Tanshinone IIA Restores Dynamic Balance of Autophagosome/Autolysosome in Doxorubicin-Induced Cardiotoxicity via Targeting Beclin1/LAMP1

 ,
,
Abstract
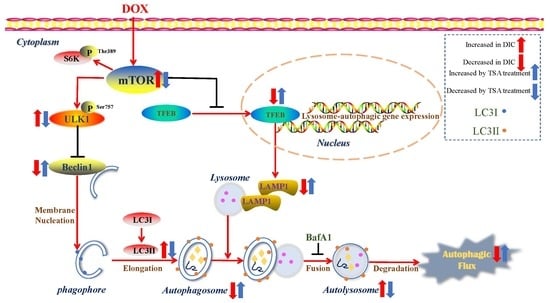
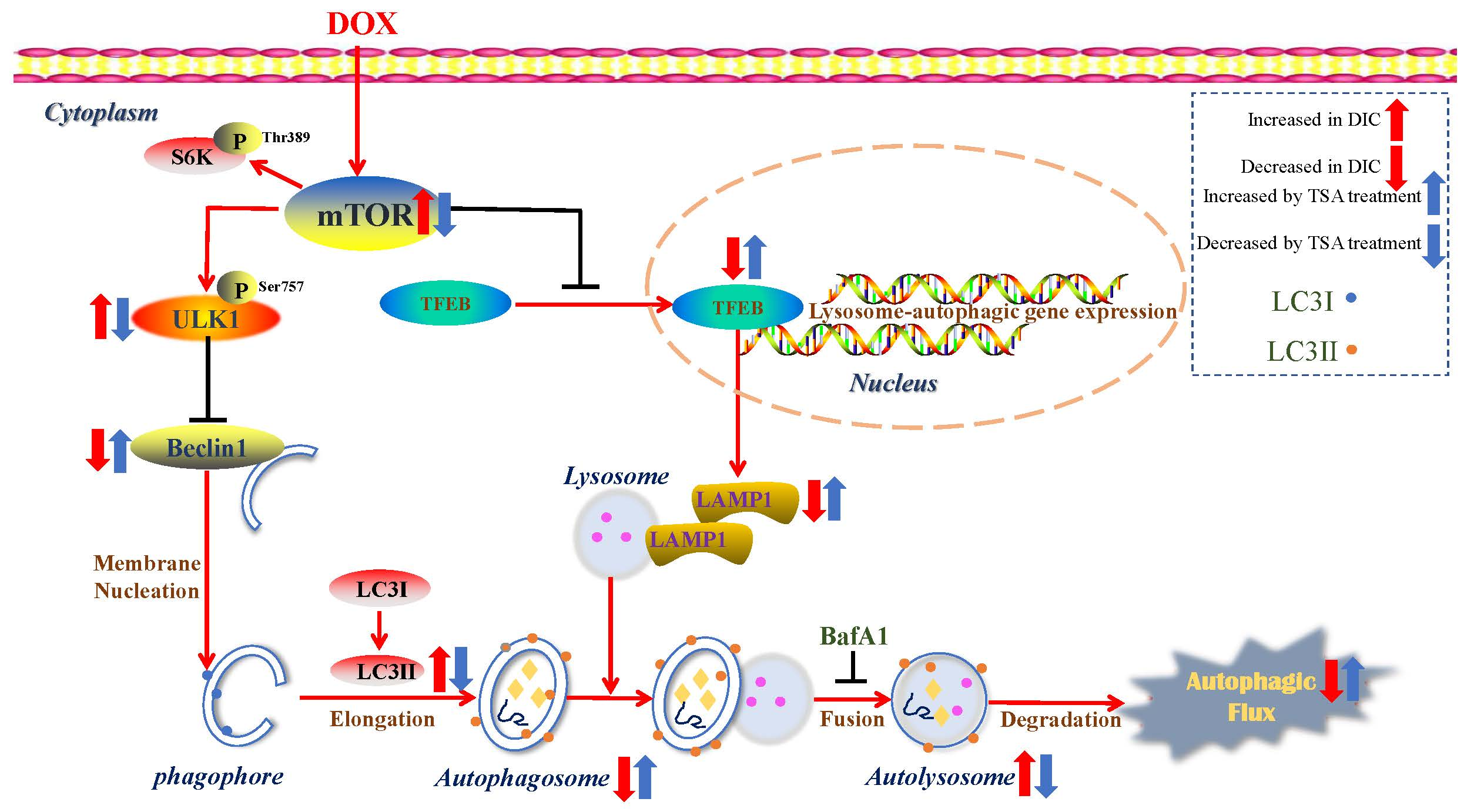
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
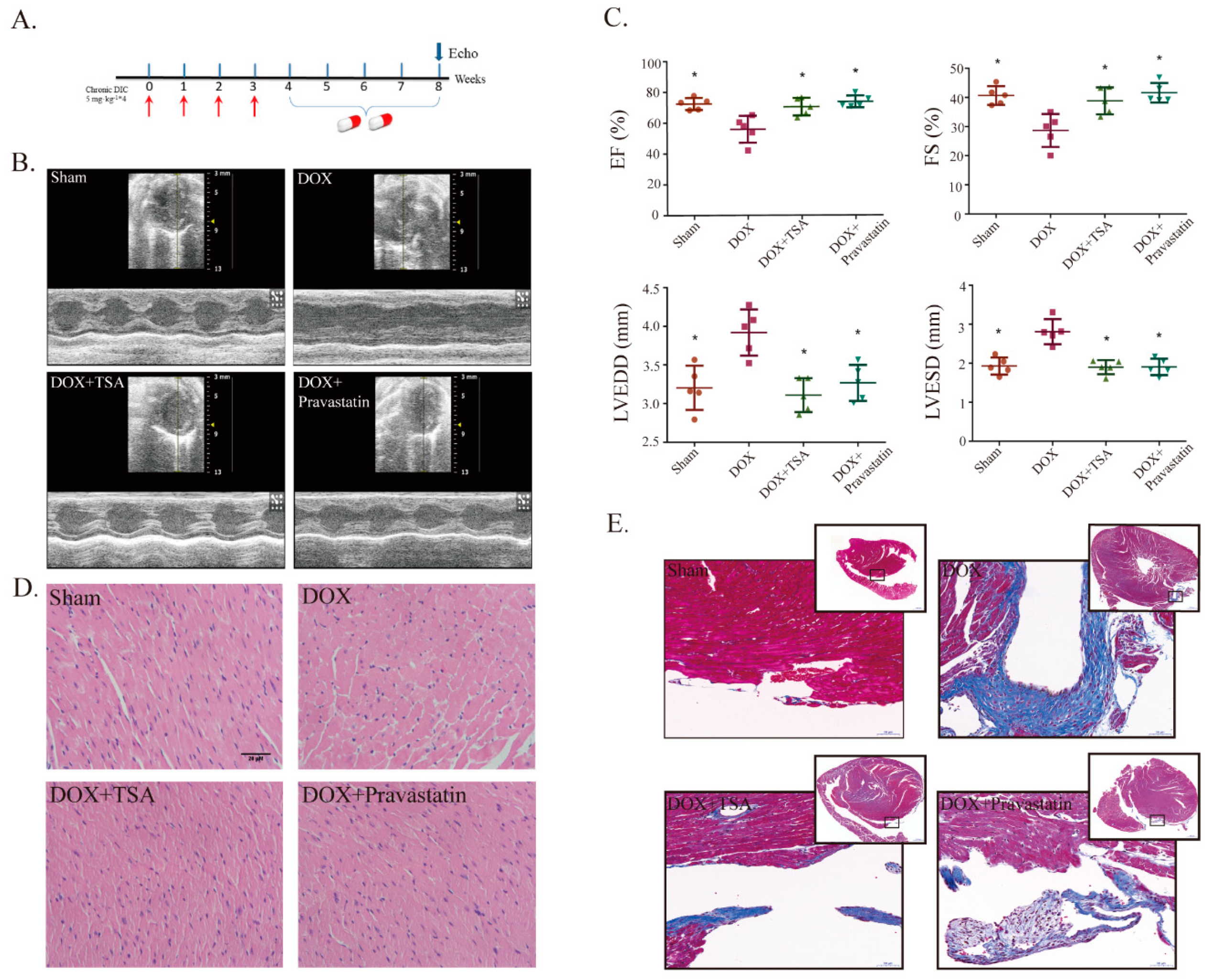
2.1. Effects of TSA on Cardiac Function and Structural Alterations in a DIC Mouse Model
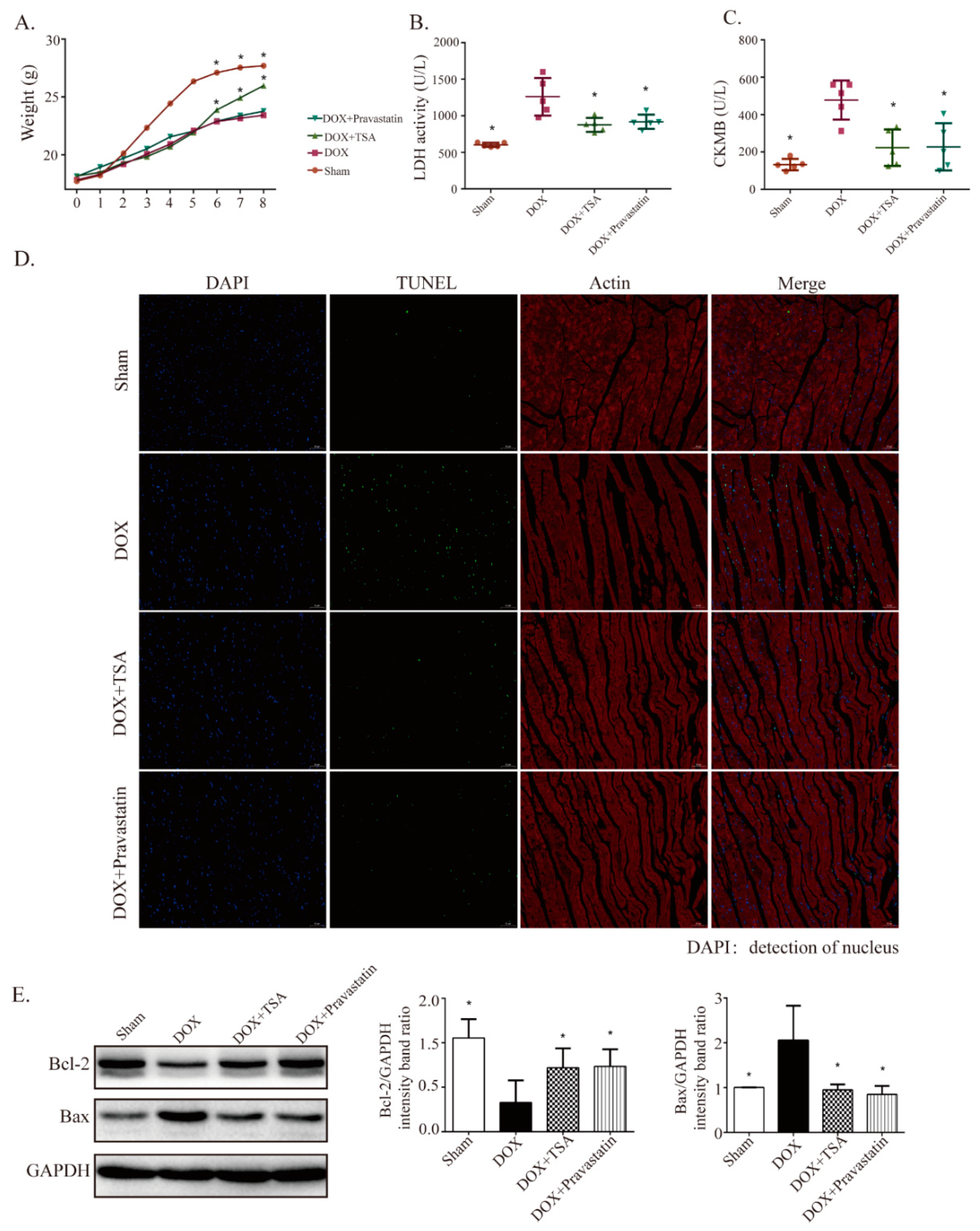
2.2. Effect of TSA on Body Weight, Serum Lactate Dehydrogenase (LDH), Creatine Kinase of Muscle–Brain Type (CKMB), and Apoptosis in the DIC Mouse Model
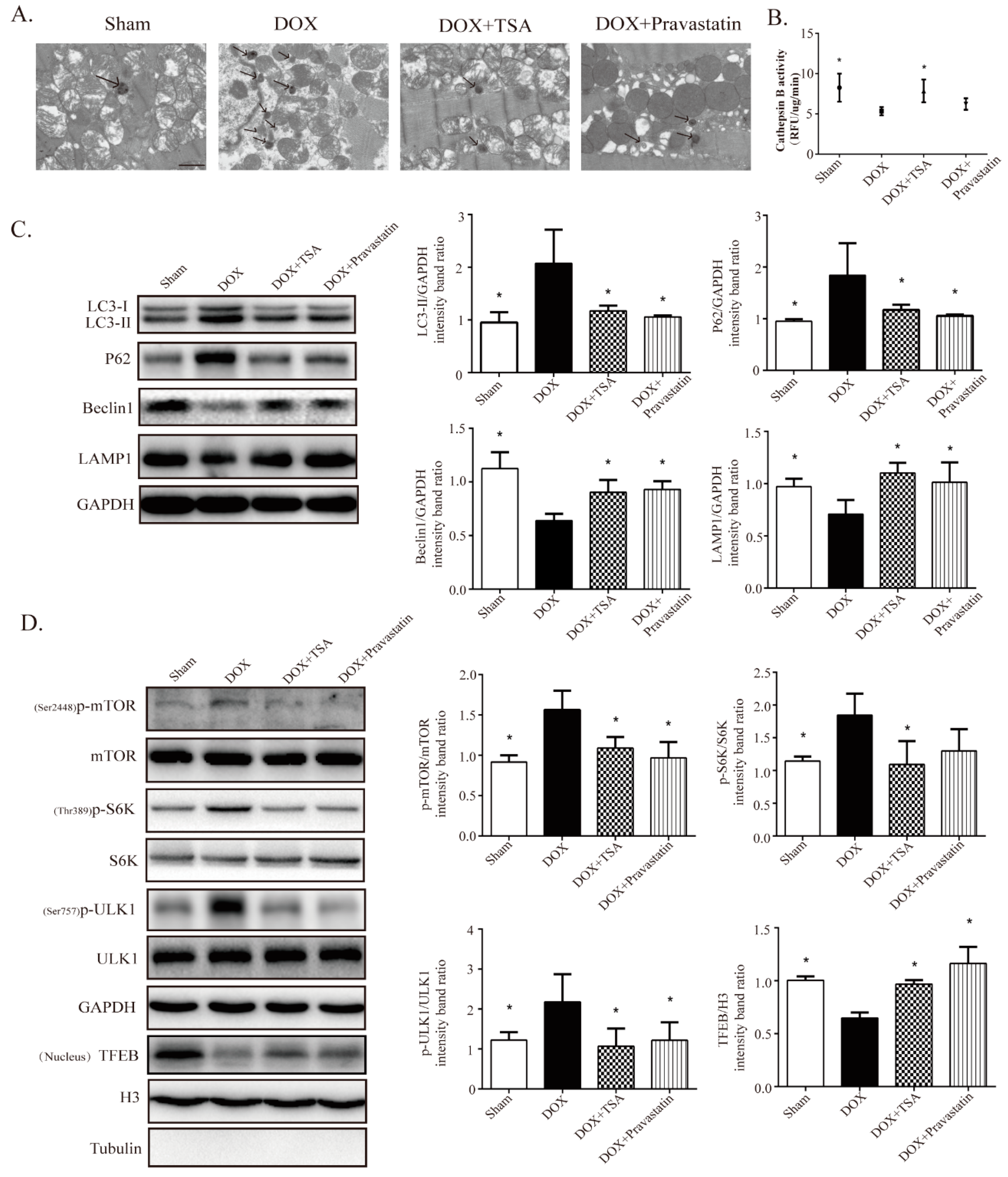
2.3. TSA Restores Autophagy in the DIC Mouse Model
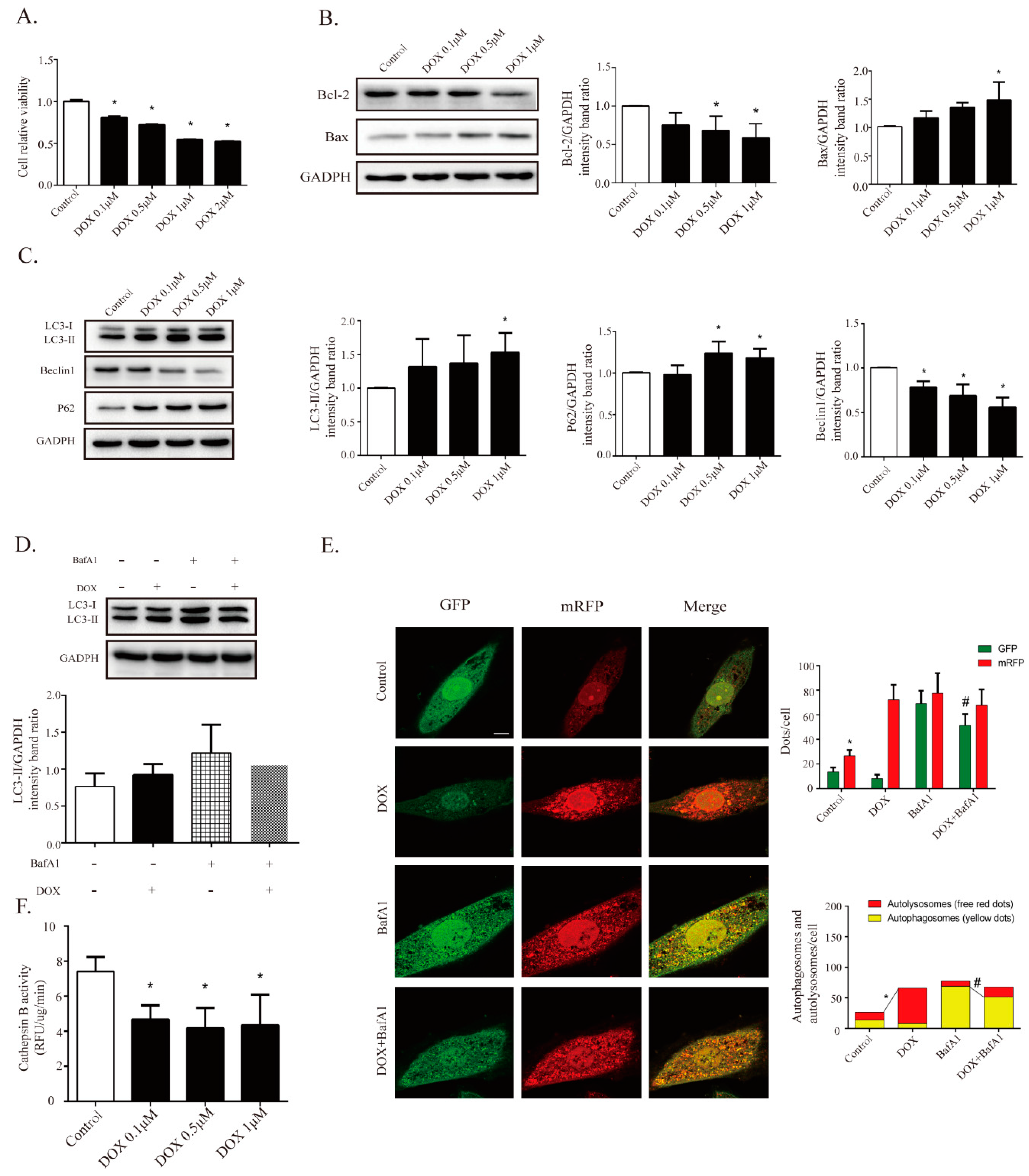
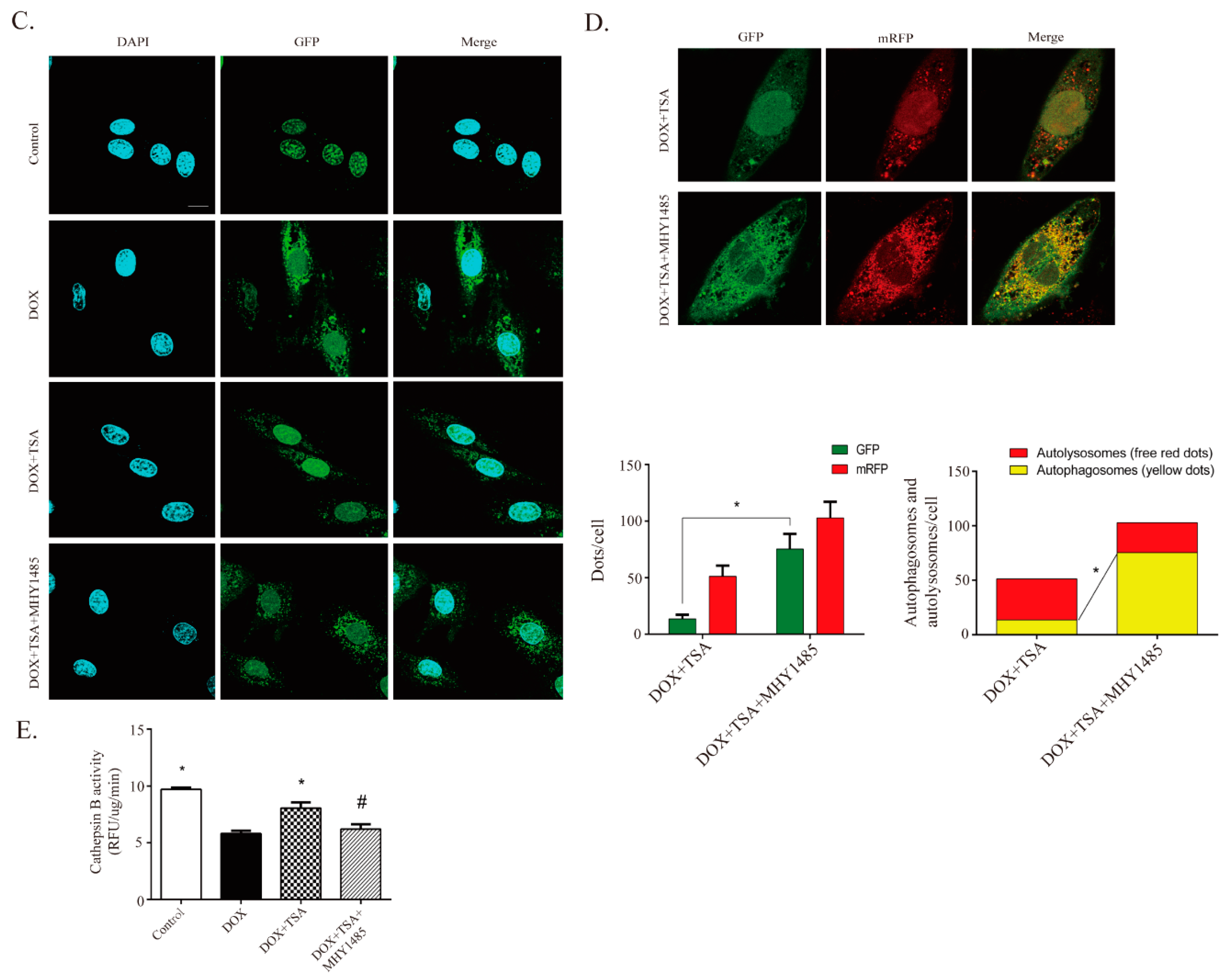
2.4. DOX Inhibits Autophagic Flux and Lysosomal Proteolysis In Vitro
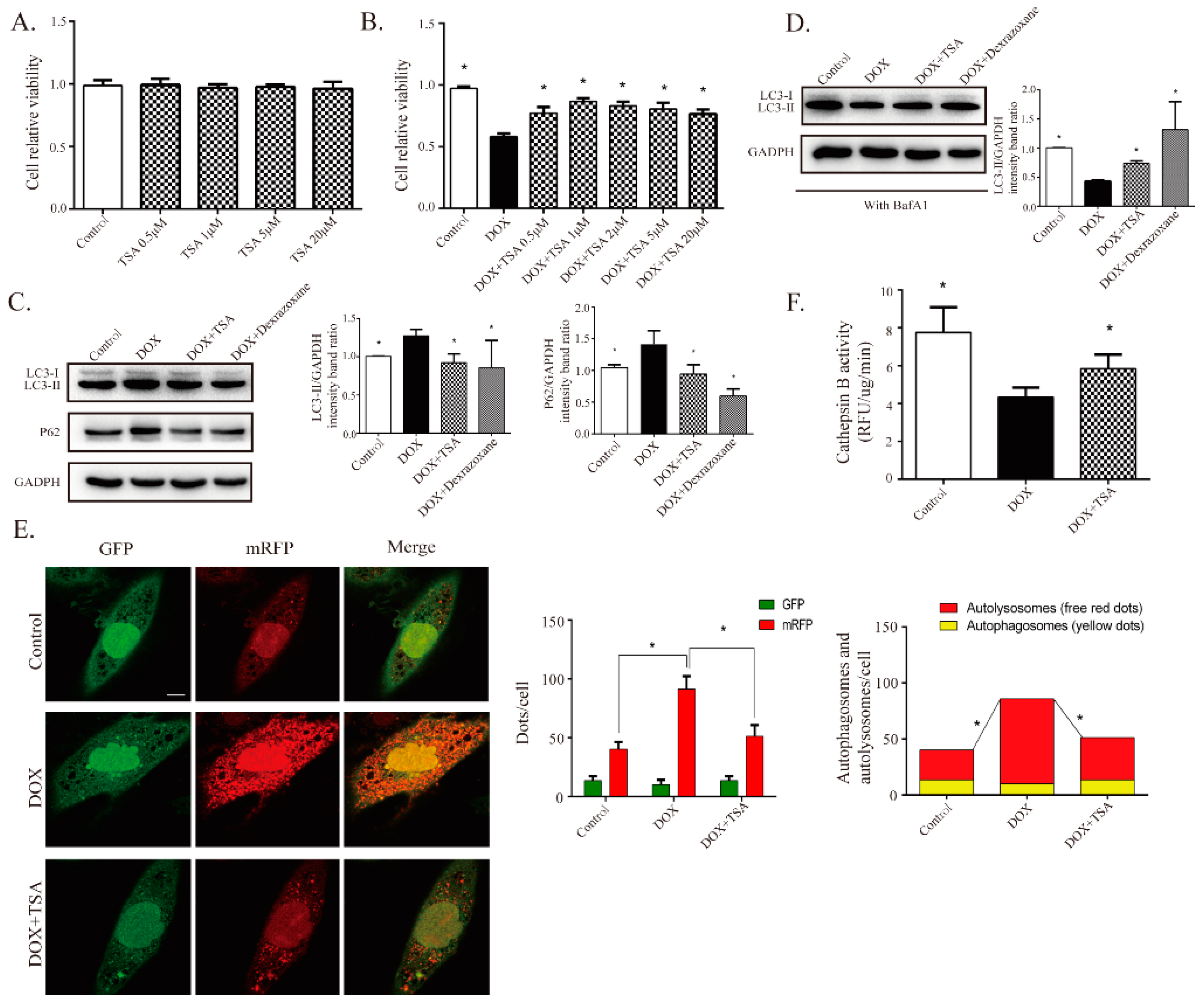
2.5. TSA Promotes Autophagosome Formation and Lysosomal Proteolysis
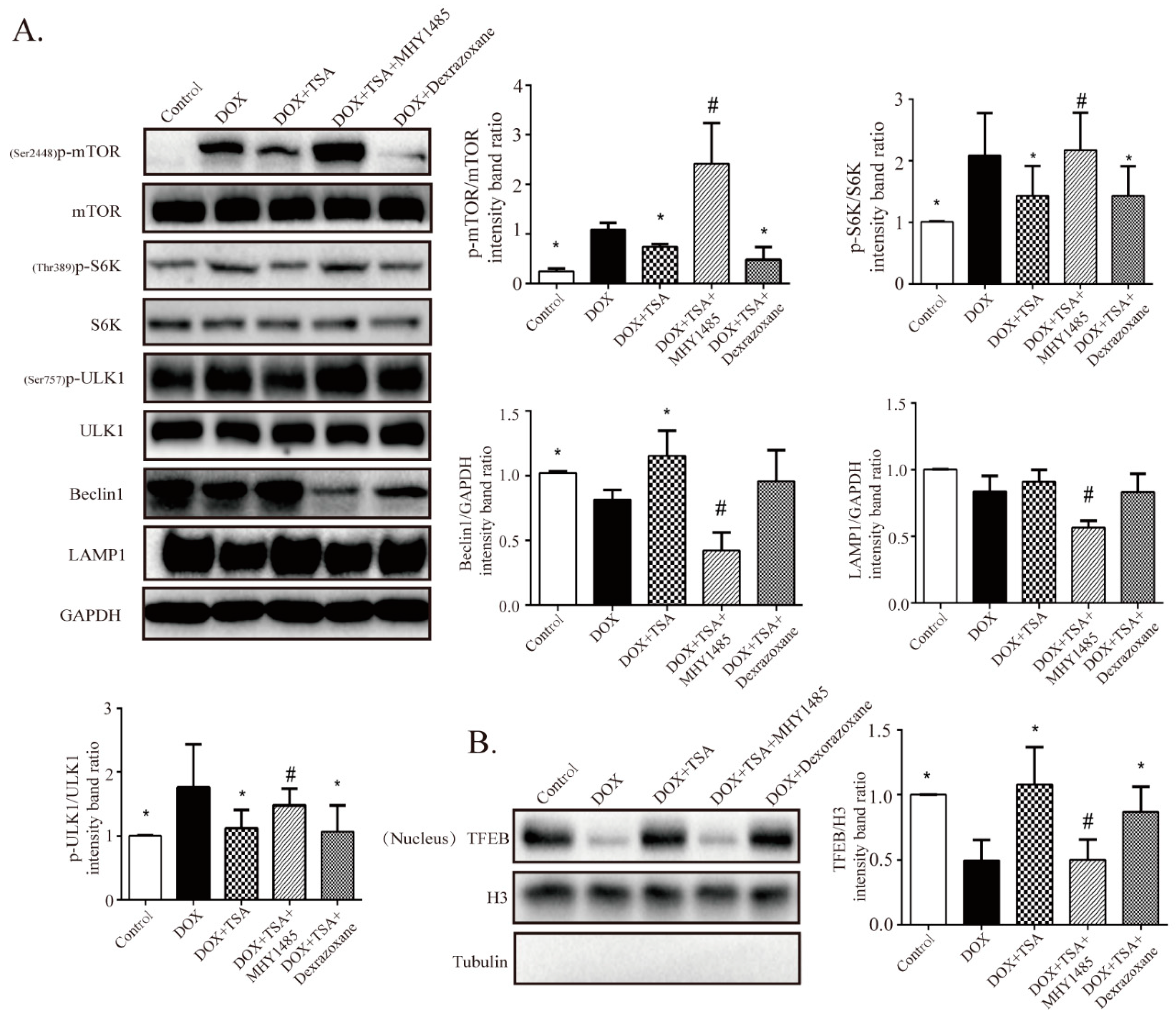
2.6. TSA Enhances Autophagic Flux by Regulating the Beclin1/LAMP1 Signaling Pathway
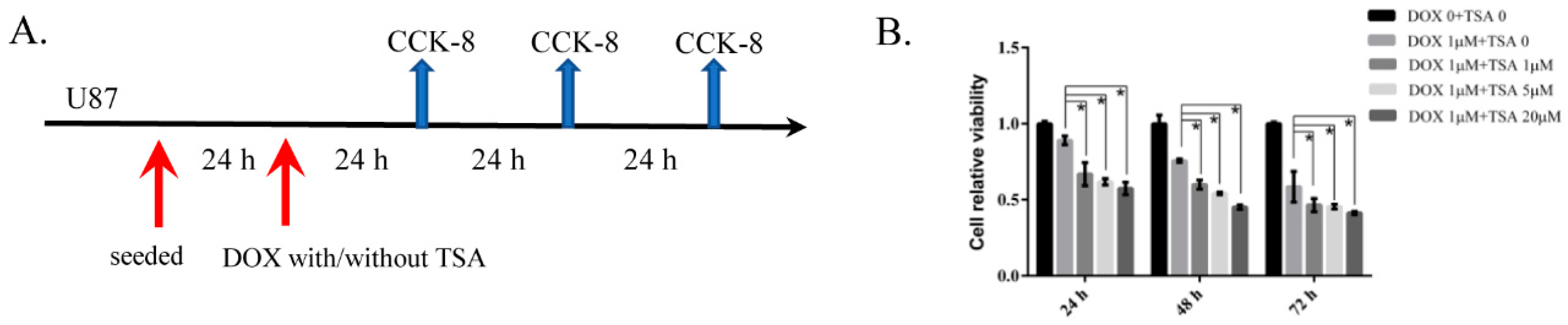
2.7. TSA Reduces Cardiotoxicity without Compromising the Antitumor Activity of DOX
3. Discussion
4. Materials and Methods
4.1. Establishment of a DIC Model in Zebrafish
4.2. Establishment of a DIC Model in Mice
4.3. Echocardiographic Assessment of Cardiac Functions
4.4. Measurement of LDH and CKMB
4.5. Histological Examination and Masson Staining
4.6. TUNEL Staining
4.7. Electron Microscopy
4.8. Western Blot Analysis
4.9. Establishment of DOX-Induced H9C2 Cell Injury Model and Cell Viability
4.10. Cell Viability Analysis for U87 Cells
4.11. Assessment of Apoptosis by a Hoechst 33258 Staining Kit
4.12. Cathepsin B Activity Assay
4.13. GFP-mRFP-LC3 Adenovirus Transfection
4.14. GFP-TFEB Lentiviral Vector Transfection
