
Glutamine-Glutamate Cycle Flux Is Similar in Cultured Astrocytes and Brain and Both Glutamate Production and Oxidation Are Mainly Catalyzed by Aspartate Aminotransferase

Abstract
:1. Introduction
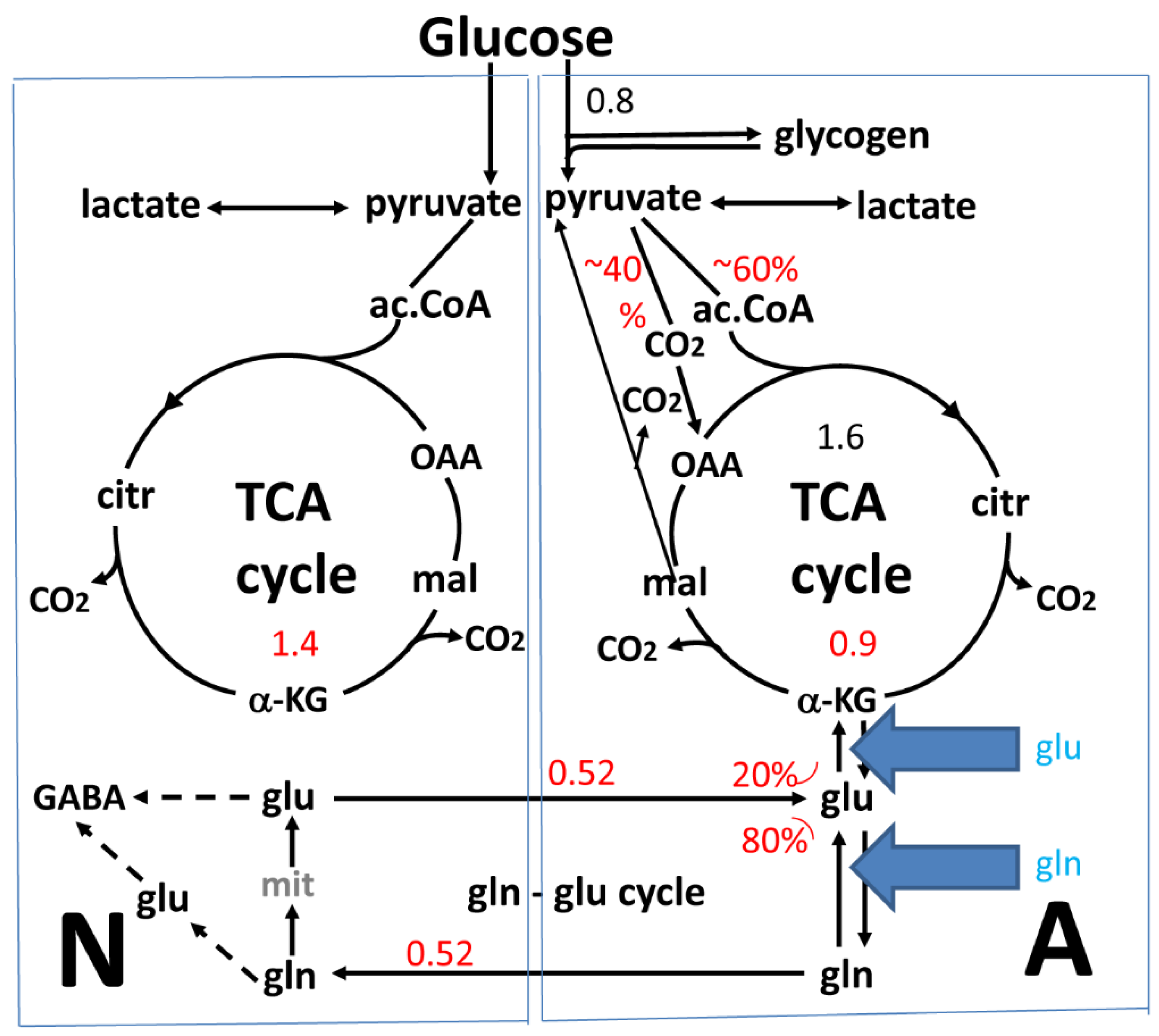
2. The Glutamate-Glutamine Cycle in the Brain In Vivo
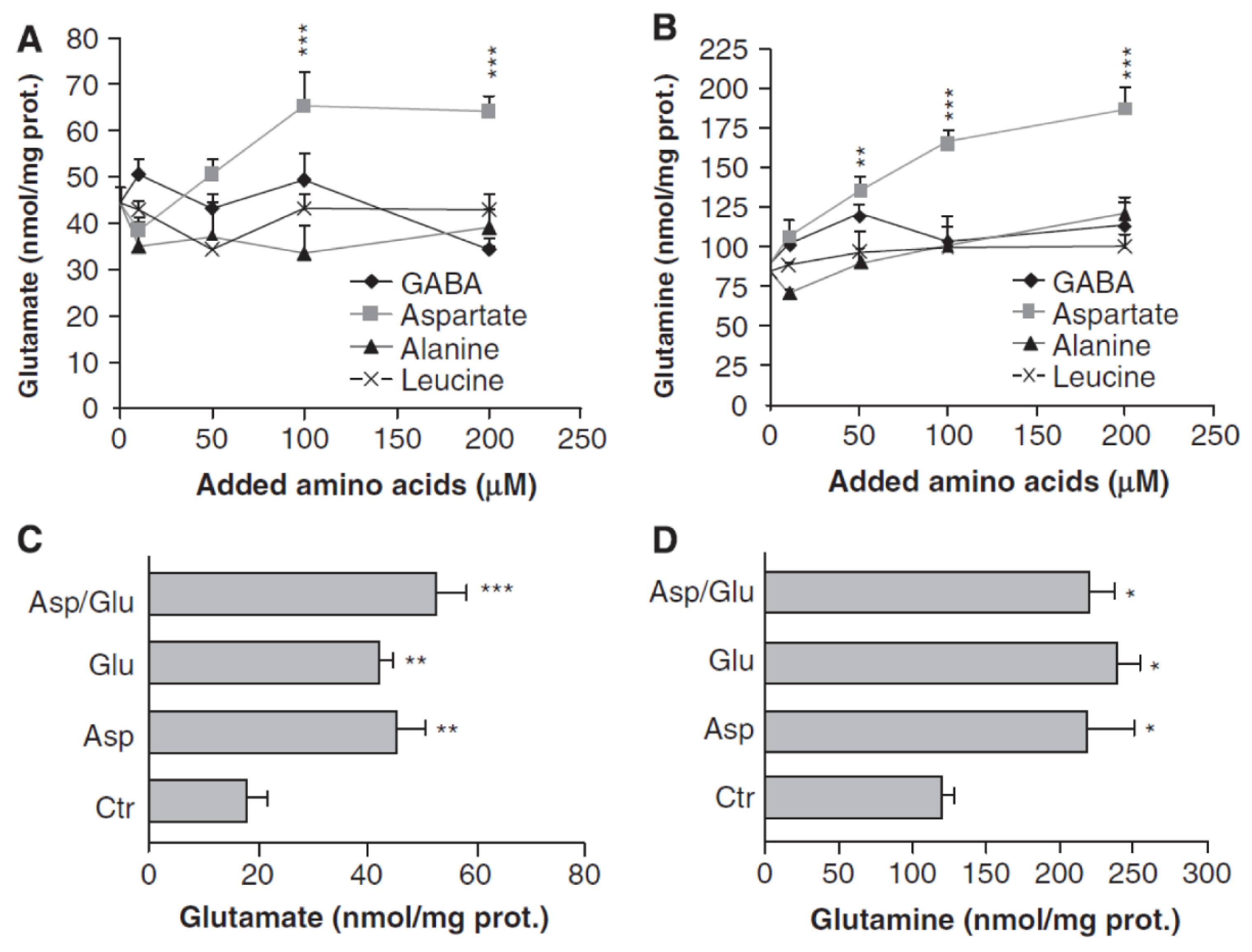
3. Glutamate and Glutamine Formation and Glutamate Oxidation in Cultured Astrocytes

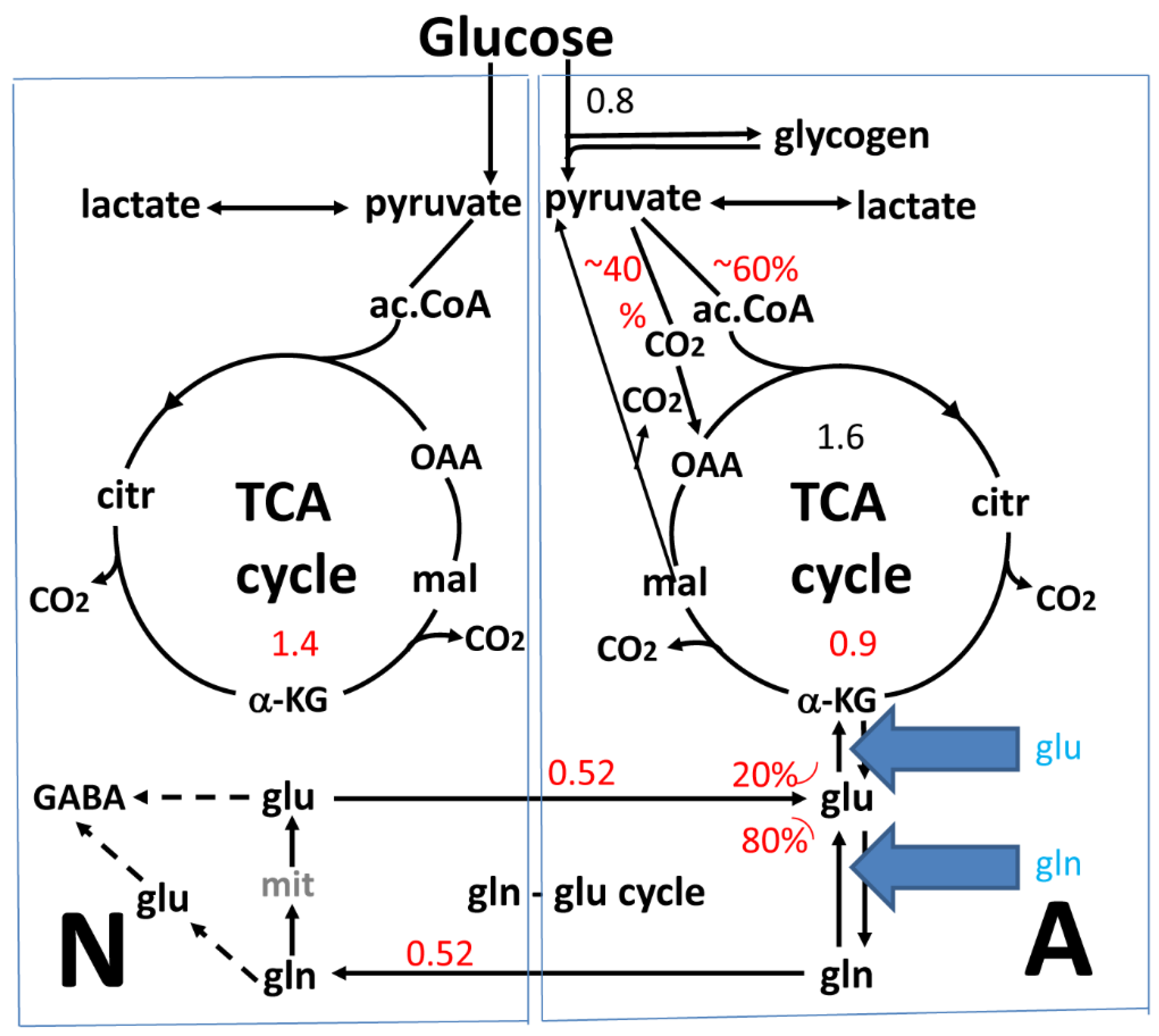{kind=link}
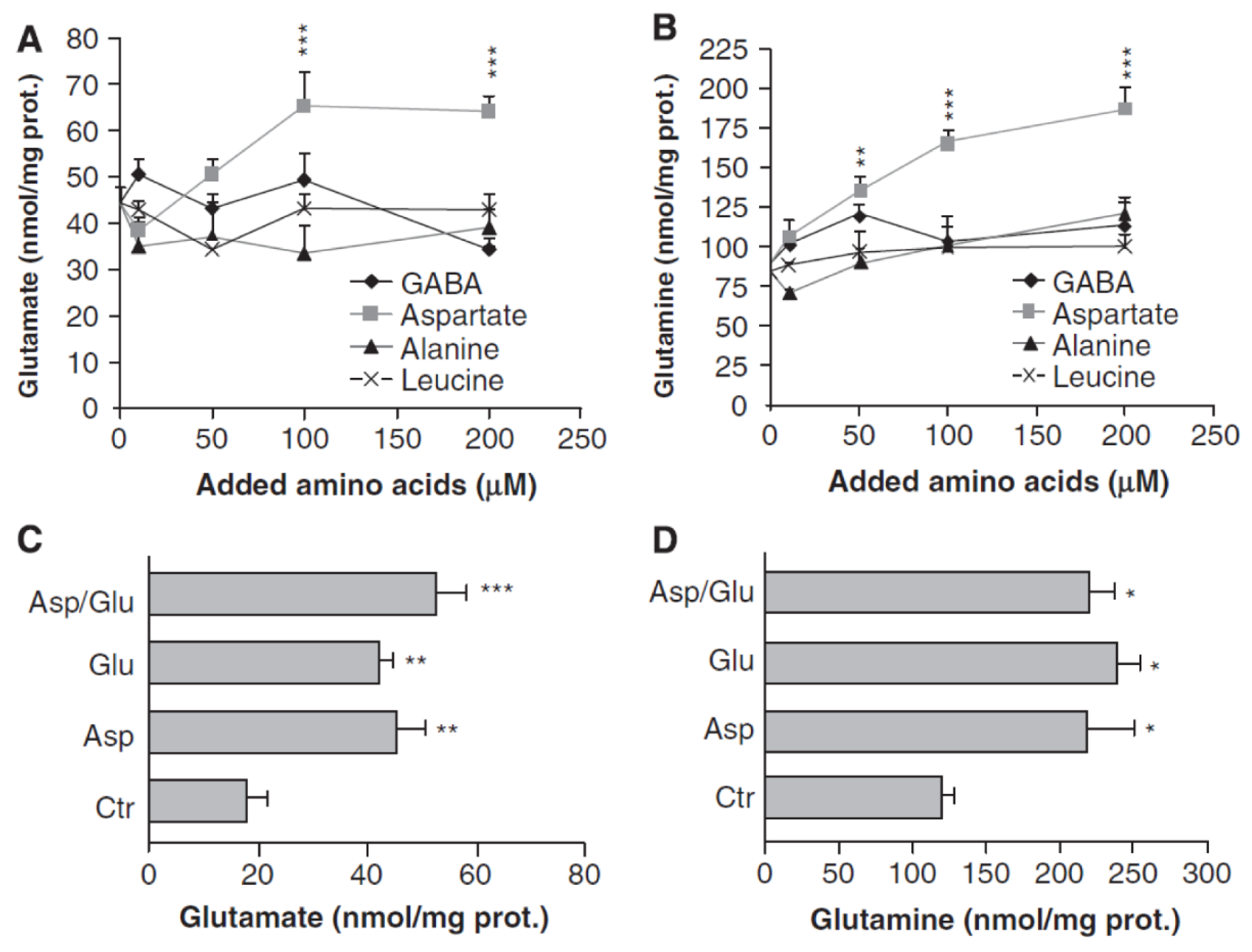{kind=link}
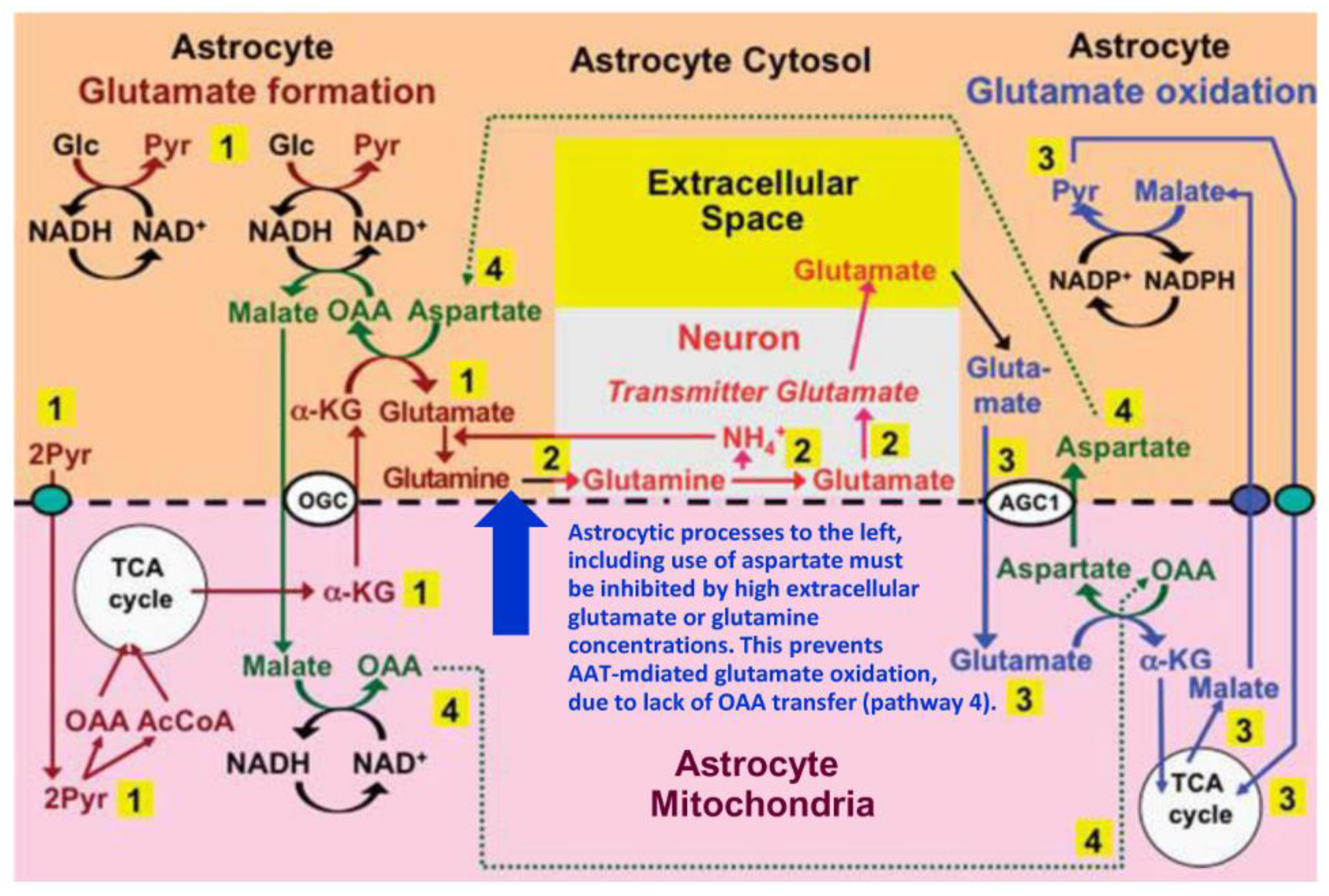{kind=link}
| Reference | Glu/Gln concentration | To glutamine (nmol/min per mg protein) | To oxaloacetate/aspartate (nmol/min per mg protein) | Glutamate to CO2 or polarographic assay (all nmol/min CO2 or O2 per mg protein) |
|---|---|---|---|---|
| Yu et al., 1982 [63] | 50 µM/2 mM | 2.4 | 1.1 | 4.1 ± 0.24 AOAA no effect |
| Yu and Hertz 1983 [66] | 50 µM/2 mM | 7.4 AOAA not tested | ||
| Yu et al. 1984 [64] | 50 µM/0.5 mM | 5.9 AOAA no additional effect when + ammonia at or above 1 mM * | ||
| Yudkoff et al., 1986 [83] | 250µM/0 mM | 9.0 | ||
| Hertz et al., 1988 [65] | 50 µM/2 mM | 5.1 AOAA not tested | ||
| Lai et al., 1989 [76] | 190 µM/0 mM | 2.7±0.22 AOAA: 0.74 ± 0.04; ammonia effect slightly less | ||
| Farinelli and Nicklas, 1992 [77] | 50 µM/0.5 mM | 1.2; 1.2 AOAA 0.1; 0.1 | 6.0; 6.9 AOAA 0.3;0.6 | |
| Huang et al., 1994 [81] | 50 µM/0.2 mM | 2.0–2.2 | ||
| Hertz and Hertz, 2003 [45] | 100 µM/0 mM no glucose | 5.0 AOAA not tested | ||
| McKenna, 2012 [46] | 1 mM/0 mM no glucose | 1.4 AOAA not tested | ||
| McKenna et al., 1996 [52] | 100–500 μM | Concentration-dependent; see text | Concentration-dependent, for details see text |
4. Suggested Pathway for Coupled Formation and Oxidation of Glutamate
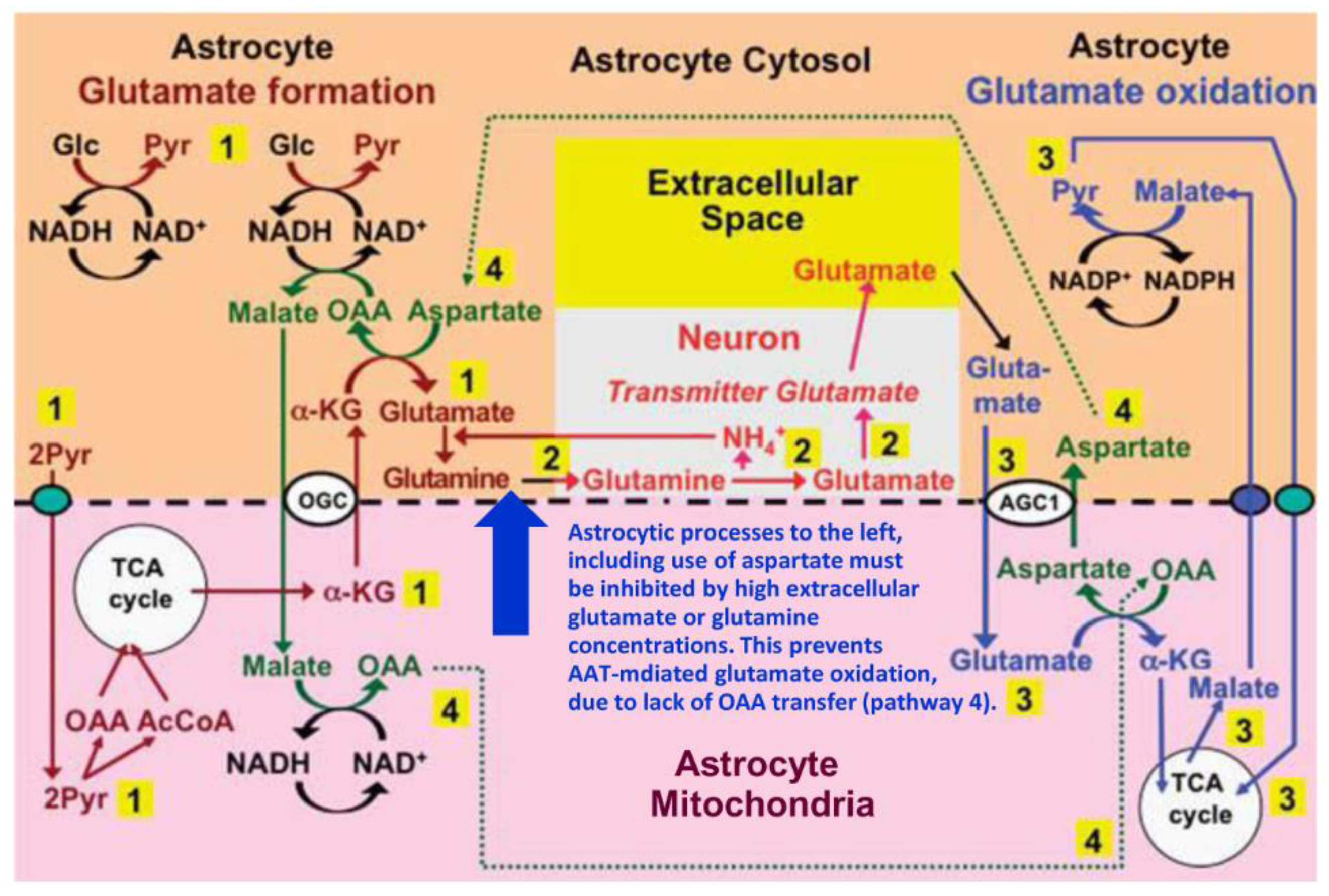
5. Evidence from GDH Knock Down/Inhibition Studies of the Roles of AAT and GDH in Glutamate Oxidation and Resynthesis
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Smith, Q.R. Transport of glutamate and other amino acids at the blood-brain barrier. J. Nutr. 2000, 130, 1016S–1022S. [Google Scholar] [PubMed]
- Patel, A.B.; de Graaf, R.A.; Rothman, D.L.; Behar, K.L. Effects of gamma-aminobutyric acid transporter 1 inhibition by tiagabine on brain glutamate and gamma-aminobutyric acid metabolism in the anesthetized rat in vivo. J. Neurosci. Res. 2015, 93, 1101–1108. [Google Scholar] [CrossRef] [PubMed]
- Danbolt, N.C.; Furness, D.N.; Zhou, Y. Neuronal vs glial glutamate uptake: Resolving the conundrum. Neurochem. Int. 2016, 98, 29–45. [Google Scholar] [CrossRef] [PubMed]
- Duarte, J.M.; Gruetter, R. Glutamatergic and gabaergic energy metabolism measured in the rat brain by 13C NMR spectroscopy at 14.1 T. J. Neurochem. 2013, 126, 579–590. [Google Scholar] [CrossRef] [PubMed]
- Schousboe, A.; Bak, L.K.; Waagepetersen, H.S. Astrocytic control of biosynthesis and turnover of the neurotransmitters glutamate and GABA. Front Endocrinol (Lausanne) 2013, 4. [Google Scholar] [CrossRef] [PubMed]
- Hertz, L.; Rothman, D.L. Glucose, lactate, β-hydroxybutyrate, acetate, GABA, and succinate as substrates for synthesis of glutamate and GABA in the glutamine-glutamate/gaba cycle. In Advances in Neurobiology; Sonnewald, U., Schousboe, A., Eds.; Springer: New York, NY, USA, 2016; pp. 9–42. [Google Scholar]
- Yu, A.C.; Drejer, J.; Hertz, L.; Schousboe, A. Pyruvate carboxylase activity in primary cultures of astrocytes and neurons. J. Neurochem. 1983, 41, 1484–1487. [Google Scholar] [CrossRef] [PubMed]
- Shank, R.P.; Bennett, G.S.; Freytag, S.O.; Campbell, G.L. Pyruvate carboxylase: An astrocyte-specific enzyme implicated in the replenishment of amino acid neurotransmitter pools. Brain Res. 1985, 329, 364–367. [Google Scholar] [CrossRef]
