPa; matrigel-coated PA gels | Soft matrix (0.1 kPa) promoted early neural differentiation. | Keung et al., 2012 [119] |
| Human ESCs | 1 kPa, 10 kPa, 3 GPa; PDMS substrates | Rigid matrix promoted cardiac differentiation. | Arshi et al., 2013 [127] |
| Mouse ESCs | 0–1.5 kPa, 3D collagen-I, Matrigel, or HA hydrogel | <0.3 kPa less neurite outgrowth and supported glial cell; 0.5 to 1 kPa more neurite outgrowth and supported neurons. | Kothapalli et al., 2013 [113] |
| Human ESCs | 0.078–1.167 MPa; PDMS substrates | Increased stiffness upregulated mesodermal differentiation. | Eroshenko et al., 2013 [128] |
| Human ESCs | 1.3 kPa, 2.1 kPa, 3.5 kPa; HA hydrogel | Stiffness of 1.2 kPa was the best to support pancreatic β-cell differentiation. | Narayanan et al., 2014 [129] |
| Human ESCs | 4–80 kPa; PA hydrogels | Stiffness of 50 kPa was the best for cardiomyocyte differentiation. Stiffness impacted the initial differentiation of hESCs to mesendoderm, while it did not impact differentiation of cardiac progenitor cells to cardiomyocytes. | Hazeltine et al., 2014 [130] |
| Human iPSCs | 19–193 kPa; 3D PCL, PET, PEKK or PCU electrospun fibers | The substrate stiffness was inversely related to the sphericity of hiPSC colonies. | Maldonado et al., 2015 [131] |
| HPSCs | 6 kPa, 10 kPa, 35 kPa; Matrigel micropatterns | High stiffness (35 kPa) induced myofibril defects of hPSC-derived cardiomyocytes and decreased mechanical output. | Ribeiro et al., 2015 [132] |
| hPSC-derived hepatocytes (hPSC-Heps) | 20, 45, 140 kPa; collagen-coated PA hydrogels substrates | On softer substrates, the hPSC-Heps formed compact colonies while on stiffer substrates they formed a diffuse monolayer. Albumin production correlated inversely with stiffness. | Mittal et al., 2016 [133] |
| Rat cortical neurons (RCN) | 5 kPa (soft), PA gels; 500 kPa (stiff), PDMS substrates; | Soft substrates enhanced cortical neurons migration. Stiff substrates increased synaptic activity. | Lantoine et al., 2016 [114] |
| Mouse ESCs and iPSCs | 300–1200 Pa; 3D PEG hydrogels | Stiffness and other biophysical effectors promoted somatic-cell reprogramming and iPSC generation; lower modulus (300–600 Pa) showed higher reprogramming efficiency. | Caiazzo et al., 2016 [134] |
| Human ESCs | 400 Pa, 60 kPa; PA hydrogels | On stiff substrates, β-catenin degradation inhibits mesodermal differentiation of human ESCs. | Przybyla et al., 2016 [135] |
| Human ESCs | 1–100 kPa; barium alginate capsules | Stiffness of 4–7 kPa supported cell proliferation and higher stiffness suppressed cell growth. Increased stiffness promoted endoderm differentiation, while suppressed pancreatic induction. About 3.9 kPa was the best for pancreatic differentiation. | Richardson et al., 2016 [136] |
| Mouse intestinal stem cells (ISC) | 300 Pa, 700 Pa, 1.3 kPa, 1.7 kPa; PEG hydrogels | Higher stiffness enhanced ISC expansion. Lower stiffness supported ISC differentiation and organoid formation. | Gjorevski et al., 2016 [137] |
| Mouse neural progenitor cells (NPC) | 0.5–50 kPa; 3D elastin-like protein hydrogels | In stiffness from 0.5 to 50 kPa, NPC stemness maintenance did not correlate with initial hydrogel stiffness. | Madl et al., 2017 [115] |
| Mouse ESCs and hiPSCs | 10–100 kPa; 3D PU scaffolds | Scaffolds with proper stiffness, Poisson’s ratio and pore structure enhanced neural differentiation of PSCs. | Yan et al., 2017 [108] |