4.15. Data and Statistical Analysis
4.16. Materials
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Li, D.L.; Wang, Z.V.; Ding, G.; Tan, W.; Luo, X.; Criollo, A.; Xie, M.; Jiang, N.; May, H.; Kyrychenko, V.; et al. Doxorubicin Blocks Cardiomyocyte Autophagic Flux by Inhibiting Lysosome Acidification. Circulation 2016, 133, 1668–1687. [Google Scholar] [CrossRef] [Green Version]
- Shaikh, F.; Dupuis, L.L.; Alexander, S.; Gupta, A.; Mertens, L.; Nathan, P.C. Cardioprotection and Second Malignant Neoplasms Associated with Dexrazoxane in Children Receiving Anthracycline Chemotherapy: A Systematic Review and Meta-Analysis. J. Natl. Cancer Inst. 2016, 108. [Google Scholar] [CrossRef]
- Tahover, E.; Segal, A.; Isacson, R.; Rosengarten, O.; Grenader, T.; Gips, M.; Cherny, N.; Heching, N.I.; Mesika, L.; Catane, R.; et al. Dexrazoxane added to doxorubicin-based adjuvant chemotherapy of breast cancer: A retrospective cohort study with a comparative analysis of toxicity and survival. Anticancer Drugs 2017, 28, 787–794. [Google Scholar] [CrossRef]
- Mitry, M.A.; Edwards, J.G. Doxorubicin induced heart failure: Phenotype and molecular mechanisms. Int. J. Cardiol. Heart Vasc. 2016, 10, 17–24. [Google Scholar] [CrossRef]
- Orogo, A.M.; Gustafsson, A.B. Therapeutic targeting of autophagy: Potential and concerns in treating cardiovascular disease. Circ. Res. 2015, 116, 489–503. [Google Scholar] [CrossRef]
- Miyamoto, S. Autophagy and cardiac aging. Cell Death Differ. 2019, 26, 653–664. [Google Scholar] [CrossRef] [Green Version]
- Bartlett, J.J.; Trivedi, P.C.; Pulinilkunnil, T. Autophagic dysregulation in doxorubicin cardiomyopathy. J. Mol. Cell Cardiol. 2017, 104, 1–8. [Google Scholar] [CrossRef]
- Gurkar, A.U.; Chu, K.; Raj, L.; Bouley, R.; Lee, S.H.; Kim, Y.B.; Dunn, S.E.; Mandinova, A.; Lee, S.W. Identification of ROCK1 kinase as a critical regulator of Beclin1-mediated autophagy during metabolic stress. Nat. Commun. 2013, 4, 2189. [Google Scholar] [CrossRef]