- Sibson, N.R.; Dhankhar, A.; Mason, G.F.; Rothman, D.L.; Behar, K.L.; Shulman, R.G. Stoichiometric coupling of brain glucose metabolism and glutamatergic neuronal activity. Proc. Natl. Acad. Sci. USA 1998, 95, 316–321. [Google Scholar] [CrossRef] [PubMed]
- Rothman, D.L.; De Feyter, H.M.; de Graaf, R.A.; Mason, G.F.; Behar, K.L. 13C MRS studies of neuroenergetics and neurotransmitter cycling in humans. NMR Biomed. 2011, 24, 943–957. [Google Scholar] [CrossRef] [PubMed]
- Hyder, F.; Fulbright, R.K.; Shulman, R.G.; Rothman, D.L. Glutamatergic function in the resting awake human brain is supported by uniformly high oxidative energy. J. Cereb. Blood Flow Metab. 2013, 33, 339–347. [Google Scholar] [CrossRef] [PubMed]
- Hertz, L. Astrocytic energy metabolism and glutamate formation—Relevance for 13C-NMR spectroscopy and importance of cytosolic/mitochondrial trafficking. Magn. Reson. Imaging 2011, 29, 1319–1329. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, E.E.; Driscoll, B.F. Carbon dioxide fixation in neuronal and astroglial cells in culture. J. Neurochem. 1992, 58, 258–262. [Google Scholar] [CrossRef] [PubMed]
- Westergaard, N.; Drejer, J.; Schousboe, A.; Sonnewald, U. Evaluation of the importance of transamination versus deamination in astrocytic metabolism of [U-13C]glutamate. Glia 1996, 17, 160–168. [Google Scholar] [CrossRef]
- Hertz, L. Brain glutamine synthesis requires neuronal aspartate: A commentary. J. Cereb. Blood Flow Metab. 2011, 31, 384–387. [Google Scholar] [CrossRef] [PubMed]
- Hertz, L. The glutamate-glutamine (GABA) Cycle: Importance of Late Postnatal Development and potential reciprocal interactions between biosynthesis and degradation. Front Endocrinol (Lausanne) 2013, 4. [Google Scholar] [CrossRef] [PubMed]
- Balazs, R. Control of glutamate metabolism: The effect of pyruvate. J. Neurochem. 1965, 12, 63–76. [Google Scholar] [CrossRef] [PubMed]
- Cooper, A.J.; Jeitner, T.M. Central role of glutamate metabolism in the maintenance of nitrogen homeostasis in normal and hyperammonemic brain. Biomolecules 2016, 6, 16. [Google Scholar] [CrossRef] [PubMed]
- Quagliariello, E.; Papa, S.; Saccone, C.; Palmieri, F.; Francavilla, A. The oxidation of glutamate by rat-liver mitochondria. Biochem. J. 1965, 95, 742–748. [Google Scholar] [CrossRef] [PubMed]
- Erecinska, M.; Pleasure, D.; Nelson, D.; Nissim, I.; Yudkoff, M. Cerebral aspartate utilization: Near-equilibrium relationships in aspartate aminotransferase reaction. J. Neurochem. 1993, 60, 1696–1706. [Google Scholar] [CrossRef] [PubMed]
- Balazs, R. Control of glutamate oxidation in brain and liver mitochondrial systems. Biochem. J. 1965, 95, 497–508. [Google Scholar] [CrossRef] [PubMed]
- Rothe, F.; Schmidt, W.; Wolf, G. Postnatal changes in the activity of glutamate dehydrogenase and aspartate aminotransferase in the rat nervous system with special reference to the glutamate transmitter metabolism. Brain Res. 1983, 313, 67–74. [Google Scholar] [CrossRef]
- Spanaki, C.; Zaganas, I.; Kounoupa, Z.; Plaitakis, A. The complex regulation of human Glud1 and Glud2 glutamate dehydrogenases and its implications in nerve tissue biology. Neurochem. Int. 2012, 61, 470–481. [Google Scholar] [CrossRef] [PubMed]
- Spanaki, C.; Kotzamani, D.; Petraki, Z.; Drakos, E.; Plaitakis, A. Heterogeneous cellular distribution of glutamate dehydrogenase in brain and in non-neural tissues. Neurochem. Res. 2014, 39, 500–515. [Google Scholar] [CrossRef] [PubMed]
- Schousboe, A.; Scafidi, S.; Bak, L.K.; Waagepetersen, H.S.; McKenna, M.C. Glutamate metabolism in the brain focusing on astrocytes. Adv. Neurobiol. 2014, 11, 13–30. [Google Scholar] [PubMed]
