The Role of Primary Cilia in the Crosstalk between the Ubiquitin–Proteasome System and Autophagy


Institute for Animal Developmental and Molecular Biology, Heinrich Heine University, 40225 Düsseldorf, Germany
*
Author to whom correspondence should be addressed.
Cells 2019, 8(3), 241; https://doi.org/10.3390/cells8030241
Submission received: 29 December 2018
/
Revised: 6 March 2019
/
Accepted: 11 March 2019
/
Published: 14 March 2019
(This article belongs to the Special Issue Proteostasis and Autophagy)
{kind=link}
{kind=link}
{kind=link}
Abstract
:Protein degradation is a pivotal process for eukaryotic development and homeostasis. The majority of proteins are degraded by the ubiquitin–proteasome system and by autophagy. Recent studies describe a crosstalk between these two main eukaryotic degradation systems which allows for establishing a kind of safety mechanism. If one of these degradation systems is hampered, the other compensates for this defect. The mechanism behind this crosstalk is poorly understood. Novel studies suggest that primary cilia, little cellular protrusions, are involved in the regulation of the crosstalk between the two degradation systems. In this review article, we summarise the current knowledge about the association between cilia, the ubiquitin–proteasome system and autophagy.
Keywords:
protein aggregation; neurodegenerative diseases; OFD1; BBS4; RPGRIP1L; hedgehog; mTOR; IFT; GLI1. Introduction
Protein aggregates are huge protein accumulations that develop as a consequence of misfolded proteins. The occurrence of protein aggregates is associated with the development of neurodegenerative diseases, such as Huntington’s disease, prion disorders, Alzheimer’s disease and Parkinson’s disease [1,2,3], demonstrating that the degradation of incorrectly folded proteins is of eminent importance for human health. In addition to the destruction of useless and dangerous proteins (protein quality control), protein degradation is an important process to regulate the cell cycle, to govern transcription and also to control intra- and intercellular signal transduction [4,5,6]. Two main protein degradation systems exist in vertebrates—the ubiquitin–proteasome system (UPS) and macroautophagy (hereafter referred to as autophagy) [7]. Their function is not only essential for vertebrate homeostasis but also for vertebrate development [4,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29]. Importantly, the UPS and autophagy are, at least partially, redundant. If one degradation system is downregulated, the other gets upregulated to prevent cell-damaging protein overload or the formation of protein aggregates, as well as to ensure the maintenance of pivotal intra- and intercellular signalling [4,7,30]. However, the proteasome-to-autophagy direction of regulation is far better documented than the autophagy-to-proteasome direction [31]. Evidence for the autophagy-to-proteasome direction is mainly provided by investigations in cancer cells and in cultured neonatal rat ventricular myocytes [32,33], while numerous studies reported findings that support the existence of the proteasome-to-autophagy direction [34,35,36,37,38,39,40,41,42,43,44,45,46,47]. In any case, a kind of crosstalk takes place between the UPS and autophagy. This article focuses on the role of primary cilia in this crosstalk. In the following sections, we will shortly introduce the UPS, autophagy and primary cilia. Afterwards, we will discuss a potential role for the UPS and autophagy in cilia-associated diseases and mechanisms underlying the UPS–autophagy crosstalk with particular regard to primary cilia.
2. The Ubiquitin–Proteasome System and Autophagy
The vast majority of the proteins (~80–90%) within the vertebrate cell are degraded by the UPS [7]. Apart from the degradation of proteins, the UPS is able to implement the proteolytic processing of particular proteins [48,49]. During this process, one or more peptide bonds of the target protein are hydrolysed. Both protein degradation and protein processing, carried out by the UPS, start with the ubiquitination of target proteins. Ubiquitin conjugation is performed by a cooperative action of ubiquitin-activating enzymes (E1), ubiquitin-conjugating enzymes (E2) and ubiquitin ligases (E3). In simplified terms, ubiquitin is activated by E1 enzymes when ATP is present and, thereafter, is transferred to E2 enzymes. Two different types of E3 ligases exist: homologous to the E6-AP carboxyl terminus (HECT) domain E3 ligases and really interesting new gene (RING) finger domain E3 ligases. The E2 enzymes pass ubiquitin onto the HECT domain E3 ligases which transfer it to the target protein [50,51]. In contrast to the HECT domain E3 ligases, the E2 enzymes do not convey ubiquitin to the RING domain E3 ligases but directly to the proteasomal substrates. The RING domain E3 ligases act as a kind of bridge between the ubiquitin-bound E2 enzymes and the substrates, thereby increasing the activity of the E2 enzymes [52]. In the context of ubiquitination, three different models exist that explain the formation of a ubiquitin chain (polyubiquitination) bound at proteasomal substrates. The first model describes the formation of the chain in a step-by-step process in which ubiquitin monomers are added sequentially to the substrate. The second model states that the ubiquitin chain might be pre-assembled on an E2 enzyme and then transferred to the substrate in a single process. The third model represents a combination of the first two models [53,54,55]. Finally, polyubiquitinated proteins are degraded or processed by the catalytic component of the UPS, the 26S proteasome. The proteasome represents a large multi-protein complex of about 1700 kDa which comprises two different kinds of subunits—the 19S subunit and the 20S subunit (Figure 1A) [56,57]. The ubiquitin chain of target proteins is recognised and bound by the 19S regulatory complex and, subsequently, the target proteins are unfolded [58]. Hereafter, these proteins get degraded or processed by the 20S subunit which harbours different protease activities (caspase-like activity, chymotrypsin-like activity, trypsin-like activity) [59]. Proteasomes were detected in the cytosol, cell nucleus, microsomes, centrosomes and at the base of primary cilia [60,61,62,63]. Since their action is of great importance for the proper transduction of numerous signalling pathways [64,65], an altered proteasomal activity provokes defects in the regular procedure of cellular signalling and associated cellular processes [66], reflecting the eminent role of the UPS in the development and function of many vertebrate organs and structures [20,21,22].
In contrast to the UPS, autophagy is able to degrade proteins but cannot process them. However, autophagy degrades intracellular pathogens, long-lived proteins, very large protein complexes and even entire cell organelles [7,67,68,69,70,71,72]. In this way, autophagy plays an important role in vertebrate development [9,10,17,18,19], participating in the development of the brain [23], eyes [24,25,26], lung [27], heart [28,29] and liver [28]. Autophagy starts with the formation of phagophores, which then elongate to develop autophagosomes (Figure 1B) [73]. Elongation of the phagophore membrane and formation of the autophagosome is dependent on the recruitment of two ubiquitin-like (Ubl) conjugation systems. To build up the first system, autophagy-related protein 12 (ATG12) gets bound to ATG5 by the action of the E1-like enzyme ATG7 and the E2-like enzyme ATG10. Afterwards, ATG12–ATG5 becomes associated with ATG16L and forms a large complex referred to as the ATG16L complex. This complex is located at the phagophore, thereby defining the site of conjugation of the second Ubl system. The second Ubl conjugation system starts with the cleavage of microtubule-associated protein 1B light chain 3 (LC3) by ATG4 to generate cytoplasmic LC3-I. With the support of ATG7 and another E2-like enzyme called ATG3, LC3-I becomes lipidated with phosphatidylethanolamine (PE) to generate membrane-tethered LC3-II [74]. Autophagosomes represent double-membraned vesicles which enclose the targets destined to be degraded. LC3-II is integrated into both the inner and outer membrane of the autophagosome where it functions in autophagy substrate selection [75,76,77]. To degrade their enclosed substrates, the autophagosomes fuse with lysosomes [73]. The formed autolysosomes release hydrolases that implement the degradation of the autophagy substrates (Figure 1B). Autophagy is regulated by mammalian target of rapamycin (also known as mechanistic target of rapamycin) complex 1 (mTORC1) which blocks the initiation of autophagy. mTOR inactivates the ULK complex by phosphorylation [78,79]. The ULK complex consists of unc-51-like kinase 1/2 (ULK1/2), focal adhesion kinase family-interacting protein of 200 kDa (FIP200) and ATG13. When mTOR is inactivated, the ULK complex is allowed to translocate to phagophores and ULK1 directly phosphorylates Beclin-1, thereby activating the pro-autophagy class III phosphoinositide 3-kinase (PI(3)K) VPS34 complex and promoting autophagy induction and maturation [80].
As with the UPS, autophagy is also able to recognise a small proportion of its target proteins by their polyubiquitination (selective autophagy). Ubiquitin has different lysine residues which participate in the generation of polyubiquitin chains [81,82]. While the UPS preferably degrades or processes proteins that have a K48-linked polyubiquitin chain, autophagy degrades proteins with K63-linked chains [83,84].