- Zhou, J.; Tan, S.H.; Nicolas, V.; Bauvy, C.; Yang, N.D.; Zhang, J.; Xue, Y.; Codogno, P.; Shen, H.M. Activation of lysosomal function in the course of autophagy via mTORC1 suppression and autophagosome-lysosome fusion. Cell Res. 2013, 23, 508–523. [Google Scholar] [CrossRef] [Green Version]
- Martina, J.A.; Chen, Y.; Gucek, M.; Puertollano, R. MTORC1 functions as a transcriptional regulator of autophagy by preventing nuclear transport of TFEB. Autophagy 2012, 8, 903–914. [Google Scholar] [CrossRef] [Green Version]
- Moruno-Manchon, J.F.; Uzor, N.E.; Kesler, S.R.; Wefel, J.S.; Townley, D.M.; Nagaraja, A.S.; Pradeep, S.; Mangala, L.S.; Sood, A.K.; Tsvetkov, A.S. TFEB ameliorates the impairment of the autophagy-lysosome pathway in neurons induced by doxorubicin. Aging 2016, 8, 3507–3519. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.; Surma, M.; Wei, L. Disruption of ROCK1 gene restores autophagic flux and mitigates doxorubicin-induced cardiotoxicity. Oncotarget 2018, 9, 12995–13008. [Google Scholar] [CrossRef] [Green Version]
- Yu, M.L.; Li, S.M.; Gao, X.; Li, J.G.; Xu, H.; Chen, K.J. Sodium Tanshinone II A Sulfonate for Coronary Heart Disease: A Systematic Review of Randomized Controlled Trials. Chin. J. Integr. Med. 2018. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, S.; Chen, X. Tanshinone IIA protects against cardiac fibrosis through inhibition of beta-tubulin expression. J. Biol. Regul. Homeost. Agents 2018, 32, 1451–1455. [Google Scholar]
- Tsai, Y.T.; Loh, S.H.; Lee, C.Y.; Lee, S.P.; Chen, Y.L.; Cheng, T.H.; Tsai, C.S. Tanshinone IIA Inhibits High Glucose-Induced Collagen Synthesis via Nuclear Factor Erythroid 2-Related Factor 2 in Cardiac Fibroblasts. Cell Physiol. Biochem. 2018, 51, 2250–2261. [Google Scholar] [CrossRef]