- Spanaki, C.; Kotzamani, D.; Plaitakis, A. Widening spectrum of cellular and subcellular expression of human Glud1 and Glud2 glutamate dehydrogenases suggests novel functions. Neurochem. Res. 2016, 42, 92–107. [Google Scholar] [CrossRef] [PubMed]
- McKenna, M.C.; Stevenson, J.H.; Huang, X.; Hopkins, I.B. Differential distribution of the enzymes glutamate dehydrogenase and aspartate aminotransferase in cortical synaptic mitochondria contributes to metabolic compartmentation in cortical synaptic terminals. Neurochem. Int. 2000, 37, 229–241. [Google Scholar] [CrossRef]
- Plaitakis, A.; Flessas, P.; Natsiou, A.B.; Shashidharan, P. Glutamate dehydrogenase deficiency in cerebellar degenerations: Clinical, biochemical and molecular genetic aspects. Can. J. Neurol. Sci. 1993, 20, S109–S116. [Google Scholar] [PubMed]
- Norenberg, M.D.; Martinez-Hernandez, A. Fine structural localization of glutamine synthetase in astrocytes of rat brain. Brain Res. 1979, 161, 303–310. [Google Scholar] [CrossRef]
- Anlauf, E.; Derouiche, A. Glutamine synthetase as an astrocytic marker: Its cell type and vesicle localization. Front Endocrinol (Lausanne) 2013, 4, 144. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Gu, L.; Hertz, L.; Peng, L. Expression of nucleoside transporter in freshly isolated neurons and astrocytes from mouse brain. Neurochem. Res. 2013, 38, 2351–2358. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.; Guo, C.; Wang, T.; Li, B.; Gu, L.; Wang, Z. Methodological limitations in determining astrocytic gene expression. Front Endocrinol (Lausanne) 2013, 4, 176. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, V.; Ambadipudi, S.; Patel, A.B. Glutamatergic and GABAergic TCA cycle and neurotransmitter cycling fluxes in different regions of mouse brain. J. Cereb. Blood Flow Metab. 2013, 33, 1523–1531. [Google Scholar] [CrossRef] [PubMed]
- Bringmann, A.; Grosche, A.; Pannicke, T.; Reichenbach, A. GABA and glutamate uptake and metabolism in retinal glial (Muller) cells. Front Endocrinol (Lausanne) 2013, 4, 48. [Google Scholar] [CrossRef] [PubMed]
- Karaca, M.; Frigerio, F.; Migrenne, S.; Martin-Levilain, J.; Skytt, D.M.; Pajecka, K.; Martin-del-Rio, R.; Gruetter, R.; Tamarit-Rodriguez, J.; Waagepetersen, H.S.; et al. GDH-dependent glutamate oxidation in the brain dictates peripheral energy substrate distribution. Cell. Rep. 2015, 13, 365–375. [Google Scholar] [CrossRef] [PubMed]
- Nissen, J.D.; Pajecka, K.; Stridh, M.H.; Skytt, D.M.; Waagepetersen, H.S. Dysfunctional TCA-cycle metabolism in glutamate dehydrogenase deficient astrocytes. Glia 2015, 63, 2313–2326. [Google Scholar] [CrossRef] [PubMed]
- Van den Berg, C.J.; Garfinkel, D. A simulation study of brain compartments: Metabolism of glutamate and related substances in mouse brain. Biochem. J. 1971, 123, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, A.M.; Quastel, J.H. Metabolism of amino acids and ammonia in rat brain cortex slices in vitro: A possible role of ammonia in brain function. J. Neurochem. 1975, 25, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, G.M.; Patel, A.B.; Mason, G.F.; Rothman, D.L.; Behar, K.L. Glutamatergic and GABAergic neurotransmitter cycling and energy metabolism in rat cerebral cortex during postnatal development. J. Cereb. Blood Flow Metab. 2007, 27, 1895–1907. [Google Scholar] [CrossRef] [PubMed]
- Erecinska, M.; Cherian, S.; Silver, I.A. Energy metabolism in mammalian brain during development. Prog. Neurobiol. 2004, 73, 397–445. [Google Scholar] [CrossRef] [PubMed]
- Toyama, H.; Ichise, M.; Liow, J.S.; Modell, K.J.; Vines, D.C.; Esaki, T.; Cook, M.; Seidel, J.; Sokoloff, L.; Green, M.V.; et al. Absolute quantification of regional cerebral glucose utilization in mice by 18F-FDG small animal pet scanning and 2-14C-DG autoradiography autoradiography. J. Nucl. Med. 2004, 45, 1398–1405. [Google Scholar] [PubMed]