3. The Primary Cilium
Primary cilia are tiny cytoplasmic protrusions (1–15 μm long) and basically contain three different compartments—the axoneme, the basal body (BB) and the transition zone (TZ) (Figure 1C). The axoneme represents the microtubule-based scaffold of the cilium and grows out of the BB, which is the modified mother centriole. The axoneme consists of nine doublet microtubules that are organised in a ring-like fashion. It stabilises the cilium and is essential for intraflagellar transport (IFT). IFT carries proteins from the base to the tip of the cilium (anterograde IFT), and then back to the base (retrograde IFT). The implementation of IFT requires the presence of motor proteins (kinesin-2 for anterograde IFT and dynein-2 for retrograde IFT) and of so-called IFT proteins. IFT proteins belonging to the IFT-B complex (e.g., IFT88) drive anterograde IFT, while IFT proteins of the IFT-A complex (e.g., IFT140) are necessary for retrograde transport [85]. The cargo that is destined for transport within the cilium is bound to IFT proteins which, in turn, are attached to motor proteins that move along the microtubules of the axoneme. The TZ, a short region of 0.5 μm, is located at the proximal end of the axoneme. It is characterised by the presence of so called Y-links, structures which appear as Y-shaped densities by transmission electron microscopy [86,87]. The TZ functions as a ciliary gatekeeper, controlling the entry and exit of proteins into and out of the cilium [87,88,89,90,91,92,93,94,95]. Several of the proteins that traverse the TZ and that are translocated through the cilium are receptors and mediators of signalling cascades such as the hedgehog (HH) pathway, the platelet-derived growth factor receptor α (PDGFRα) pathway and the transforming growth factor β (TGFβ) pathway [96,97,98,99,100,101,102,103]. HH signalling is one of the best studied cilia-mediated signalling pathways. In the absence of HH ligand, the activation of smoothened (SMO) is inhibited by the HH receptor patched (PTC), which is located in the ciliary membrane [101]. In this case, the HH mediator proteins glioblastoma 2 (GLI2) and glioblastoma 3 (GLI3) are proteolytically processed into GLI2-R and GLI3-R, two transcriptional repressors of HH target gene expression (Figure 2A). In the presence of HH, the HH ligand binds to PTC and the HH/PTC complex is translocated out of the cilium. Subsequently, SMO is activated, enters the cilium and induces the generation of GLI2-A and GLI3-A, two transcriptional activators of HH target gene expression [98,104,105] (Figure 2A). PDGFRα signalling starts with the binding of the ligand platelet-derived growth factor AA (PDGF-AA) to its ciliary membrane-bound receptor PDGFRα, which subsequently dimerises and undergoes phosphorylation [100]. As a consequence, the transduction of the mitogen-activated protein kinases 1/2 (MEK1/2)–extracellular signal-regulated kinases 1/2 (ERK1/2) and protein kinase B (AKT/PKB) pathways is initiated (Figure 2B). The importance of primary cilia in mediating PDGFRα signalling is reflected by the fact that the loss of cilia completely blocks PDGFRα signalling [100]. In the case of TGFβ signalling, both receptors of the pathway, TGFβ-RI and TGFβ-RII, localise to cilia [102]. By ligand binding, TGFβ-RI and TGFβ-RII form a heterotetrameric receptor complex which, in turn, activates the mediator proteins Sma- and Mad-related Protein 2 (SMAD2) and Sma- and Mad-related protein 3 (SMAD3) at the base of primary cilia (Figure 2B). Then, SMAD2 and SMAD3 join with Sma- and Mad-related protein 4 (SMAD4) at the ciliary base and the SMAD2–3–4 complex leaves the cilium in order to enter the nucleus and to induce TGFβ target gene expression [102]. Cilia-mediated signalling cascades are essential for the regulation of cellular processes during the entire development of an organism [103,106,107,108,109]. Consequently, many severe human diseases are associated with dysfunctional primary cilia and their number is permanently increasing [110]. The diseases caused by ciliary dysfunctions are commonly referred to as ciliopathies. Ciliopathies comprise many life-threatening diseases such as polycystic kidney disease, Meckel–Gruber syndrome, Joubert syndrome, Bardet–Biedl syndrome, Leber congenital amaurosis, Senior–Løken syndrome, orofaciodigital syndrome type 1, Alström syndrome, Jeune asphyxiating thoracic dystrophy, Ellis–van Creveld syndrome and Sensenbrenner syndrome [111,112].
4. Do the UPS and Autophagy Play a Role in the Development of Ciliopathies?
Current treatment of ciliopathies is limited to symptomatic therapies, as curative medication against ciliopathies is not yet available [113]. For this reason, cilia research is focused on the investigation of molecular mechanisms underlying ciliopathies as well as on developing curative therapies against these severe diseases [66,114,115,116,117,118,119,120,121]. There are promising approaches to tackle ciliopathies ranging from gene therapy to the use of small molecules, but none have yet successfully gone through clinical trials [122,123,124]. According to several studies, reduced activity of the UPS and/or of autophagy might be involved in the development of ciliopathies. For instance, it was reported that the ciliopathy phenotype of Bardet-Biedl syndrome (bbs) and Oral-Facial-Digital Syndrome 1 (ofd1) morphant zebrafish embryos is ameliorated by injecting human proteasomal subunit component (RPN10, RPN13, or RPT6) mRNA or by injecting the proteasome activators sulforaphane (SFN) and mevalonolactone (alias mevalonic acid lactone, mevalonate, and (±)-β-hydroxy-β-methyl-δ-valerolactone and abbreviated MVA), respectively [66]. Regarding autophagy, many data were collected in the context of polycystic kidney disease. In zebrafish embryos, it was demonstrated that a decreased autophagic activity causes polycystic kidney disease and that a specific inducer Beclin-1 peptide and the autophagy activators rapamycin (RAP) as well as carbamazepine (CBZ) and minoxidil ameliorates cyst formation and restores kidney function [125,126]. While the macrolide RAP activates autophagy by inhibiting mTORC1 signalling [127,128,129] which is known to block autophagy [130,131,132,133], carbamazepine (CBZ) and minoxidil exert their autophagy-activating function independently of mTOR [134,135]. Interestingly, in utero application of RAP also markedly attenuated cyst formation in mouse embryos suffering from polycystic kidney disease [136]. Moreover, treatment of adult mice suffering from polycystic kidney disease with RAP reduces renal cystogenesis [137,138]. Furthermore, the treatment of rats displaying polycystic kidney disease with RAP as well as with the mTOR inhibitor and autophagy activator PP242 blocks renal enlargement and cystogenesis [139,140,141,142]. In polycystic kidney patients, the application of RAP slows kidney growth and prevents the worsening of renal function [137,143,144,145]. In mice, it was shown that a proper dose of RAP is essential for its positive effect on polycystic kidney disease [146,147]. Based on these studies, it is conceivable that a decreased proteasomal and/or autophagic activity might be involved in the development of ciliopathies.
5. Which Role Does the Primary Cilium Play in the Crosstalk between the UPS and Autophagy?
For many years, crosstalk between the UPS and autophagy was negated since the general view was that the two main degradation systems have different substrate preferences [148,149]. In the last 10 years, interplay between the UPS and autophagy was the subject of intense research. The key finding was that the UPS and autophagy are at least partially redundant, a result that is based on the fact that both degradation systems partially share the same substrates, and that if one degradation system is downregulated, the other gets upregulated. It was shown that numerous proteins participate in this crosstalk [150]. Due to existing excellent review articles about the crosstalk between these degradation systems [31,150,151,152], we will concentrate on those interplay mechanisms in which the primary cilium is obviously involved.
It is unquestionable that the primary cilium takes part in the crosstalk between the UPS and autophagy, but it is difficult to define its exact role since different relationships between ciliary proteins and both degradation systems have been elucidated (Figure 3). BBS4 and OFD1 are good examples for explaining this difficulty. Both proteins positively regulate proteasomal activity since the loss of BBS4 (in the kidney, liver, brain and retina) and OFD1 (in mouse embryonic stem cells) results in a reduced proteasomal activity, respectively [66]. Moreover, both proteins are influenced by autophagy. In mouse embryonic fibroblasts (MEFs), BBS4 is recruited to primary cilia by autophagy, most likely via an indirect mechanism, and OFD1 is an autophagic substrate which is degraded at the ciliary base [153]. Considering that ciliary BBS4 positively regulates proteasomal activity, autophagy would promote proteasomal activity by the ciliary recruitment of BBS4. By contrast, autophagy seems to inhibit proteasomal activity via degrading OFD1. Furthermore, the autophagy-dependent degradation of OFD1 at the ciliary base promotes ciliogenesis and ciliary elongation [153,154]. Consequently, autophagy ensures the formation of cilia. In turn, ciliary presence is essential for the proteolytic processing of GLI3 in mice [99]. Investigations in MEFs revealed that GLI3 processing is implemented by the cilia-regulated proteasome, a kind of proteasome that localises to the ciliary base and is controlled differently from all other proteasomes within the cell [63]. The absence of retinitis pigmentosa GTPase regulator-interacting protein 1-like (RPGRIP1L) leads to a reduced catalytic activity of the cilia-regulated proteasome, while the activity of all other proteasomes within the cell is not affected [63]. The proteolytic processing of GLI3 gives rise to GLI3-R, which inhibits HH signalling [48]. Cilia-mediated HH signalling is able to activate autophagy in MEFs and mouse kidney epithelial cells [155]. Thus, the cilia-regulated proteasome negatively regulates autophagy. As mentioned before, the catalytic activity of the cilia-regulated proteasome is positively governed by RPGRIP1L in MEFs, suggesting that RPGRIP1L negatively regulates autophagy via its positive regulation of the cilia-regulated proteasome. However, RPGRIP1L positively regulates both the activity of the cilia-regulated proteasome and autophagy in MEFs. Additionally, it was demonstrated that RPGRIP1L controls both activities independently of each other in MEFs [156]. Remarkably, RPGRIP1L negatively controls the proteasome-based degradation of dishevelled (DSH) in Madin–Darby canine kidney (MDCK) cells [157], suggesting that RPGRIP1L governs proteasomal activity in a cell type-specific manner. Moreover, RPGRIP1L controls the assembly of the TZ by ensuring the proper amount of other proteins at the ciliary base in MEFs, mouse embryonic kidneys and mouse embryonic limbs [158]. One of these proteins is BBS4. In MEFs, autophagy recruits BBS4 to cilia [153] and RPGRIP1L positively regulates autophagic activity [156]. Thus, it is conceivable that RPGRIP1L ensures the ciliary amount of BBS4 via regulating autophagy. Considering that BBS4 positively regulates proteasomal activity [66], it is possible that RPGRIP1L controls proteasomal activity by affecting the ciliary amount of BBS4. However, BBS4 seems to control overall cellular proteasomal activity [66] while RPGRIP1L governs proteasomal activity exclusively at the ciliary base [63]. Furthermore, it was demonstrated that RPGRIP1L regulates the activity of the cilia-regulated proteasome by interacting with proteasome 26S non-ATPase regulatory subunit 2 (PSMD2) [63], arguing for a BBS4-independent control of proteasomal activity by RPGRIP1L. Another protein whose abundance at the TZ is governed by RPGRIP1L represents nephrocystin 4 (NPHP4), which also participates in the regulation of protein degradation. NPHP4 interacts with an E3 ligase named Jade-1, which targets β-catenin, the main mediator of the canonical WNT pathway, to the proteasome. By this interaction, NPHP4 stabilises Jade-1 and enhances its capability to promote the proteasomal degradation of β-catenin in a human embryonic kidney cell line (HEK293 cells) [159]. A link between NPHP4 and autophagy has not been shown. The E3 ligases c-CBL and CBL-b also interact with a ciliary protein, the intraflagellar transport protein 20 (IFT20). It stabilises c-CBL and CBL-b by inhibiting their autoubiquitination and proteasomal degradation. In this way, IFT20 supports the ubiquitination and internalization of PDGFRα thereby preventing aberrant PDGFRα signalling in immortalised MEFs (NIH3T3 cells) [160]. In the context of autophagy, analyses in MEFs demonstrated that IFT20 is an autophagic substrate and a positive regulator of autophagy suggesting “a novel mechanism for self-containment of the autophagic process” [155].