- Meng, Z.J.; Wang, C.; Meng, L.T.; Bao, B.H.; Wu, J.H.; Hu, Y.Q. Sodium tanshinone IIA sulfonate attenuates cardiac dysfunction and improves survival of rats with cecal ligation and puncture-induced sepsis. Chin. J. Nat. Med. 2018, 16, 846–855. [Google Scholar] [CrossRef]
- He, Z.; Sun, C.; Xu, Y.; Cheng, D. Reduction of atrial fibrillation by Tanshinone IIA in chronic heart failure. Biomed. Pharmacother. 2016, 84, 1760–1767. [Google Scholar] [CrossRef]
- Wang, C.; Li, H.; Zhou, K.; Luo, C.; Li, Y.; Xie, L.; Hua, Y. Sodium tanshinone IIA sulfonate and sodium danshensu open the placental barrier through down-regulation of placental P-glycoprotein in mice: Implications in the transplacental digoxin treatment for fetal heart failure. Int. J. Cardiol. 2014, 176, 1331–1333. [Google Scholar] [CrossRef]
- Guo, Z.; Yan, M.; Chen, L.; Fang, P.; Li, Z.; Wan, Z.; Cao, S.; Hou, Z.; Wei, S.; Li, W.; et al. Nrf2-dependent antioxidant response mediated the protective effect of tanshinone IIA on doxorubicin-induced cardiotoxicity. Exp. Ther. Med. 2018, 16, 3333–3344. [Google Scholar] [CrossRef]
- Jiang, B.; Zhang, L.; Wang, Y.; Li, M.; Wu, W.; Guan, S.; Liu, X.; Yang, M.; Wang, J.; Guo, D.A. Tanshinone IIA sodium sulfonate protects against cardiotoxicity induced by doxorubicin in vitro and in vivo. Food Chem. Toxicol. 2009, 47, 1538–1544. [Google Scholar] [CrossRef]
- Hamada, M.; Shigematsu, Y.; Ohtani, T.; Ikeda, S. Elevated Cardiac Enzymes in Hypertrophic Cardiomyopathy Patients With Heart Failure-A 20-Year Prospective Follow-up Study. Circ. J. 2016, 80, 218–226. [Google Scholar] [CrossRef]
- Dickey, J.S.; Gonzalez, Y.; Aryal, B.; Mog, S.; Nakamura, A.J.; Redon, C.E.; Baxa, U.; Rosen, E.; Cheng, G.; Zielonka, J.; et al. Mito-tempol and dexrazoxane exhibit cardioprotective and chemotherapeutic effects through specific protein oxidation and autophagy in a syngeneic breast tumor preclinical model. PLoS ONE 2013, 8, e70575. [Google Scholar] [CrossRef]