- Sutherland, G.R.; Tyson, R.L.; Auer, R.N. Truncation of the Krebs cycle during hypoglycemic coma. Med. Chem. 2008, 4, 379–385. [Google Scholar] [CrossRef] [PubMed]
- Lebon, V.; Petersen, K.F.; Cline, G.W.; Shen, J.; Mason, G.F.; Dufour, S.; Behar, K.L.; Shulman, G.I.; Rothman, D.L. Astroglial contribution to brain energy metabolism in humans revealed by 13C nuclear magnetic resonance spectroscopy: Elucidation of the dominant pathway for neurotransmitter glutamate repletion and measurement of astrocytic oxidative metabolism. J. Neurosci. 2002, 22, 1523–1531. [Google Scholar] [PubMed]
- Sonnewald, U. Glutamate synthesis has to be matched by its degradation—Where do all the carbons go? J. Neurochem. 2014, 131, 399–406. [Google Scholar] [CrossRef] [PubMed]
- Hertz, L.; Hertz, E. Cataplerotic TCA cycle flux determined as glutamate-sustained oxygen consumption in primary cultures of astrocytes. Neurochem. Int. 2003, 43, 355–361. [Google Scholar] [CrossRef]
- McKenna, M.C. Substrate competition studies demonstrate oxidative metabolism of glucose, glutamate, glutamine, lactate and 3-hydroxybutyrate in cortical astrocytes from rat brain. Neurochem. Res. 2012, 37, 2613–2626. [Google Scholar] [CrossRef] [PubMed]
- McKenna, M.C. Glutamate pays its own way in astrocytes. Front Endocrinol (Lausanne) 2013, 4, 191. [Google Scholar] [CrossRef] [PubMed]
- Hertz, L.; Peng, L.; Dienel, G.A. Energy metabolism in astrocytes: High rate of oxidative metabolism and spatiotemporal dependence on glycolysis/glycogenolysis. J. Cereb. Blood Flow Metab. 2007, 27, 219–249. [Google Scholar] [CrossRef] [PubMed]
- Kurz, G.M.; Wiesinger, H.; Hamprecht, B. Purification of cytosolic malic enzyme from bovine brain, generation of monoclonal antibodies, and immunocytochemical localization of the enzyme in glial cells of neural primary cultures. J. Neurochem. 1993, 60, 1467–1474. [Google Scholar] [CrossRef] [PubMed]
- Lo, A.S.; Liew, C.T.; Ngai, S.M.; Tsui, S.K.; Fung, K.P.; Lee, C.Y.; Waye, M.M. Developmental regulation and cellular distribution of human cytosolic malate dehydrogenase (MDH1). J. Cell. Biochem. 2005, 94, 763–773. [Google Scholar] [CrossRef] [PubMed]
- Sonnewald, U.; Westergaard, N.; Hassel, B.; Müller, T.B.; Unsgård, G.; Fonnum, F.; Hertz, L.; Schousboe, A.; Petersen, S.B. NMR spectroscopic studies of 13C acetate and 13C glucose metabolism in neocortical astrocytes: Evidence for mitochondrial heterogeneity. Dev. Neurosci. 1993, 15, 351–358. [Google Scholar] [CrossRef] [PubMed]
- McKenna, M.C.; Sonnewald, U.; Huang, X.; Stevenson, J.; Zielke, H.R. Exogenous glutamate concentration regulates the metabolic fate of glutamate in astrocytes. J. Neurochem. 1996, 66, 386–393. [Google Scholar] [CrossRef] [PubMed]
- Hertz, L.; O’Dowd, B.S.; Ng, K.T.; Gibbs, M.E. Reciprocal changes in forebrain contents of glycogen and of glutamate/glutamine during early memory consolidation in the day-old chick. Brain Res. 2003, 994, 226–233. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, M.E.; Lloyd, H.G.; Santa, T.; Hertz, L. Glycogen is a preferred glutamate precursor during learning in 1-day-old chick: Biochemical and behavioral evidence. J. Neurosci. Res. 2007, 85, 3326–3333. [Google Scholar] [CrossRef] [PubMed]
- Bjørnsen, L.P.; Eid, T.; Holmseth, S.; Danbolt, N.C.; Spencer, D.D.; de Lanerolle, N.C. Changes in glial glutamate transporters in human epileptogenic hippocampus: Inadequate explanation for high extracellular glutamate during seizures. Neurobiol. Dis. 2007, 25, 319–330. [Google Scholar] [CrossRef] [PubMed]
