
The Glutamate Dehydrogenase Pathway and Its Roles in Cell and Tissue Biology in Health and Disease

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. The GDH Enzymes
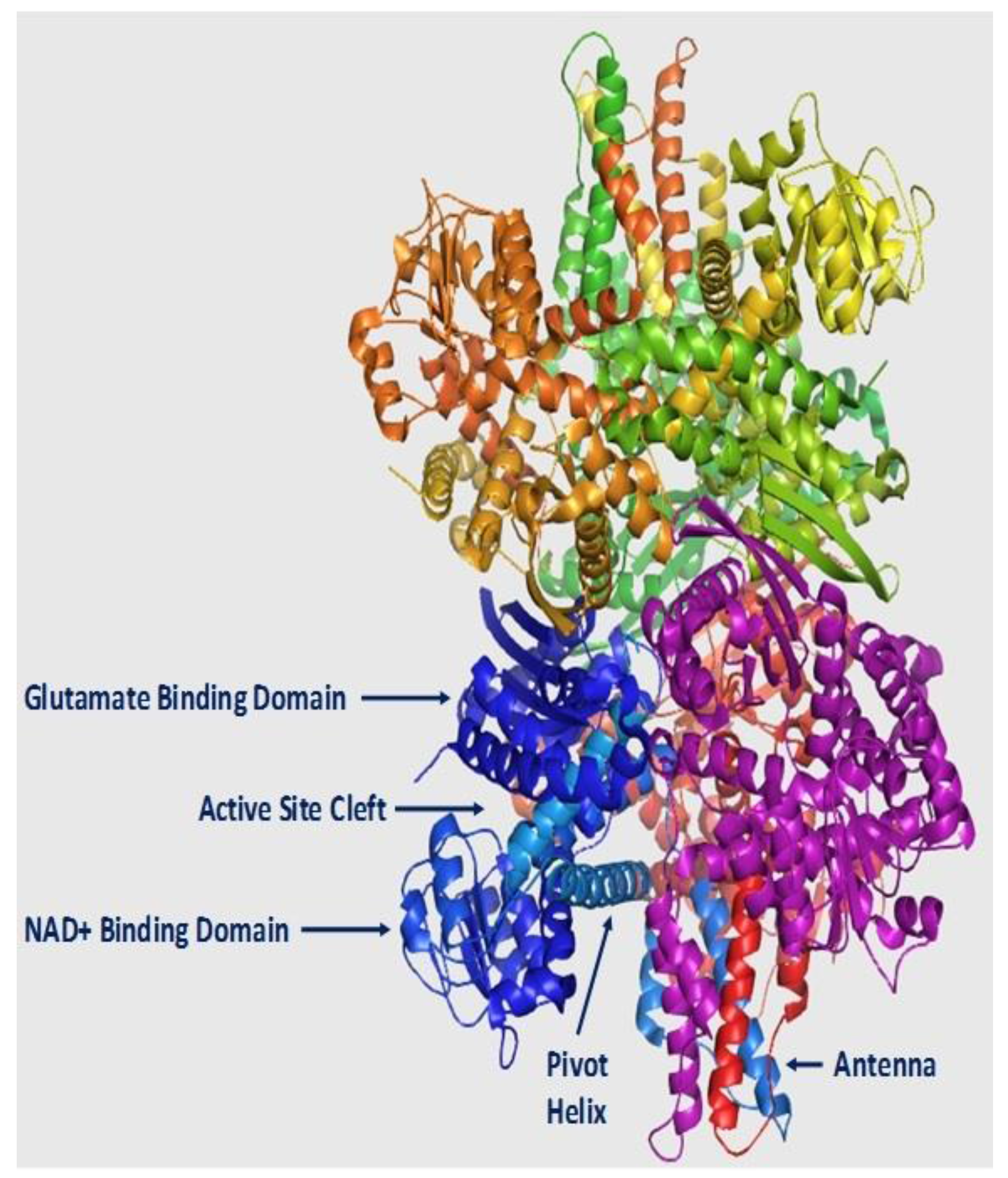
2. Structure
3. The GDH Catalysis
4. GDH Regulation