- Kan, S.; Cheung, W.M.; Zhou, Y.; Ho, W.S. Enhancement of doxorubicin cytotoxicity by tanshinone IIA in HepG2 human hepatoma cells. Planta Med. 2014, 80, 70–76. [Google Scholar] [CrossRef]
- Liu, X.; Wang, Y.; Ma, C.; Zhang, L.; Wu, W.; Guan, S.; Yang, M.; Wang, J.; Jiang, B.; Guo, D.A. Proteomic assessment of tanshinone IIA sodium sulfonate on doxorubicin induced nephropathy. Am. J. Chin. Med. 2011, 39, 395–409. [Google Scholar] [CrossRef]
- Hong, H.J.; Liu, J.C.; Chen, P.Y.; Chen, J.J.; Chan, P.; Cheng, T.H. Tanshinone IIA prevents doxorubicin-induced cardiomyocyte apoptosis through Akt-dependent pathway. Int. J. Cardiol. 2012, 157, 174–179. [Google Scholar] [CrossRef]
- Wang, X.; Cui, T. Autophagy modulation: A potential therapeutic approach in cardiac hypertrophy. Am. J. Physiol. Heart Circ. Physiol. 2017, 313, H304–H319. [Google Scholar] [CrossRef]
- Bartlett, J.J.; Trivedi, P.C.; Yeung, P.; Kienesberger, P.C.; Pulinilkunnil, T. Doxorubicin impairs cardiomyocyte viability by suppressing transcription factor EB expression and disrupting autophagy. Biochem. J. 2016, 473, 3769–3789. [Google Scholar] [CrossRef] [Green Version]
- Lai, L.; Chen, J.; Wang, N.; Zhu, G.; Duan, X.; Ling, F. MiRNA-30e mediated cardioprotection of ACE2 in rats with Doxorubicin-induced heart failure through inhibiting cardiomyocytes autophagy. Life Sci. 2017, 169, 69–75. [Google Scholar] [CrossRef]
- Park, J.H.; Choi, S.H.; Kim, H.; Ji, S.T.; Jang, W.B.; Kim, J.H.; Baek, S.H.; Kwon, S.M. Doxorubicin Regulates Autophagy Signals via Accumulation of Cytosolic Ca2+ in Human Cardiac Progenitor Cells. Int. J. Mol. Sci. 2016, 17, 1680. [Google Scholar] [CrossRef]