- Peca, S.; Carni, M.; Di Bonaventura, C.; Aprile, T.; Hagberg, G.E.; Giallonardo, A.T.; Manfredi, M.; Mangia, S.; Garreffa, G.; Maraviglia, B.; et al. Metabolic correlatives of brain activity in a fos epilepsy patient. NMR Biomed. 2010, 23, 170–178. [Google Scholar] [CrossRef] [PubMed]
- Mangia, S.; Giove, F.; Dinuzzo, M. Metabolic pathways and activity-dependent modulation of glutamate concentration in the human brain. Neurochem. Res. 2012, 37, 2554–2561. [Google Scholar] [CrossRef] [PubMed]
- Abdallah, C.G.; Niciu, M.J.; Fenton, L.R.; Fasula, M.K.; Jiang, L.; Black, A.; Rothman, D.L.; Mason, G.F.; Sanacora, G. Decreased occipital cortical glutamate levels in response to successful cognitive-behavioral therapy and pharmacotherapy for major depressive disorder. Psychother. Psychosom. 2014, 83, 298–307. [Google Scholar] [CrossRef] [PubMed]
- Hertz, E.; Hertz, L. Polarographic measurement of oxygen uptake by astrocytes in primary cultures using the tissue-culture flask as the respirometer chamber. In Vitro 1979, 15, 429–436. [Google Scholar] [CrossRef] [PubMed]
- Olson, J.; Holtzman, D. Respiration and cell volume of primary cultured cerebral astrocytes in media of various osmolarities. Brain Res. 1982, 246, 273–279. [Google Scholar] [CrossRef]
- Katzman, R.; Pappius, H.M. Brain Electrolytes and Fluid Metabolism; Williams & Wilkins: Baltimore, MD, USA, 1973. [Google Scholar]
- Dittmann, L.; Sensenbrenner, M.; Hertz, L.; Mandel, P. Respiration by cultivated astrocytes and neurons from the cerebral hemispheres. J. Neurochem. 1973, 21, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Yu, A.C.; Schousboe, A.; Hertz, L. Metabolic fate of 14C-labeled glutamate in astrocytes in primary cultures. J. Neurochem. 1982, 39, 954–960. [Google Scholar] [CrossRef] [PubMed]
- Yu, A.C.; Schousboe, A.; Hertz, L. Influence of pathological concentrations of ammonia on metabolic fate of 14C-labeled glutamate in astrocytes in primary cultures. J. Neurochem. 1984, 42, 594–597. [Google Scholar] [CrossRef] [PubMed]
- Hertz, L.; Drejer, J.; Schousboe, A. Energy metabolism in glutamatergic neurons, gabaergic neurons and astrocytes in primary cultures. Neurochem. Res. 1988, 13, 605–610. [Google Scholar] [CrossRef] [PubMed]
- Yu, A.C.H.; Hertz, L. Metabolic source of energy in astrocytes. In Glutamine, Glutamate and GABA in the Central Nervous System; Hertz, L., Kvamme, E., McGeer, E.G., Schousboe, A., Eds.; Alan R. Liss: New York, NY, USA, 1983. [Google Scholar]
- Sonnewald, U.; White, L.R.; Ødegård, E.; Westergaard, N.; Bakken, I.J.; Aasly, J.; Unsgård, G.; Schousboe, A. MRS study of glutamate metabolism in cultured neurons/glia. Neurochem. Res. 1996, 21, 987–993. [Google Scholar] [CrossRef] [PubMed]
- Sonnewald, U.; Westergaard, N.; Schousboe, A. Glutamate transport and metabolism in astrocytes. Glia 1997, 21, 56–63. [Google Scholar] [CrossRef]
- Hertz, L.; Chen, Y.; Song, D. Astrocyte cultures mimicking brain astrocytes in gene expression, signaling, metabolism and K+ uptake and showing astrocytic gene expression overlooked by immunohistochemistry and in situ hybridization. Neurochem. Res. 2017, 42, 254–271. [Google Scholar] [CrossRef] [PubMed]
- Hyman, A.A.; Brangwynne, C.P. Beyond stereospecificity: Liquids and mesoscale organization of cytoplasm. Dev. Cell. 2011, 21, 14–16. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.L. The cytoophidium and its kind: Filamentation and compartmentation of metabolic enzymes. Annu. Rev. Cell Dev. Biol. 2016, 32, 349–372. [Google Scholar] [CrossRef] [PubMed]
- Krebs, H.A. Metabolism of amino-acids: The synthesis of glutamine from glutamic acid and ammonia, and the enzymic hydrolysis of glutamine in animal tissues. Biochem. J. 1935, 29, 1951–1969. [Google Scholar] [CrossRef] [PubMed]