Apart from HH signalling, another signal transduction cascade links cilia to the UPS and autophagy. The mTOR signalling pathway is mediated by primary cilia in a human kidney proximal tubular epithelial cell line (HK2 cells) and in MEFs [154,156] and among many other processes is deeply involved in the regulation of both the UPS and autophagy [161]. It is known that mTOR signalling inhibits autophagy [130,131,132,133,161]. In addition, mTOR signalling negatively regulates proteasomal activity in MEFs and in HEK293 cells [40]. Regarding proteasomal activity, the opposite is also observed, namely that mTOR signalling positively governs proteasomal activity as was shown in MEFs and in HEK293 cells [162], leading to an unresolved situation [163,164].
6. Conclusions
The primary cilium plays a vital role in the crosstalk between the UPS and autophagy, but it is impossible to define a blanket function of the primary cilium in this crosstalk since it is a signalling hub and houses numerous proteins. As we have outlined in this article, various ciliary proteins and cilia-mediated signalling pathways have different effects on the two degradation systems. It is the beginning of an enormous puzzle where the bulk of the pieces are missing and future studies have to be undertaken to yield a clear picture.
Funding
This research was funded by the Deutsche Forschungsgemeinschaft (Sonderforschungsbereiche 590 and 612) to U.R.
Conflicts of Interest
The authors declare that they have no competing interests.
References
- Irvine, G.; El-Agnaf, O.; Shankar, G.; Walsh, D. Protein aggregation in the brain: The molecular basis for Alzheimer’s and Parkinson’s diseases. Mol. Med. 2008, 14, 451–464. [Google Scholar] [CrossRef]
- Cox, D.; Raeburn, C.; Sui, X.; Hatters, D. Protein aggregation in cell biology: An aggregomics perspective of health and disease. In Seminars in Cell & Developmental Biology; pii: S1084-9521; Academic Press: New York, NY, USA, 2018. [Google Scholar]
- Soto, C.; Pritzkow, S. Protein misfolding, aggregation, and conformational strains in neurodegenerative diseases. Nat. Neurosci. 2018, 21, 1332–1340. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Urbé, S.; Clague, M. Selective protein degradation in cell signalling. Semin. Cell Dev. Biol. 2012, 23, 509–514. [Google Scholar] [CrossRef]
- Koepp, D. Cell cycle regulation by protein degradation. Methods Mol. Biol. 2014, 1170, 61–73. [Google Scholar] [PubMed]
- Yao, T.; Ndoja, A. Regulation of gene expression by the ubiquitin-proteasome system. Semin. Cell Dev. Biol. 2012, 23, 523–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lilienbaum, A. Relationship between the proteasomal system and autophagy. Int. J. Biochem. Mol. Biol. 2013, 4, 1–26. [Google Scholar]
- Gerhardt, C.; Leu, T.; Lier, J.; Rüther, U. The cilia-regulated proteasome and its role in the development of ciliopathies and cancer. Cilia 2016, 5, 14. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Levine, B. Autophagy in mammalian development and differentiation. Nat. Cell Biol. 2010, 12, 823–830. [Google Scholar] [CrossRef] [Green Version]
- Cecconi, F.; Levine, B. The role of autophagy in mammalian development: Cell makeover rather than cell death. Dev. Cell 2008, 15, 344–357. [Google Scholar] [CrossRef]
- Le Bot, N. Autophagy: A new regulator of development. Nat. Cell Biol. 2007, 9, 741. [Google Scholar] [CrossRef]
- Ryter, S.; Cloonan, S.; Choi, A. Autophagy: A critical regulator of cellular metabolism and homeostasis. Mol. Cells 2013, 36, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Gerhardt, C.; Wiegering, A.; Leu, T.; Rüther, U. Control of Hedgehog signalling by the cilia-regulated proteasome. J. Dev. Biol. 2016, 4, 27. [Google Scholar] [CrossRef] [PubMed]
- Lippai, M.; Szatmári, Z. Autophagy-from molecular mechanisms to clinical relevance. Cell Biol. Toxicol. 2017, 33, 145–168. [Google Scholar] [CrossRef] [PubMed]
- Mizumura, K.; Choi, A.; Ryter, S. Emerging role of selective autophagy in human diseases. Front. Pharmacol. 2014, 5, 244. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.; Finley, D. Regulation of proteasome activity in health and disease. Biochim. Biophys. Acta 2014, 1843, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Aburto, M.; Hurlé, J.; Varela-Nieto, I.; Magariños, M. Autophagy during vertebrate development. Cells 2012, 1, 428–448. [Google Scholar] [CrossRef]
- Di Bartolomeo, S.; Nazio, F.; Cecconi, F. The role of autophagy during development in higher eukaryotes. Traffic 2010, 11, 1280–1289. [Google Scholar] [CrossRef]
- Yue, Z.; Jin, S.; Yang, C.; Levine, A.; Heintz, N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc. Natl. Acad. Sci. USA 2003, 100, 15077–15082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubinsztein, D. The roles of intracellular protein-degradation pathways in neurodegeneration. Nature 2006, 443, 780–786. [Google Scholar] [CrossRef]
- Breusing, N.; Arndt, J.; Voss, P.; Bresgen, N.; Wiswedel, I.; Gardemann, A.; Siems, W.; Grune, T. Inverse correlation of protein oxidation and proteasome activity in liver and lung. Mech. Ageing Dev. 2009, 130, 748–753. [Google Scholar] [CrossRef]
- Wang, X.; Robbins, J. Proteasomal and lysosomal protein degradation and heart disease. J. Mol. Cell Cardiol. 2014, 71, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Fimia, G.; Stoykova, A.; Romagnoli, A.; Giunta, L.; Di Bartolomeo, S.; Nardacci, R.; Corazzari, M.; Fuoco, C.; Ucar, A.; Schwartz, P.; et al. Ambra1 regulates autophagy and development of the nervous system. Nature 2007, 447, 1121–1125. [Google Scholar] [PubMed]
- Costello, M.; Brennan, L.; Basu, S.; Chauss, D.; Mohamed, A.; Gilliland, K.; Johnsen, S.; Menko, A.; Kantorow, M. Autophagy and mitophagy participate in ocular lens organelle degradation. Exp. Eye Res. 2013, 116, 141–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basu, S.; Rajakaruna, S.; Reyes, B.; Van Bockstaele, E.; Menko, A. Suppression of MAPK/JNK-MTORC1 signaling leads to premature loss of organelles and nuclei by autophagy during terminal differentiation of lens fiber cells. Autophagy 2014, 10, 1193–1211. [Google Scholar] [CrossRef] [PubMed]
- Mellén, M.; de la Rosa, E.; Boya, P. The autophagic machinery is necessary for removal of cell corpses from the developing retinal neuroepithelium. Cell Death Differ. 2008, 15, 1279–1290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qu, X.; Zou, Z.; Sun, Q.; Luby-Phelps, K.; Cheng, P.; Hogan, R.; Gilpin, C.; Levine, B. Autophagy gene-dependent clearance of apoptotic cells during embryonic development. Cell 2007, 128, 931–946. [Google Scholar] [CrossRef] [PubMed]
- Gan, B.; Peng, X.; Nagy, T.; Alcaraz, A.; Gu, H.; Guan, J. Role of FIP200 in cardiac and liver development and its regulation of TNFalpha and TSC-mTOR signaling pathways. J. Cell Biol. 2006, 175, 121–133. [Google Scholar] [CrossRef]
- Lee, E.; Koo, Y.; Ng, A.; Wei, Y.; Luby-Phelps, K.; Juraszek, A.; Xavier, R.; Cleaver, O.; Levine, B.; Amatruda, J. Autophagy is essential for cardiac morphogenesis during vertebrate development. Autophagy 2014, 10, 572–587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levine, B.; Kroemer, G. Autophagy in the pathogenesis of disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef]
- Dikic, I. Proteasomal and Autophagic Degradation Systems. Annu. Rev. Biochem. 2017, 86, 193–224. [Google Scholar] [CrossRef]
- Wang, X.; Yu, J.; Wong, S.; Cheng, A.; Chan, F.; Ng, S.; Cho, C.; Sung, J.; Wu, W. A novel crosstalk between two major protein degradation systems. Autophagy 2013, 9, 1500–1508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tannous, P.; Zhu, H.; Nemchenko, A.; Berry, J.; Johnstone, J.; Shelton, J.; Miller, F.J.; Rothermel, B.; Hill, J. Intracellular protein aggregation is a proximal trigger of cardiomyocyte autophagy. Circulation 2008, 117, 3070–3078. [Google Scholar] [CrossRef]