- Park, J.H.; Kim, K.P.; Ko, J.J.; Park, K.S. PI3K/Akt/mTOR activation by suppression of ELK3 mediates chemosensitivity of MDA-MB-231 cells to doxorubicin by inhibiting autophagy. Biochem. Biophys. Res. Commun. 2016, 477, 277–282. [Google Scholar] [CrossRef]
- Pizarro, M.; Troncoso, R.; Martinez, G.J.; Chiong, M.; Castro, P.F.; Lavandero, S. Basal autophagy protects cardiomyocytes from doxorubicin-induced toxicity. Toxicology 2016, 370, 41–48. [Google Scholar] [CrossRef]
- Sun, A.; Cheng, Y.; Zhang, Y.; Zhang, Q.; Wang, S.; Tian, S.; Zou, Y.; Hu, K.; Ren, J.; Ge, J. Aldehyde dehydrogenase 2 ameliorates doxorubicin-induced myocardial dysfunction through detoxification of 4-HNE and suppression of autophagy. J. Mol. Cell Cardiol. 2014, 71, 92–104. [Google Scholar] [CrossRef]
- Wang, X.; Wang, X.L.; Chen, H.L.; Wu, D.; Chen, J.X.; Wang, X.X.; Li, R.L.; He, J.H.; Mo, L.; Cen, X.; et al. Ghrelin inhibits doxorubicin cardiotoxicity by inhibiting excessive autophagy through AMPK and p38-MAPK. Biochem. Pharmacol. 2014, 88, 334–350. [Google Scholar] [CrossRef]
- Napolitano, G.; Ballabio, A. TFEB at a glance. J. Cell Sci. 2016, 129, 2475–2481. [Google Scholar] [CrossRef] [Green Version]
- Settembre, C.; Zoncu, R.; Medina, D.L.; Vetrini, F.; Erdin, S.; Erdin, S.; Huynh, T.; Ferron, M.; Karsenty, G.; Vellard, M.C.; et al. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J. 2012, 31, 1095–1108. [Google Scholar] [CrossRef] [Green Version]
- Song, T.; Yao, Y.; Wang, T.; Huang, H.; Xia, H. Tanshinone IIA ameliorates apoptosis of myocardiocytes by up-regulation of miR-133 and suppression of Caspase-9. Eur. J. Pharmacol. 2017, 815, 343–350. [Google Scholar] [CrossRef]
- Maejima, Y.; Isobe, M.; Sadoshima, J. Regulation of autophagy by Beclin 1 in the heart. J. Mol. Cell Cardiol. 2016, 95, 19–25. [Google Scholar] [CrossRef]
- Wang, F.; Jia, J.; Rodrigues, B. Autophagy, Metabolic Disease, and Pathogenesis of Heart Dysfunction. Can. J. Cardiol. 2017, 33, 850–859. [Google Scholar] [CrossRef]