- Engel, P.C.; Dalziel, K. The equilibrium constants of the glutamate dehydrogenase systems. Biochem. J. 1967, 105, 691–695. [Google Scholar] [CrossRef] [PubMed]
- Cooper, A.J.L. L-glutamate (2-oxoglutarate) aminotransferases. In Glutamine and Glutamate in Mammals; Kvamme, E., Ed.; CRC Press: Boca Raton, FL, USA, 1988; Volume I, pp. 124–145. [Google Scholar]
- Cooper, A.J.; McDonald, J.M.; Gelbard, A.S.; Gledhill, R.F.; Duffy, T.E. The metabolic fate of 13N-labeled ammonia in rat brain. J. Biol. Chem. 1979, 254, 4982–4992. [Google Scholar] [PubMed]
- Lai, J.C.; Murthy, C.R.; Cooper, A.J.; Hertz, E.; Hertz, L. Differential effects of ammonia and beta-methylene-DL-aspartate on metabolism of glutamate and related amino acids by astrocytes and neurons in primary culture. Neurochem. Res. 1989, 14, 377–389. [Google Scholar] [CrossRef] [PubMed]
- Farinelli, S.E.; Nicklas, W.J. Glutamate metabolism in rat cortical astrocyte cultures. J. Neurochem. 1992, 58, 1905–1915. [Google Scholar] [CrossRef] [PubMed]
- Pardo, B.; Rodrigues, T.B.; Contreras, L.; Garzon, M.; Llorente-Folch, I.; Kobayashi, K.; Saheki, T.; Cerdan, S.; Satrustegui, J. Brain glutamine synthesis requires neuronal-born aspartate as amino donor for glial glutamate formation. J. Cereb. Blood Flow Metab. 2011, 31, 90–101. [Google Scholar] [CrossRef] [PubMed]
- Lieth, E.; LaNoue, K.F.; Berkich, D.A.; Xu, B.; Ratz, M.; Taylor, C.; Hutson, S.M. Nitrogen shuttling between neurons and glial cells during glutamate synthesis. J. Neurochem. 2001, 76, 1712–1723. [Google Scholar] [CrossRef] [PubMed]
- McKenna, M.C.; Hopkins, I.B.; Lindauer, S.L.; Bamford, P. Aspartate aminotransferase in synaptic and nonsynaptic mitochondria: Differential effect of compounds that influence transient hetero-enzyme complex (metabolon) formation. Neurochem. Int. 2006, 48, 629–636. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.; Kala, G.; Murthy, R.K.; Hertz, L. Effects of chronic exposure to ammonia on glutamate and glutamine interconversion and compartmentation in homogeneous primary cultures of mouse astrocytes. Neurochem. Res. 1994, 19, 257–265. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; McNeill, J.R.; Hajek, I.; Hertz, L. Effect of vasopressin on brain swelling at the cellular level: Do astrocytes exhibit a furosemide--vasopressin-sensitive mechanism for volume regulation? Can. J. Physiol. Pharmacol. 1992, 70, S367–S373. [Google Scholar] [CrossRef] [PubMed]
- Yudkoff, M.; Nissim, I.; Hummeler, K.; Medow, M.; Pleasure, D. Utilization of [15N]glutamate by cultured astrocytes. Biochem. J. 1986, 234, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Balcar, V.J.; Hauser, K.L. Transport of [3H]L-Glutamate and [3H]L-Glutamine by Dissociated Glial and Neuronal Cells in Primary Culture; Verlag Chemie: Weinheim, Germany, 1978. [Google Scholar]
- Hertz, L.; Schousboe, A.; Boechler, N.; Mukerji, S.; Fedoroff, S. Kinetic characteristics of the glutamate uptake into normal astrocytes in cultures. Neurochem. Res. 1978, 3, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Hertz, L.; Bock, E.; Schousboe, A. Gfa content, glutamate uptake and activity of glutamate metabolizing enzymes in differentiating mouse astrocytes in primary cultures. Dev. Neurosci. 1978, 1, 226–238. [Google Scholar] [CrossRef]
- Teixeira, A.P.; Santos, S.S.; Carinhas, N.; Oliveira, R.; Alves, P.M. Combining metabolic flux analysis tools and 13C NMR to estimate intracellular fluxes of cultured astrocytes. Neurochem. Int. 2008, 52, 478–486. [Google Scholar] [CrossRef] [PubMed]
- Pellerin, L.; Magistretti, P.J. Glutamate uptake into astrocytes stimulates aerobic glycolysis: A mechanism coupling neuronal activity to glucose utilization. Proc. Natl. Acad. Sci. USA 1994, 91, 10625–10629. [Google Scholar] [CrossRef] [PubMed]