- Demishtein, A.; Fraiberg, M.; Berko, D.; Tirosh, B.; Elazar, Z.; Navon, A. SQSTM1/p62-mediated autophagy compensates for loss of proteasome polyubiquitin recruiting capacity. Autophagy 2017, 13, 1697–1708. [Google Scholar] [CrossRef] [PubMed]
- Fan, T.; Huang, Z.; Wang, W.; Zhang, B.; Xu, Y.; Mao, Z.; Chen, L.; Hu, H.; Geng, Q. Proteasome inhibition promotes autophagy and protects from endoplasmic reticulum stress in rat alveolar macrophages exposed to hypoxia-reoxygenation injury. J. Cell. Physiol. 2018, 233, 6748–6758. [Google Scholar] [CrossRef] [PubMed]
- Ge, P.; Zhang, J.; Wang, X.; Meng, F.; Li, W.; Luan, Y.; Ling, F.; Luo, Y. Inhibition of autophagy induced by proteasome inhibition increases cell death in human SHG-44 glioma cells. Acta Pharmacol. Sin. 2009, 30, 1046–1052. [Google Scholar] [CrossRef]
- Jiang, S.; Park, D.; Gao, Y.; Ravi, S.; Darley-Usmar, V.; Abraham, E.; Zmijewski, J. Participation of proteasome-ubiquitin protein degradation in autophagy and the activation of AMP-activated protein kinase. Cell. Signal. 2015, 27, 1186–1197. [Google Scholar] [CrossRef] [Green Version]
- Kyrychenko, V.; Nagibin, V.; Tumanovska, L.; Pashevin, D.; Gurianova, V.; Moibenko, A.; Dosenko, V.; Klionsky, D. Knockdown of PSMB7 induces autophagy in cardiomyocyte cultures: Possible role in endoplasmic reticulum stress. Pathobiology 2014, 81, 8–14. [Google Scholar] [CrossRef]
- Zhao, J.; Brault, J.; Schild, A.; Cao, P.; Sandri, M.; Schiaffino, S.; Lecker, S.; Goldberg, A. FoxO3 coordinately activates protein degradation by the autophagic/lysosomal and proteasomal pathways in atrophying muscle cells. Cell Metab. 2007, 6, 472–483. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Zhai, B.; Gygi, S.; Goldberg, A. mTOR inhibition activates overall protein degradation by the ubiquitin proteasome system as well as by autophagy. Proc. Natl. Acad. Sci. USA 2015, 112, 15790–15797. [Google Scholar] [CrossRef] [Green Version]
- Selimovic, D.; Porzig, B.; El-Khattouti, A.; Badura, H.; Ahmad, M.; Ghanjati, F.; Santourlidis, S.; Haikel, Y.; Hassan, M. Bortezomib/proteasome inhibitor triggers both apoptosis and autophagy-dependent pathways in melanoma cells. Cell. Signal. 2013, 25, 308–318. [Google Scholar] [CrossRef]
- Sha, Z.; Schnell, H.; Ruoff, K.; Goldberg, A. Rapid induction of p62 and GABARAPL1 upon proteasome inhibition promotes survival before autophagy activation. J. Cell Biol. 2018, 217, 1757–1776. [Google Scholar] [CrossRef]
- Sun, A.; Li, C.; Chen, R.; Huang, Y.; Chen, Q.; Cui, X.; Liu, H.; Thrasher, J.; Li, B. GSK-3β controls autophagy by modulating LKB1-AMPK pathway in prostate cancer cells. Prostate 2016, 76, 172–183. [Google Scholar] [CrossRef] [PubMed]
- Tang, B.; Cai, J.; Sun, L.; Li, Y.; Qu, J.; Snider, B.; Wu, S. Proteasome inhibitors activate autophagy involving inhibition of PI3K-Akt-mTOR pathway as an anti-oxidation defense in human RPE cells. PLoS ONE 2014, 9, e103364. [Google Scholar] [CrossRef]
- Wu, W.; Wu, Y.; Yu, L.; Li, Z.; Sung, J.; Cho, C. Induction of autophagy by proteasome inhibitor is associated with proliferative arrest in colon cancer cells. Biochem. Biophys. Res. Commun. 2008, 374, 258–263. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Wang, S.; Viollet, B.; Zou, M. Regulation of the proteasome by AMPK in endothelial cells: The role of O-GlcNAc transferase (OGT). PLoS ONE 2012, 7, e36717. [Google Scholar] [CrossRef] [PubMed]
- Zhu, K.; Dunner, K.J.; McConkey, D. Proteasome inhibitors activate autophagy as a cytoprotective response in human prostate cancer cells. Oncogene 2010, 29, 451–462. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Fallon, J.; Beachy, P. Hedgehog-regulated processing of Gli3 produces an anterior/posterior repressor gradient in the developing vertebrate limb. Cell 2000, 100, 423–434. [Google Scholar] [CrossRef]
- Schrader, E.; Harstad, K.; Holmgren, R.; Matouschek, A. A three-part signal governs differential processing of Gli1 and Gli3 proteins by the proteasome. J. Biol. Chem. 2011, 286, 39051–39058. [Google Scholar] [CrossRef] [PubMed]
- Weissman, A. Themes and variations on ubiquitylation. Nat. Rev. Mol. Cell Biol. 2001, 2, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Pickart, C. Mechanisms underlying ubiquitination. Annu. Rev. Biochem. 2001, 70, 503–533. [Google Scholar] [CrossRef] [PubMed]
- Metzger, M.; Hristova, V.; Weissman, A. HECT and RING finger families of E3 ubiquitin ligases at a glance. J. Cell Sci. 2012, 125, 531–537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Tu, D.; Brunger, A.; Ye, Y. A ubiquitin ligase transfers preformed polyubiquitin chains from a conjugating enzyme to a substrate. Nature 2007, 446, 333–337. [Google Scholar] [CrossRef] [PubMed]
- Ravid, T.; Hochstrasser, M. Autoregulation of an E2 enzyme by ubiquitin-chain assembly on its catalytic residue. Nat. Cell Biol. 2007, 9, 422–427. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Rape, M. Building ubiquitin chains: E2 enzymes at work. Nat. Rev. Mol. Cell Biol. 2009, 10, 755–764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolf, D.; Hilt, W. The proteasome: A proteolytic nanomachine of cell regulation and waste disposal. Biochim. Biophys. Acta 2004, 1695, 19–31. [Google Scholar] [CrossRef]
- Jung, T.; Grune, T. Structure of the proteasome. Prog. Mol. Biol. Transl. Sci. 2012, 109, 1–39. [Google Scholar]
- Liu, C.; Jacobson, A. Functions of the 19S complex in proteasomal degradation. Trends Biochem. Sci. 2013, 38, 103–110. [Google Scholar] [CrossRef]
- Coux, O.; Tanaka, K.; Goldberg, A. Structure and functions of the 20S and 26S proteasomes. Annu. Rev. Biochem. 1996, 65, 801–847. [Google Scholar] [CrossRef]
- Gerdes, J.; Liu, Y.; Zaghloul, N.; Leitch, C.; Lawson, S.; Kato, M.; Beachy, P.; Beales, P.; DeMartino, G.; Fisher, S.; et al. Disruption of the basal body compromises proteasomal function and perturbs intracellular Wnt response. Nat. Genet. 2007, 39, 1350–1360. [Google Scholar] [CrossRef]
- Brooks, P.; Fuertes, G.; Murray, R.; Bose, S.; Knecht, E.; Rechsteiner, M.; Hendil, K.; Tanaka, K.; Dyson, J.; Rivett, J. Subcellular localization of proteasomes and their regulatory complexes in mammalian cells. Biochem. J. 2000, 346, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Wigley, W.; Fabunmi, R.; Lee, M.; Marino, C.; Muallem, S.; DeMartino, G.; Thomas, P. Dynamic association of proteasomal machinery with the centrosome. J. Cell Biol. 1999, 145, 481–490. [Google Scholar] [CrossRef] [PubMed]
- Gerhardt, C.; Lier, J.; Burmühl, S.; Struchtrup, A.; Deutschmann, K.; Vetter, M.; Leu, T.; Reeg, S.; Grune, T.; Rüther, U. The transition zone protein Rpgrip1l regulates proteasomal activity at the primary cilium. J. Cell Biol. 2015, 210, 115–133. [Google Scholar] [CrossRef] [PubMed]
- Jurek, A.; Amagasaki, K.; Gembarska, A.; Heldin, C.; Lennartsson, J. Negative and positive regulation of MAPK phosphatase 3 controls platelet-derived growth factor-induced Erk activation. J. Biol. Chem. 2009, 284, 4626–4634. [Google Scholar] [CrossRef] [PubMed]
- Voutsadakis, I. The ubiquitin-proteasome system and signal transduction pathways regulating Epithelial Mesenchymal transition of cancer. J. Biomed. Sci. 2012, 19, 67. [Google Scholar] [CrossRef]
- Liu, Y.; Tsai, I.; Morleo, M.; Oh, E.; Leitch, C.; Massa, F.; Lee, B.; Parker, D.; Finley, D.; Zaghloul, N.; et al. Ciliopathy proteins regulate paracrine signaling by modulating proteasomal degradation of mediators. J. Clin. Investig. 2014, 124, 2059–2070. [Google Scholar] [CrossRef] [Green Version]
- Farré, J.; Manjithaya, R.; Mathewson, R.; Subramani, S. PpAtg30 tags peroxisomes for turnover by selective autophagy. Dev. Cell 2008, 14, 365–376. [Google Scholar] [CrossRef]
- Kraft, C.; Deplazes, A.; Sohrmann, M.; Peter, M. Mature ribosomes are selectively degraded upon starvation by an autophagy pathway requiring the Ubp3p/Bre5p ubiquitin protease. Nat. Cell Biol. 2008, 10, 602–610. [Google Scholar] [CrossRef]