- Kim, S.H.; Kim, G.; Han, D.H.; Lee, M.; Kim, I.; Kim, B.; Kim, K.H.; Song, Y.M.; Yoo, J.E.; Wang, H.J.; et al. Ezetimibe ameliorates steatohepatitis via AMP activated protein kinase-TFEB-mediated activation of autophagy and NLRP3 inflammasome inhibition. Autophagy 2017, 13, 1767–1781. [Google Scholar] [CrossRef]
- Zhu, Z.; Yang, C.; Iyaswamy, A.; Krishnamoorthi, S.; Sreenivasmurthy, S.G.; Liu, J.; Wang, Z.; Tong, B.C.; Song, J.; Lu, J.; et al. Balancing mTOR Signaling and Autophagy in the Treatment of Parkinson’s Disease. Int. J. Mol. Sci. 2019, 20, 728. [Google Scholar] [CrossRef]
- Sha, Y.; Rao, L.; Settembre, C.; Ballabio, A.; Eissa, N.T. STUB1 regulates TFEB-induced autophagy-lysosome pathway. EMBO J. 2017, 36, 2544–2552. [Google Scholar] [CrossRef]
- Settembre, C.; Fraldi, A.; Medina, D.L.; Ballabio, A. Signals from the lysosome: A control centre for cellular clearance and energy metabolism. Nat. Rev. Mol. Cell Biol. 2013, 14, 283–296. [Google Scholar] [CrossRef]
- Sasaki, Y.; Ikeda, Y.; Iwabayashi, M.; Akasaki, Y.; Ohishi, M. The Impact of Autophagy on Cardiovascular Senescence and Diseases. Int. Heart J. 2017, 58, 666–673. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.H.; Rodeo, S.A.; Fortier, L.A.; Lu, C.; Erisken, C.; Mao, J.J. Protein-releasing polymeric scaffolds induce fibrochondrocytic differentiation of endogenous cells for knee meniscus regeneration in sheep. Sci. Transl. Med. 2014, 6, 266. [Google Scholar] [CrossRef]
- Kostogrys, R.B.; Franczyk-Zarow, M.; Gasior-Glogowska, M.; Kus, E.; Jasztal, A.; Wrobel, T.P.; Baranska, M.; Czyzynska-Cichon, I.; Drahun, A.; Manterys, A.; et al. Anti-atherosclerotic effects of pravastatin in brachiocephalic artery in comparison with en face aorta and aortic roots in ApoE/LDLR(-/-) mice. Pharmacol. Rep. 2017, 69, 112–118. [Google Scholar] [CrossRef]