- Keiding, S.; Sørensen, M.; Bender, D.; Munk, O.L.; Ott, P.; Vilstrup, H. Brain metabolism of 13n-ammonia during acute hepatic encephalopathy in cirrhosis measured by positron emission tomography. Hepatology 2006, 43, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Hertz, L.; Song, D.; Peng, L.; Chen, Y. Multifactorial effects on different types of brain cells contribute to ammonia toxicity. Neurochem. Res. 2016. [Google Scholar] [CrossRef] [PubMed]
- Cooper, A.J.; Plum, F. Biochemistry and physiology of brain ammonia. Physiol. Rev. 1987, 67, 440–519. [Google Scholar] [PubMed]
- Fitzpatrick, S.M.; Hetherington, H.P.; Behar, K.L.; Shulman, R.G. Effects of acute hyperammonemia on cerebral amino acid metabolism and phi in vivo, measured by 1H and 31P nuclear magnetic resonance. J. Neurochem. 1989, 52, 741–749. [Google Scholar] [CrossRef] [PubMed]
- Cudalbu, C.; Lanz, B.; Duarte, J.M.; Morgenthaler, F.D.; Pilloud, Y.; Mlynarik, V.; Gruetter, R. Cerebral glutamine metabolism under hyperammonemia determined in vivo by localized 1H and 15N NMR spectroscopy. J. Cereb. Blood Flow Metab. 2012, 32, 696–708. [Google Scholar] [CrossRef] [PubMed]
- Dejong, C.H.; Kampman, M.T.; Deutz, N.E.; Soeters, P.B. Cerebral cortex ammonia and glutamine metabolism during liver insufficiency-induced hyperammonemia in the rat. J. Neurochem. 1992, 59, 1071–1079. [Google Scholar] [CrossRef] [PubMed]
- Nagaraja, T.N.; Brookes, N. Intracellular acidification induced by passive and active transport of ammonium ions in astrocytes. Am. J. Physiol. 1998, 274, C883–C891. [Google Scholar] [PubMed]
- Kanamori, K.; Ross, B.D. Kinetics of glial glutamine efflux and the mechanism of neuronal uptake studied in vivo in mildly hyperammonemic rat brain. J. Neurochem. 2006, 99, 1103–1113. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, G.; Peterson, A.; Iverfeldt, K.; Walum, E. Sodium-dependent glutamate uptake as an activator of oxidative metabolism in primary astrocyte cultures from newborn rat. Glia 1995, 15, 152–156. [Google Scholar] [CrossRef] [PubMed]
- Kvamme, E.; Lenda, K. Regulation of glutaminase by exogenous glutamate, ammonia and 2-oxoglutarate in synaptosomal enriched preparation from rat brain. Neurochem. Res. 1982, 7, 667–678. [Google Scholar] [CrossRef] [PubMed]
- Cavus, I.; Romanyshyn, J.C.; Kennard, J.T.; Farooque, P.; Williamson, A.; Eid, T.; Spencer, S.S.; Duckrow, R.; Dziura, J.; Spencer, D.D. Elevated basal glutamate and unchanged glutamine and GABA in refractory epilepsy: Microdialysis study of 79 patients at the Yale epilepsy surgery program. Ann. Neurol. 2016, 80, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Dennis, S.C.; Lai, J.C.; Clark, J.B. Comparative studies on glutamate metabolism in synpatic and non-synaptic rat brain mitochondria. Biochem. J. 1977, 164, 727–736. [Google Scholar] [CrossRef] [PubMed]
- Lovatt, D.; Sonnewald, U.; Waagepetersen, H.S.; Schousboe, A.; He, W.; Lin, J.H.; Han, X.; Takano, T.; Wang, S.; Sim, F.J.; et al. The transcriptome and metabolic gene signature of protoplasmic astrocytes in the adult murine cortex. J. Neurosci. 2007, 27, 12255–12266. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Hertz, L.; Peng, L. Aralar mRNA and protein levels in neurons and astrocytes freshly isolated from young and adult mouse brain and in maturing cultured astrocytes. Neurochem. Int. 2012, 61, 1325–1332. [Google Scholar] [CrossRef] [PubMed]
- Ramos, M.; del Arco, A.; Pardo, B.; Martinez-Serrano, A.; Martinez-Morales, J.R.; Kobayashi, K.; Yasuda, T.; Bogonez, E.; Bovolenta, P.; Saheki, T.; et al. Developmental changes in the Ca2+-regulated mitochondrial aspartate-glutamate carrier aralar1 in brain and prominent expression in the spinal cord. Brain Res. Dev. Brain Res. 2003, 143, 33–46. [Google Scholar] [CrossRef]