| Human iPSCs | 3–168 kPa; PDMS substrates | Elasticity of substrates significantly affected cell colony formation. Intermediate substrate elasticity of about 9 kPa is preferable to reach an EB-like aggregation and optimal for cardiac differentiation. | Wang et al., 2018 [138] |
| Mouse ESCs | 3.4 kPa, 64 kPa, 144 kPa; PEGDA or PEG hydrogel substrates | Soft hydrogel (3.4 kPa) showed strong cell attachment and a growth pattern similar to 2D surface. Stiff hydrogel (144 kPa) supported a 3D aggregation. | Dorsey et al., 2018 [139] |
| Mouse iPSCs | 0–2.4 MPa; PDMS substrates | Stiffer substrate supported pluripotency of iPSCs. Softer substrate promoted cardiac differentiation. | Fu et al., 2018 [140] |
| Neural crest stem cells (NCSCs) from hiPSCs | 1kPa, 15 kPa, 1 GPa; PA gel substrates | >50 kPa promoted smooth muscle cells from NCSCs, <15 kPa promoted glial cells from NCSCs. | Zhu et al., 2018 [116] |
| Mouse hippocampal neurons | 2.13 kPa, 22.1 kPa; PDMS substrates | Stiff substrate enhanced voltage-gated Ca2+ channel currents in neurons. | Wen et al., 2018 [117] |
| Neural crest stem cells (NCSCs) derived from hESCs | 3.3 kPa, 1.7 MPa, 1 GPa; PDMS substrates | Soft substrate increased differentiation of ectodermal mesenchymal stem cells (MSCs) from NCSCs via CD44 mediated PDGFR signaling. | Srinivasan et al., 2018 [118] |
| iPSCs and neonatal rat cardiomyocytes | 9, 20, 180 kPa; PA gel substrates | Cardiac differentiation preferred rigid substrates, and beating behavior preferred soft substrate. | Hirata et al., 2018 [141] |
| Human iPSCs | About 24 Pa, fibrin-based gel substrates (human platelet lysate gel); >1 GPa, tissue culture plastics | Soft substrates did not impact on differentiation of iPSCs into MSCs. | Goetzke et al., 2018 [142] |
| Human ESCs | 118 ± 51 Pa, 800 ± 180 Pa, 5600 ± 1100 Pa, and 8900 ± 1500 Pa; decellularized fibroblast-derived matrices crosslinked by genipin | Soft matrix supported cell migration and induced EMT of hPSC. Stiff matrix supported cell pluripotency and suppressed EMT of hPSCs. | Kim et al., 2018 [143] |
Note: PA, polyacrylamide; vmIPNs, variable moduli interpenetrating polymer networks; MAC, methacrylamide chitosan; PDMS, polydimethylsiloxane; HA, hyaluronic acid; PLLA, poly-L-lactide acid; PLGA: poly(lactic co-glycolic acid); PCL, polycaprolactone; PEGDA, polyethylene glycol diacrylate; PET, polyrethylene terephthalate; PEKK, poly(etherketoneketone); PCU, polycarbonate-urethane; GAG, glycosaminoglycan; PEG, polyethylene glycol; PU, polyurethane; PDGFR, platelet-derived growth factor receptor beta (PDGFRβ) signaling; EMT, epithelial-mesenchymal-transition.
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Yan, Y.; Bejoy, J.; Marzano, M.; Li, Y. The Use of Pluripotent Stem Cell-Derived Organoids to Study Extracellular Matrix Development during Neural Degeneration. Cells 2019, 8, 242. https://doi.org/10.3390/cells8030242
AMA Style
Yan Y, Bejoy J, Marzano M, Li Y. The Use of Pluripotent Stem Cell-Derived Organoids to Study Extracellular Matrix Development during Neural Degeneration. Cells. 2019; 8(3):242. https://doi.org/10.3390/cells8030242
Chicago/Turabian StyleYan, Yuanwei, Julie Bejoy, Mark Marzano, and Yan Li. 2019. "The Use of Pluripotent Stem Cell-Derived Organoids to Study Extracellular Matrix Development during Neural Degeneration" Cells 8, no. 3: 242. https://doi.org/10.3390/cells8030242
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.