- Orvedahl, A.; Sumpter, R.J.; Xiao, G.; Ng, A.; Zou, Z.; Tang, Y.; Narimatsu, M.; Gilpin, C.; Sun, Q.; Roth, M.; et al. Image-based genome-wide siRNA screen identifies selective autophagy factors. Nature 2011, 480, 113–117. [Google Scholar] [CrossRef]
- Rott, R.; Szargel, R.; Haskin, J.; Bandopadhyay, R.; Lees, A.; Shani, V.; Engelender, S. α-Synuclein fate is determined by USP9X-regulated monoubiquitination. Proc. Natl. Acad. Sci. USA 2011, 108, 18666–18671. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Klionsky, D. Mitochondria removal by autophagy. Autophagy 2011, 7, 297–300. [Google Scholar] [CrossRef] [Green Version]
- Thurston, T.; Wandel, M.; von Muhlinen, N.; Foeglein, A.; Randow, F. Galectin 8 targets damaged vesicles for autophagy to defend cells against bacterial invasion. Nature 2012, 482, 414–418. [Google Scholar] [CrossRef]
- Arstila, A.; Trump, B. Studies on cellular autophagocytosis. The formation of autophagic vacuoles in the liver after glucagon administration. Am. J. Pathol. 1968, 53, 687–733. [Google Scholar]
- Geng, J.; Klionsky, D. The Atg8 and Atg12 ubiquitin-like conjugation systems in macroautophagy. ’Protein modifications: Beyond the usual suspects’ review series. EMBO Rep. 2008, 9, 859–864. [Google Scholar] [CrossRef]
- Kabeya, Y.; Mizushima, N.; Ueno, T.; Yamamoto, A.; Kirisako, T.; Noda, T.; Kominami, E.; Ohsumi, Y.; Yoshimori, T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000, 19, 5720–5728. [Google Scholar] [CrossRef] [Green Version]
- Mizushima, N.; Yamamoto, A.; Hatano, M.; Kobayashi, Y.; Kabeya, Y.; Suzuki, K.; Tokuhisa, T.; Ohsumi, Y.; Yoshimori, T. Dissection of autophagosome formation using Apg5-deficient mouse embryonic stem cells. J. Cell Biol. 2001, 152, 657–668. [Google Scholar] [CrossRef]
- Barth, S.; Glick, D.; Macleod, K. Autophagy: Assays and artifacts. J. Pathol. 2010, 221, 117–124. [Google Scholar] [CrossRef]
- Wesselborg, S.; Stork, B. Autophagy signal transduction by ATG proteins: From hierarchies to networks. Cell. Mol. Life Sci. 2015, 72, 4721–4757. [Google Scholar] [CrossRef]
- Mizushima, N.; Yoshimori, T.; Ohsumi, Y. The role of Atg proteins in autophagosome formation. Annu. Rev. Cell Dev. Biol. 2011, 27, 107–132. [Google Scholar] [CrossRef]
- Russell, R.; Tian, Y.; Yuan, H.; Park, H.; Chang, Y.; Kim, J.; Kim, H.; Neufeld, T.; Dillin, A.; Guan, K. ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat. Cell Biol. 2013, 15, 741–750. [Google Scholar] [CrossRef]
- Pickart, C.; Eddins, M. Ubiquitin: Structures, functions, mechanisms. Biochim. Biophys. Acta 2004, 1695, 55–72. [Google Scholar] [CrossRef]
- Sun, F.; Kanthasamy, A.; Anantharam, V.; Kanthasamy, A. Environmental neurotoxic chemicals-induced ubiquitin proteasome system dysfunction in the pathogenesis and progression of Parkinson’s disease. Pharmacol. Ther. 2007, 114, 327–344. [Google Scholar] [CrossRef]
- Thrower, J.; Hoffman, L.; Rechsteiner, M.; Pickart, C. Recognition of the polyubiquitin proteolytic signal. EMBO J. 2000, 19, 94–102. [Google Scholar] [CrossRef] [Green Version]
- Tan, J.; Wong, E.; Kirkpatrick, D.; Pletnikova, O.; Ko, H.; Tay, S.; Ho, M.; Troncoso, J.; Gygi, S.; Lee, M.; et al. Lysine 63-linked ubiquitination promotes the formation and autophagic clearance of protein inclusions associated with neurodegenerative diseases. Hum. Mol. Genet. 2008, 17, 431–439. [Google Scholar] [CrossRef]
- Hao, L.; Scholey, J. Intraflagellar transport at a glance. J. Cell Sci. 2009, 122, 889–892. [Google Scholar] [CrossRef] [Green Version]
- Gilula, N.; Satir, P. The ciliary necklace. A ciliary membrane specialization. J. Cell Biol. 1972, 53, 494–509. [Google Scholar] [CrossRef]
- Reiter, J.; Blacque, O.; Leroux, M. The base of the cilium: Roles for transition fibres and the transition zone in ciliary formation, maintenance and compartmentalization. EMBO Rep. 2012, 13, 608–618. [Google Scholar] [CrossRef]
- Czarnecki, P.G.; Shah, J.V. The ciliary transition zone: From morphology and molecules to medicine. Trends Cell Biol. 2012, 22, 201–210. [Google Scholar] [CrossRef]
- Garcia-Gonzalo, F.; Reiter, J. Scoring a backstage pass: Mechanisms of ciliogenesis and ciliary access. J. Cell Biol. 2012, 197, 697–709. [Google Scholar] [CrossRef] [Green Version]
- Omran, H. NPHP proteins: Gatekeepers of the ciliary compartment. J. Cell Biol. 2010, 190, 715–717. [Google Scholar] [CrossRef]
- Benzing, T.; Schermer, B. Transition zone proteins and cilia dynamics. Nat. Genet. 2011, 43, 723–724. [Google Scholar] [CrossRef]
- Garcia-Gonzalo, F.; Reiter, J. Open Sesame: How Transition Fibers and the Transition Zone Control Ciliary Composition. Cold Spring Harb. Perspect. Biol. 2017, 9, a028134. [Google Scholar] [CrossRef]
- Jensen, V.; Leroux, M. Gates for soluble and membrane proteins, and two trafficking systems (IFT and LIFT), establish a dynamic ciliary signaling compartment. Curr. Opin. Cell Biol. 2017, 47, 83–91. [Google Scholar] [CrossRef]
- Betleja, E.; Cole, D. Ciliary trafficking: CEP290 guards a gated community. Curr. Biol. 2010, 20, R928–R931. [Google Scholar] [CrossRef]
- Craige, B.; Tsao, C.; Diener, D.; Hou, Y.; Lechtreck, K.; Rosenbaum, J.; Witman, G. CEP290 tethers flagellar transition zone microtubules to the membrane and regulates flagellar protein content. J. Cell Biol. 2010, 190, 927–940. [Google Scholar] [CrossRef] [Green Version]
- Berbari, N.; O’Connor, A.; Haycraft, C.; Yoder, B. The primary cilium as a complex signaling center. Curr. Biol. 2009, 19, R526–R535. [Google Scholar] [CrossRef]
- Eggenschwiler, J.; Anderson, K. Cilia and developmental signaling. Annu. Rev. Cell Dev. Biol. 2007, 23, 345–373. [Google Scholar] [CrossRef]
- Corbit, K.; Aanstad, P.; Singla, V.; Norman, A.; Stainier, D.; Reiter, J. Vertebrate Smoothened functions at the primary cilium. Nature 2005, 437, 1018–1021. [Google Scholar] [CrossRef]
- Haycraft, C.; Banizs, B.; Aydin-Son, Y.; Zhang, Q.; Michaud, E.; Yoder, B. Gli2 and Gli3 localize to cilia and require the intraflagellar transport protein polaris for processing and function. PLoS Genet. 2005, 1, e53. [Google Scholar] [CrossRef]
- Schneider, L.; Clement, C.; Teilmann, S.; Pazour, G.; Hoffmann, E.; Satir, P.; Christensen, S. PDGFRalphaalpha signaling is regulated through the primary cilium in fibroblasts. Curr. Biol. 2005, 15, 1861–1866. [Google Scholar] [CrossRef]
- Rohatgi, R.; Milenkovic, L.; Scott, M. Patched1 regulates hedgehog signaling at the primary cilium. Science 2007, 317, 372–376. [Google Scholar] [CrossRef]
- Clement, C.; Ajbro, K.; Koefoed, K.; Vestergaard, M.; Veland, I.; Henriques de Jesus, M.; Pedersen, L.; Benmerah, A.; Andersen, C.; Larsen, L.; et al. TGF-β signaling is associated with endocytosis at the pocket region of the primary cilium. Cell Rep. 2013, 3, 1806–1814. [Google Scholar] [CrossRef] [PubMed]
- Satir, P.; Pedersen, L.; Christensen, S. The primary cilium at a glance. J. Cell Sci. 2010, 123, 499–503. [Google Scholar] [Green Version]
- Chen, M.; Wilson, C.; Li, Y.; Law, K.; Lu, C.; Gacayan, R.; Zhang, X.; Hui, C.; Chuang, P. Cilium-independent regulation of Gli protein function by Sufu in Hedgehog signaling is evolutionarily conserved. Genes Dev. 2009, 23, 1910–1928. [Google Scholar] [CrossRef] [PubMed]
- Humke, E.; Dorn, K.; Milenkovic, L.; Scott, M.; Rohatgi, R. The output of Hedgehog signaling is controlled by the dynamic association between Suppressor of Fused and the Gli proteins. Genes Dev. 2010, 24, 670–682. [Google Scholar] [CrossRef] [Green Version]
- Christensen, S.; Pedersen, S.; Satir, P.; Veland, I.; Schneider, L. The primary cilium coordinates signaling pathways in cell cycle control and migration during development and tissue repair. Curr. Top. Dev. Biol. 2008, 85, 261–301. [Google Scholar]