- Berkich, D.A.; Ola, M.S.; Cole, J.; Sweatt, A.J.; Hutson, S.M.; LaNoue, K.F. Mitochondrial transport proteins of the brain. J. Neurosci. Res. 2007, 85, 3367–3377. [Google Scholar] [CrossRef] [PubMed]
- Contreras, L. Role of AGC1/aralar in the metabolic synergies between neuron and glia. Neurochem. Int. 2015, 88, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Hindfelt, B.; Plum, F.; Duffy, T.E. Effect of acute ammonia intoxication on cerebral metabolism in rats with portacaval shunts. J. Clin Invest. 1977, 59, 386–396. [Google Scholar] [CrossRef] [PubMed]
- Lavoie, J.; Giguere, J.F.; Layrargues, G.P.; Butterworth, R.F. Amino acid changes in autopsied brain tissue from cirrhotic patients with hepatic encephalopathy. J. Neurochem. 1987, 49, 692–697. [Google Scholar] [CrossRef] [PubMed]
- Massucci, F.A.; DiNuzzo, M.; Giove, F.; Maraviglia, B.; Castillo, I.P.; Marinari, E.; De Martino, A. Energy metabolism and glutamate-glutamine cycle in the brain: A stoichiometric modeling perspective. BMC Syst. Biol. 2013, 7, 103. [Google Scholar] [CrossRef] [PubMed]
- Frigerio, F.; Karaca, M.; De Roo, M.; Mlynarik, V.; Skytt, D.M.; Carobbio, S.; Pajecka, K.; Waagepetersen, H.S.; Gruetter, R.; Muller, D.; et al. Deletion of glutamate dehydrogenase 1 (Glud1) in the central nervous system affects glutamate handling without altering synaptic transmission. J. Neurochem. 2012, 123, 342–348. [Google Scholar] [CrossRef] [PubMed]
- Skytt, D.M.; Klawonn, A.M.; Stridh, M.H.; Pajecka, K.; Patruss, Y.; Quintana-Cabrera, R.; Bolanos, J.P.; Schousboe, A.; Waagepetersen, H.S. siRNA knock down of glutamate dehydrogenase in astrocytes affects glutamate metabolism leading to extensive accumulation of the neuroactive amino acids glutamate and aspartate. Neurochem. Int. 2012, 61, 490–497. [Google Scholar] [CrossRef] [PubMed]
- Hertz, L.; Schousboe, A. Ion and energy metabolism of the brain at the cellular level. Int. Rev. Neurobiol. 1975, 18, 141–211. [Google Scholar] [PubMed]
- Okada, Y.; Lipton, P. Glucose, oxidative energy metabolism, and neural function in brain slices—Glycolysis plays a key role in neural activity. In Handbook of Neurochemistry and Molecular Biology, 3rd ed.; Lajtha, A., Ed.; Springer: New York, NY, USA, 2007; pp. 17–39. [Google Scholar]
- Patel, A.B.; Lai, J.C.; Chowdhury, G.M.; Hyder, F.; Rothman, D.L.; Shulman, R.G.; Behar, K.L. Direct evidence for activity-dependent glucose phosphorylation in neurons with implications for the astrocyte-to-neuron lactate shuttle. Proc. Natl. Acad. Sci. USA 2014, 111, 5385–5390. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.B.; Jackson, J.G. Astroglial glutamate transporters coordinate excitatory signaling and brain energetics. Neurochem. Int. 2016, 98, 56–71. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hertz, L.; Rothman, D.L. Glutamine-Glutamate Cycle Flux Is Similar in Cultured Astrocytes and Brain and Both Glutamate Production and Oxidation Are Mainly Catalyzed by Aspartate Aminotransferase. Biology 2017, 6, 17. https://doi.org/10.3390/biology6010017
Hertz L, Rothman DL. Glutamine-Glutamate Cycle Flux Is Similar in Cultured Astrocytes and Brain and Both Glutamate Production and Oxidation Are Mainly Catalyzed by Aspartate Aminotransferase. Biology. 2017; 6(1):17. https://doi.org/10.3390/biology6010017
Chicago/Turabian StyleHertz, Leif, and Douglas L Rothman. 2017. "Glutamine-Glutamate Cycle Flux Is Similar in Cultured Astrocytes and Brain and Both Glutamate Production and Oxidation Are Mainly Catalyzed by Aspartate Aminotransferase" Biology 6, no. 1: 17. https://doi.org/10.3390/biology6010017