- Huang, L.; Zhu, J.; Zheng, M.; Zou, R.; Zhou, Y.; Zhu, M. Tanshinone IIA protects against subclinical lipopolysaccharide induced cardiac fibrosis in mice through inhibition of NADPH oxidase. Int. Immunopharmacol. 2018, 60, 59–63. [Google Scholar] [CrossRef]
- Curtis, M.J.; Bond, R.A.; Spina, D.; Ahluwalia, A.; Alexander, S.P.; Giembycz, M.A.; Gilchrist, A.; Hoyer, D.; Insel, P.A.; Izzo, A.A.; et al. Experimental design and analysis and their reporting: New guidance for publication in BJP. Br. J. Pharmacol. 2015, 172, 3461–3471. [Google Scholar] [CrossRef]
- QuanJun, Y.; GenJin, Y.; LiLi, W.; YongLong, H.; Yan, H.; Jie, L.; JinLu, H.; Jin, L.; Run, G.; Cheng, G. Protective Effects of Dexrazoxane against Doxorubicin-Induced Cardiotoxicity: A Metabolomic Study. PLoS ONE 2017, 12, e0169567. [Google Scholar] [CrossRef]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, X.; Li, C.; Wang, Q.; Li, W.; Guo, D.; Zhang, X.; Shao, M.; Chen, X.; Ma, L.; Zhang, Q.; et al. Tanshinone IIA Restores Dynamic Balance of Autophagosome/Autolysosome in Doxorubicin-Induced Cardiotoxicity via Targeting Beclin1/LAMP1. Cancers 2019, 11, 910. https://doi.org/10.3390/cancers11070910
Wang X, Li C, Wang Q, Li W, Guo D, Zhang X, Shao M, Chen X, Ma L, Zhang Q, et al. Tanshinone IIA Restores Dynamic Balance of Autophagosome/Autolysosome in Doxorubicin-Induced Cardiotoxicity via Targeting Beclin1/LAMP1. Cancers. 2019; 11(7):910. https://doi.org/10.3390/cancers11070910
Chicago/Turabian StyleWang, Xiaoping, Chun Li, Qiyan Wang, Weili Li, Dongqing Guo, Xuefeng Zhang, Mingyan Shao, Xu Chen, Lin Ma, Qian Zhang, and et al. 2019. "Tanshinone IIA Restores Dynamic Balance of Autophagosome/Autolysosome in Doxorubicin-Induced Cardiotoxicity via Targeting Beclin1/LAMP1" Cancers 11, no. 7: 910. https://doi.org/10.3390/cancers11070910