- Yun, S.; Lee, M.; Ryu, J.; Song, C.; Han, H. Role of HIF-1alpha and VEGF in human mesenchymal stem cell proliferation by 17beta-estradiol: Involvement of PKC, PI3K/Akt, and MAPKs. Am. J. Physiol. Cell Physiol. 2009, 296, 317–326. [Google Scholar] [CrossRef]
- Schild, C.; Wirth, M.; Reichert, M.; Schmid, R.; Saur, D.; Schneider, G. PI3K signaling maintains c-myc expression to regulate transcription of E2F1 in pancreatic cancer cells. Mol. Carcinog. 2009, 48, 1149–1158. [Google Scholar] [CrossRef]
- Massagué, J. TGFβ signalling in context. Nat. Rev. Mol. Cell Biol. 2012, 13, 616–630. [Google Scholar] [CrossRef]
- Reiter, J.; Leroux, M. Genes and molecular pathways underpinning ciliopathies. Nat. Rev. Mol. Cell Biol. 2017, 18, 533–547. [Google Scholar] [CrossRef] [PubMed]
- Hildebrandt, F.; Benzing, T.; Katsanis, N. Ciliopathies. N. Engl. J. Med. 2011, 364, 1533–1543. [Google Scholar] [CrossRef] [Green Version]
- Waters, A.; Beales, P. Ciliopathies: An expanding disease spectrum. Pediatr. Nephrol. 2011, 26, 1039–1056. [Google Scholar] [CrossRef]
- McIntyre, J.; Williams, C.; Martens, J. Smelling the roses and seeing the light: Gene therapy for ciliopathies. Trends Biotechnol. 2013, 31, 355–363. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Kim, S.; Jung, Y.; Jung, E.; Kwon, H.; Kim, J. Eupatilin rescues ciliary transition zone defects to ameliorate ciliopathy-related phenotypes. J. Clin. Investig. 2018, 128, 3642–3648. [Google Scholar] [CrossRef] [PubMed]
- McIntyre, J.; Davis, E.; Joiner, A.; Williams, C.; Tsai, I.; Jenkins, P.; McEwen, D.; Zhang, L.; Escobado, J.; Thomas, S.; et al. Gene therapy rescues cilia defects and restores olfactory function in a mammalian ciliopathy model. Nat. Med. 2012, 18, 1423–1428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, C.; Uytingco, C.; Green, W.; McIntyre, J.; Ukhanov, K.; Zimmerman, A.; Shively, D.; Zhang, L.; Nishimura, D.; Sheffield, V.; et al. Gene Therapeutic Reversal of Peripheral Olfactory Impairment in Bardet-Biedl Syndrome. Mol. Ther. 2017, 25, 904–916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruan, G.; Barry, E.; Yu, D.; Lukason, M.; Cheng, S.; Scaria, A. CRISPR/Cas9-Mediated Genome Editing as a Therapeutic Approach for Leber Congenital Amaurosis 10. Mol. Ther. 2017, 25, 331–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Ran, J.; Liu, M.; Li, D.; Li, Y.; Shi, X.; Meng, D.; Pan, J.; Ou, G.; Aneja, R.; et al. CYLD mediates ciliogenesis in multiple organs by deubiquitinating Cep70 and inactivating HDAC6. Cell Res. 2014, 24, 1342–1353. [Google Scholar] [CrossRef] [Green Version]
- Mockel, A.; Obringer, C.; Hakvoort, T.; Seeliger, M.; Lamers, W.; Stoetzel, C.; Dollfus, H.; Marion, V. Pharmacological modulation of the retinal unfolded protein response in Bardet-Biedl syndrome reduces apoptosis and preserves light detection ability. J. Biol. Chem. 2012, 287, 37483–37494. [Google Scholar] [CrossRef]
- Ramsbottom, S.; Molinari, E.; Srivastava, S.; Silberman, F.; Henry, C.; Alkanderi, S.; Devlin, L.; White, K.; Steel, D.; Saunier, S.; et al. Targeted exon skipping of a CEP290 mutation rescues Joubert syndrome phenotypes in vitro and in a murine model. Proc. Natl. Acad. Sci. USA 2018, 115, 12489–12494. [Google Scholar]
- Green, W.; Uytingco, C.; Ukhanov, K.; Kolb, Z.; Moretta, J.; McIntyre, J.; Martens, J. Peripheral Gene Therapeutic Rescue of an Olfactory Ciliopathy Restores Sensory Input, Axonal Pathfinding, and Odor-Guided Behavior. J. Neurosci. 2018, 38, 7462–7475. [Google Scholar] [CrossRef]
- Shivanna, M.; Anand, M.; Chakrabarti, S.; Khanna, H. Ocular Ciliopathies: Genetic and mechanistic insights into developing therapies. Curr. Med. Chem. 2018. [Google Scholar] [CrossRef]
- Stayner, C.; Brooke, D.; Bates, M.; Eccles, M. Targeted Therapies for Autosomal Dominant Polycystic Kidney Disease. Curr. Med. Chem. 2018. [Google Scholar] [CrossRef]
- Uytingco, C.; Green, W.; Martens, J. Olfactory loss and dysfunction in ciliopathies: Molecular mechanisms and potential therapies. Curr. Med. Chem. 2018. [Google Scholar] [CrossRef]
- Zhu, P.; Sieben, C.; Xu, X.; Harris, P.; Lin, X. Autophagy activators suppress cystogenesis in an autosomal dominant polycystic kidney disease model. Hum. Mol. Genet. 2017, 26, 158–172. [Google Scholar] [CrossRef]
- Tobin, J.; Beales, P. Restoration of renal function in zebrafish models of ciliopathies. Pediatr. Nephrol. 2008, 23, 2095–2099. [Google Scholar] [CrossRef] [Green Version]
- Heitman, J.; Movva, N.; Hall, M. Targets for cell cycle arrest by the immunosuppressant rapamycin in yeast. Science 1991, 253, 905–909. [Google Scholar] [CrossRef]
- Kunz, J.; Henriquez, R.; Schneider, U.; Deuter-Reinhard, M.; Movva, N.; Hall, M. Target of rapamycin in yeast, TOR2, is an essential phosphatidylinositol kinase homolog required for G1 progression. Cell 1993, 73, 585–596. [Google Scholar] [CrossRef]
- Brown, E.; Albers, M.; Shin, T.; Ichikawa, K.; Keith, C.; Lane, W.; Schreiber, S. A mammalian protein targeted by G1-arresting rapamycin-receptor complex. Nature 1994, 369, 756–758. [Google Scholar] [CrossRef]
- Chang, Y.; Juhász, G.; Goraksha-Hicks, P.; Arsham, A.; Mallin, D.; Muller, L.; Neufeld, T. Nutrient-dependent regulation of autophagy through the target of rapamycin pathway. Biochem. Soc. Trans. 2009, 37, 232–236. [Google Scholar] [CrossRef]
- Ganley, I.; Lam, D.; Wang, J.; Ding, X.; Chen, S.; Jiang, X. ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J. Biol. Chem. 2009, 284, 12297–12305. [Google Scholar] [CrossRef]
- Jung, C.; Jun, C.; Ro, S.; Kim, Y.; Otto, N.; Cao, J.; Kundu, M.; Kim, D. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol. Biol. Cell 2009, 20, 1992–2003. [Google Scholar] [CrossRef]
- Hosokawa, N.; Hara, T.; Kaizuka, T.; Kishi, C.; Takamura, A.; Miura, Y.; Iemura, S.; Natsume, T.; Takehana, K.; Yamada, N.; et al. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol. Biol. Cell 2009, 20, 1981–1991. [Google Scholar] [CrossRef]
- Sarkar, S.; Floto, R.; Berger, Z.; Imarisio, S.; Cordenier, A.; Pasco, M.; Cook, L.; Rubinsztein, D. Lithium induces autophagy by inhibiting inositol monophosphatase. J. Cell Biol. 2005, 170, 1101–1111. [Google Scholar] [CrossRef] [Green Version]
- Williams, A.; Sarkar, S.; Cuddon, P.; Ttofi, E.; Saiki, S.; Siddiqi, F.; Jahreiss, L.; Fleming, A.; Pask, D.; Goldsmith, P.; et al. Novel targets for Huntington’s disease in an mTOR-independent autophagy pathway. Nat. Chem. Biol. 2008, 4, 295–305. [Google Scholar] [CrossRef]
- Stayner, C.; Shields, J.; Slobbe, L.; Shillingford, J.; Weimbs, T.; Eccles, M. Rapamycin-mediated suppression of renal cyst expansion in del34 Pkd1-/- mutant mouse embryos: An investigation of the feasibility of renal cyst prevention in the foetus. Nephrology (Carlton) 2012, 17, 739–747. [Google Scholar] [CrossRef]
- Shillingford, J.; Murcia, N.; Larson, C.; Low, S.; Hedgepeth, R.; Brown, N.; Flask, C.; Novick, A.; Goldfarb, D.; Kramer-Zucker, A.; et al. The mTOR pathway is regulated by polycystin-1, and its inhibition reverses renal cystogenesis in polycystic kidney disease. Proc. Natl. Acad. Sci. USA 2006, 103, 5466–5471. [Google Scholar] [CrossRef] [Green Version]
- Gattone, V.N.; Sinders, R.; Hornberger, T.; Robling, A. Late progression of renal pathology and cyst enlargement is reduced by rapamycin in a mouse model of nephronophthisis. Kidney Int. 2009, 76, 178–182. [Google Scholar] [CrossRef] [Green Version]
- Ravichandran, K.; Zafar, I.; Ozkok, A.; Edelstein, C. An mTOR kinase inhibitor slows disease progression in a rat model of polycystic kidney disease. Nephrol. Dial. Transplant. 2015, 30, 45–53. [Google Scholar] [CrossRef]
- Tao, Y.; Kim, J.; Schrier, R.; Edelstein, C. Rapamycin markedly slows disease progression in a rat model of polycystic kidney disease. J. Am. Soc. Nephrol. 2005, 16, 46–51. [Google Scholar] [CrossRef]
- Wahl, P.; Serra, A.; Le Hir, M.; Molle, K.; Hall, M.; Wüthrich, R. Inhibition of mTOR with sirolimus slows disease progression in Han:SPRD rats with autosomal dominant polycystic kidney disease (ADPKD). Nephrol. Dial. Transplant. 2006, 21, 598–604. [Google Scholar] [CrossRef]
- Zhang, T.; Wang, L.; Xiong, X.; Mao, Z.; Wang, L.; Mei, C. Mycophenolate mofetil versus Rapamycin in Han: SPRD rats with Polycystic Kidney Disease. Biol. Res. 2009, 42, 437–444. [Google Scholar] [CrossRef]
- Liu, Y.; Shao, Y.; He, Q. Sirolimus for treatment of autosomal-dominant polycystic kidney disease: A meta-analysis of randomized controlled trials. Transplant. Proc. 2014, 46, 66–74. [Google Scholar] [CrossRef]
- Stallone, G.; Infante, B.; Grandaliano, G.; Bristogiannis, C.; Macarini, L.; Mezzopane, D.; Bruno, F.; Montemurno, E.; Schirinzi, A.; Sabbatini, M.; et al. Rapamycin for treatment of type I autosomal dominant polycystic kidney disease (RAPYD-study): A randomized, controlled study. Nephrol. Dial. Transplant. 2012, 27, 3560–3567. [Google Scholar] [CrossRef]
- Anandh, U.; Chandrasekar, G.; Agarwal, V. Mammalian target of rapamycin inhibitors in a patient with polycystic kidney disease-1-tuberous sclerosis-2 contiguous gene syndrome. Saudi J. Kidney Dis. Transpl. 2018, 29, 1475–1479. [Google Scholar] [CrossRef]
- Li, A.; Fan, S.; Xu, Y.; Meng, J.; Shen, X.; Mao, J.; Zhang, L.; Zhang, X.; Moeckel, G.; Wu, D.; et al. Rapamycin treatment dose-dependently improves the cystic kidney in a new ADPKD mouse model via the mTORC1 and cell-cycle-associated CDK1/cyclin axis. J. Cell. Mol. Med. 2017, 21, 1619–1635. [Google Scholar] [CrossRef]
- Novalic, Z.; van der Wal, A.; Leonhard, W.; Koehl, G.; Breuning, M.; Geissler, E.; de Heer, E.; Peters, D. Dose-dependent effects of sirolimus on mTOR signaling and polycystic kidney disease. J. Am. Soc. Nephrol. 2012, 23, 842–853. [Google Scholar] [CrossRef]
- Korolchuk, V.; Menzies, F.; Rubinsztein, D. Mechanisms of cross-talk between the ubiquitin-proteasome and autophagy-lysosome systems. FEBS Lett. 2010, 584, 1393–1398. [Google Scholar] [CrossRef]
- Korolchuk, V.; Menzies, F.; Rubinsztein, D. A novel link between autophagy and the ubiquitin-proteasome system. Autophagy 2009, 5, 862–863. [Google Scholar] [CrossRef] [Green Version]
- Nam, T.; Han, J.; Devkota, S.; Lee, H. Emerging Paradigm of Crosstalk between Autophagy and the Ubiquitin-Proteasome System. Mol. Cells 2017, 40, 897–905. [Google Scholar]
- Ji, C.; Kwon, Y. Crosstalk and Interplay between the Ubiquitin-Proteasome System and Autophagy. Mol. Cells 2017, 40, 441–449. [Google Scholar] [Green Version]
- Kocaturk, N.; Gozuacik, D. Crosstalk Between Mammalian Autophagy and the Ubiquitin-Proteasome System. Front. Cell Dev. Biol. 2018, 6, 128. [Google Scholar] [CrossRef]
- Tang, Z.; Lin, M.; Stowe, T.; Chen, S.; Zhu, M.; Stearns, T.; Franco, B.; Zhong, Q. Autophagy promotes primary ciliogenesis by removing OFD1 from centriolar satellites. Nature 2013, 502, 254–257. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Livingston, M.; Su, Y.; Dong, Z. Reciprocal regulation of cilia and autophagy via the MTOR and proteasome pathways. Autophagy 2015, 11, 607–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pampliega, O.; Orhon, I.; Patel, B.; Sridhar, S.; Díaz-Carretero, A.; Beau, I.; Codogno, P.; Satir, B.; Satir, P.; Cuervo, A. Functional interaction between autophagy and ciliogenesis. Nature 2013, 502, 194–200. [Google Scholar] [CrossRef]
- Struchtrup, A.; Wiegering, A.; Stork, B.; Rüther, U.; Gerhardt, C. The ciliary protein RPGRIP1L governs autophagy independently of its proteasome-regulating function at the ciliary base in mouse embryonic fibroblasts. Autophagy 2018, 14, 567–583. [Google Scholar] [CrossRef] [Green Version]
- Mahuzier, A.; Gaudé, H.; Grampa, V.; Anselme, I.; Silbermann, F.; Leroux-Berger, M.; Delacour, D.; Ezan, J.; Montcouquiol, M.; Saunier, S.; et al. Dishevelled stabilization by the ciliopathy protein Rpgrip1l is essential for planar cell polarity. J. Cell Biol. 2012, 198, 927–940. [Google Scholar] [CrossRef] [Green Version]
- Wiegering, A.; Dildrop, R.; Kalfhues, L.; Spychala, A.; Kuschel, S.; Lier, J.; Zobel, T.; Dahmen, S.; Leu, T.; Struchtrup, A.; et al. Cell type-specific regulation of ciliary transition zone assembly in vertebrates. EMBO J. 2018, 37, e97791. [Google Scholar] [CrossRef]
- Borgal, L.; Habbig, S.; Hatzold, J.; Liebau, M.; Dafinger, C.; Sacarea, I.; Hammerschmidt, M.; Benzing, T.; Schermer, B. The Ciliary Protein Nephrocystin-4 Translocates the Canonical Wnt-Regulator Jade-1 to the Nucleus to Negatively Regulate Beta-Catenin Signaling. J. Biol. Chem. 2012, 287, 25370–25380. [Google Scholar] [CrossRef]
- Schmid, F.; Schou, K.; Vilhelm, M.; Holm, M.; Breslin, L.; Farinelli, P.; Larsen, L.; Andersen, J.; Pedersen, L.; Christensen, S. IFT20 modulates ciliary PDGFRα signaling by regulating the stability of Cbl E3 ubiquitin ligases. J. Cell Biol. 2018, 217, 151–161. [Google Scholar] [CrossRef]
- Saxton, R.; Sabatini, D. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Nicholatos, J.; Dreier, J.; Ricoult, S.; Widenmaier, S.; Hotamisligil, G.; Kwiatkowski, D.; Manning, B. Coordinated regulation of protein synthesis and degradation by mTORC1. Nature 2014, 513, 440–443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Manning, B. Zhang & Manning reply. Nature 2016, 529, E2–E3. [Google Scholar] [PubMed] [Green Version]
- Zhao, J.; Garcia, G.; Goldberg, A. Control of proteasomal proteolysis by mTOR. Nature 2016, 529, E1–E2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
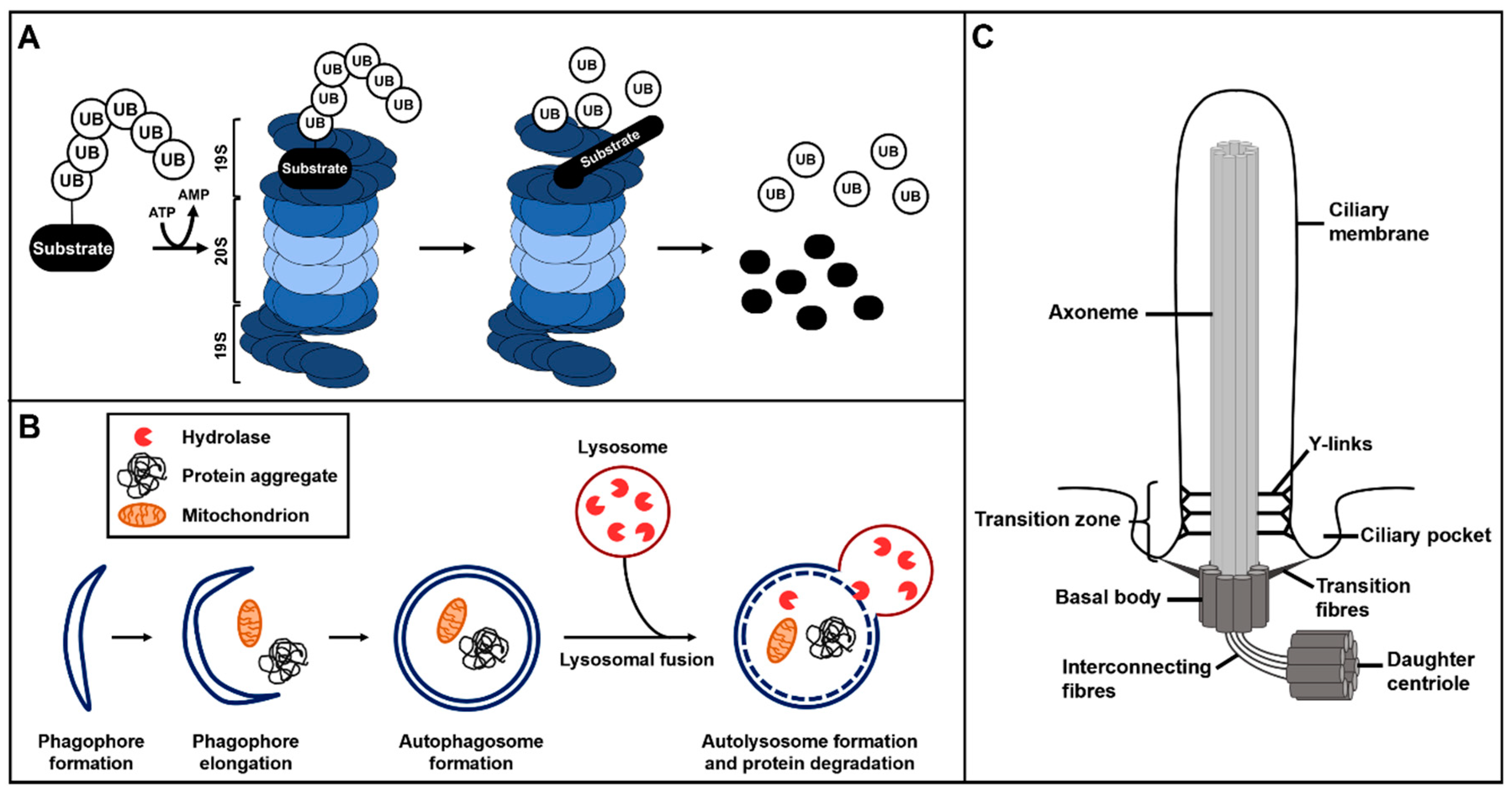
Figure 1.
Overview of ubiquitin–proteasome system (UPS) protein degradation, autophagy and primary cilium structure. (A) The ubiquitin–proteasome system. The ubiquitinated substrate is recognised by the 19S regulatory subunit of the 26S proteasome and gets degraded or proteolytically processed by the 20S subunit of the 26S proteasome. (B) Autophagy starts with the formation of phagophores which subsequently elongate to finally develop into autophagosomes. During autophagosome formation, target proteins and structures become enclosed in the autophagosomes. These autophagosomes fuse with lysosomes and the hydrolases of the lysosomes degrade the target proteins and structures. (C) Primary cilia consist of a microtubule scaffold called axoneme. The axoneme is surrounded by the ciliary membrane and grows out of the basal body. The basal body is a modified mother centriole that is connected to the daughter centriole by interconnecting fibres. The basal body is attached to the ciliary membrane in the region of the ciliary pocket via transition fibres. The transition zone, with its Y-links, is located at the proximal part of the axoneme.
Figure 1.
Overview of ubiquitin–proteasome system (UPS) protein degradation, autophagy and primary cilium structure. (A) The ubiquitin–proteasome system. The ubiquitinated substrate is recognised by the 19S regulatory subunit of the 26S proteasome and gets degraded or proteolytically processed by the 20S subunit of the 26S proteasome. (B) Autophagy starts with the formation of phagophores which subsequently elongate to finally develop into autophagosomes. During autophagosome formation, target proteins and structures become enclosed in the autophagosomes. These autophagosomes fuse with lysosomes and the hydrolases of the lysosomes degrade the target proteins and structures. (C) Primary cilia consist of a microtubule scaffold called axoneme. The axoneme is surrounded by the ciliary membrane and grows out of the basal body. The basal body is a modified mother centriole that is connected to the daughter centriole by interconnecting fibres. The basal body is attached to the ciliary membrane in the region of the ciliary pocket via transition fibres. The transition zone, with its Y-links, is located at the proximal part of the axoneme.

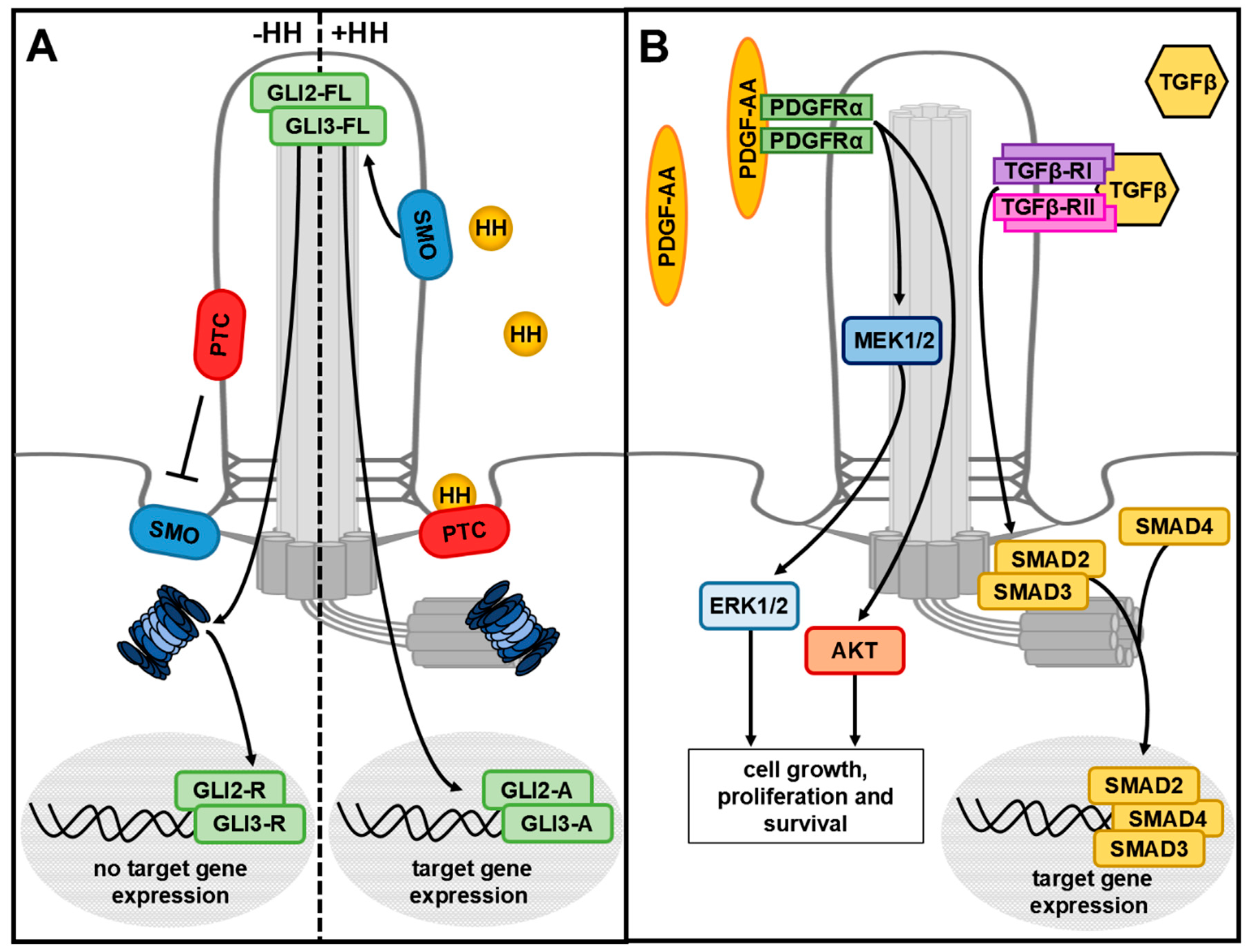
Figure 2.
Cilia-mediated HH, PDGFRα and TGFβ signalling. (A) Without the HH ligand, the receptor PTC inhibits ciliary entry of SMO. GLI2-FL and GLI3-FL are proteolytically processed into the repressor forms GLI2-R and GLI3-R by the ciliary proteasome. They translocate into the nucleus and inhibit HH target gene expression. In the presence of HH, HH binds to its receptor PTC and the HH/PTC complex leaves the cilium. SMO enters the cilium and GLI2-FL and GLI3-FL become activated and, in turn, initiate HH target gene expression. (B) PDGF-AA binds to the ciliary receptor PDGFRα and activates AKT signalling or the MEK1/2–ERK1/2 signalling cascade. TGFβ binds to a heterotetrameric receptor composed of TGFβ-RI and TGFβ-RII in the ciliary membrane. At the base of cilia, the signal is transduced via different SMAD proteins.
Figure 2.
Cilia-mediated HH, PDGFRα and TGFβ signalling. (A) Without the HH ligand, the receptor PTC inhibits ciliary entry of SMO. GLI2-FL and GLI3-FL are proteolytically processed into the repressor forms GLI2-R and GLI3-R by the ciliary proteasome. They translocate into the nucleus and inhibit HH target gene expression. In the presence of HH, HH binds to its receptor PTC and the HH/PTC complex leaves the cilium. SMO enters the cilium and GLI2-FL and GLI3-FL become activated and, in turn, initiate HH target gene expression. (B) PDGF-AA binds to the ciliary receptor PDGFRα and activates AKT signalling or the MEK1/2–ERK1/2 signalling cascade. TGFβ binds to a heterotetrameric receptor composed of TGFβ-RI and TGFβ-RII in the ciliary membrane. At the base of cilia, the signal is transduced via different SMAD proteins.

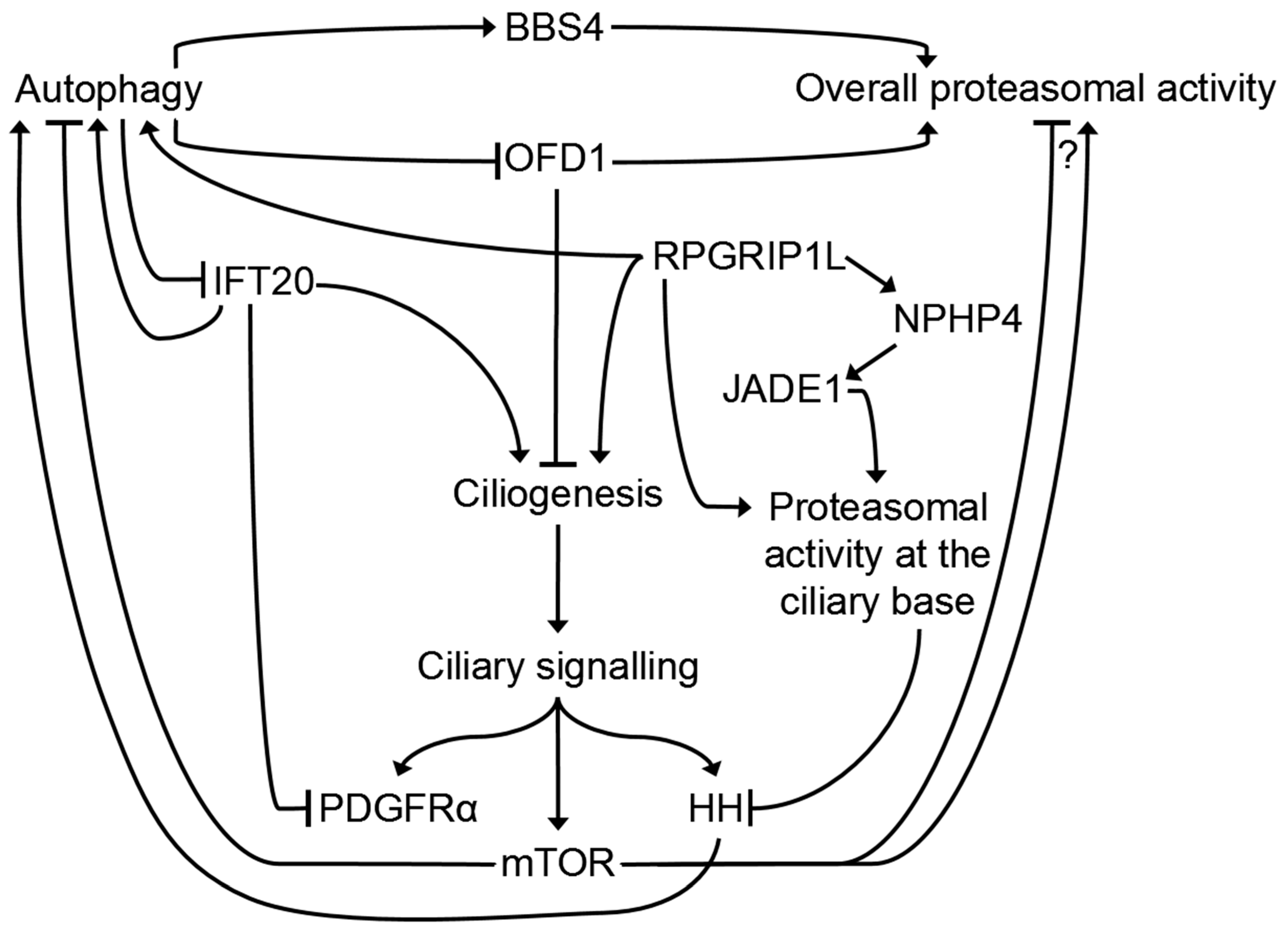
Figure 3.
Complex network of the cilia-related crosstalk between the UPS and autophagy. Autophagy is able to regulate overall proteasomal activity in a positive or negative manner via BBS4 or OFD1. In addition, autophagy regulates ciliogenesis via OFD1 and/or IFT20 and thereby affects ciliary signalling. In turn, several cilia-mediated signalling cascades, like HH and mTOR signalling, modulate autophagy. Moreover, mTOR signalling regulates the overall proteasomal activity either positively or negatively (which is a matter of fierce debate). Additional cilia-associated proteins, like IFT20 or RPGRIP1L, regulate ciliogenesis as well as autophagy, whereby RPGRIP1L is also able to regulate the activity of the ciliary proteasome.
Figure 3.
Complex network of the cilia-related crosstalk between the UPS and autophagy. Autophagy is able to regulate overall proteasomal activity in a positive or negative manner via BBS4 or OFD1. In addition, autophagy regulates ciliogenesis via OFD1 and/or IFT20 and thereby affects ciliary signalling. In turn, several cilia-mediated signalling cascades, like HH and mTOR signalling, modulate autophagy. Moreover, mTOR signalling regulates the overall proteasomal activity either positively or negatively (which is a matter of fierce debate). Additional cilia-associated proteins, like IFT20 or RPGRIP1L, regulate ciliogenesis as well as autophagy, whereby RPGRIP1L is also able to regulate the activity of the ciliary proteasome.

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Wiegering, A.; Rüther, U.; Gerhardt, C. The Role of Primary Cilia in the Crosstalk between the Ubiquitin–Proteasome System and Autophagy. Cells 2019, 8, 241. https://doi.org/10.3390/cells8030241
AMA Style
Wiegering A, Rüther U, Gerhardt C. The Role of Primary Cilia in the Crosstalk between the Ubiquitin–Proteasome System and Autophagy. Cells. 2019; 8(3):241. https://doi.org/10.3390/cells8030241
Chicago/Turabian StyleWiegering, Antonia, Ulrich Rüther, and Christoph Gerhardt. 2019. "The Role of Primary Cilia in the Crosstalk between the Ubiquitin–Proteasome System and Autophagy" Cells 8, no. 3: 241. https://doi.org/10.3390/cells8030241
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.