5. GDH Subcellular Localization
6. Transport of Human GDHs in S. Cerevisiae Mitochondria
7. Expression of GDH in Mammalian Organs
8. Expression of hGDH1 and hGDH2 in Non-Neural Human Tissues
9. Expression of GDH in Nerve Tissue of Experimental Animals
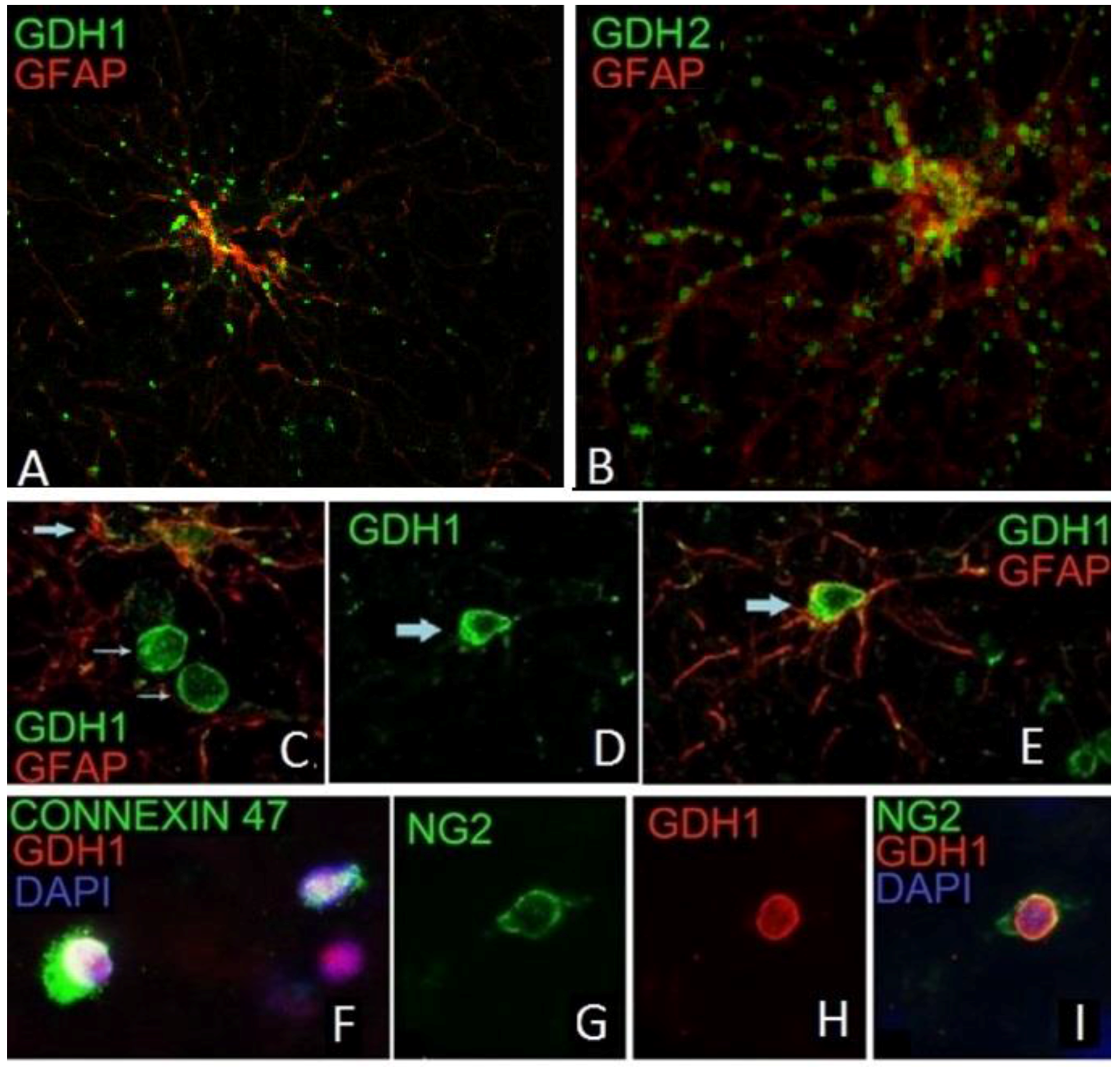
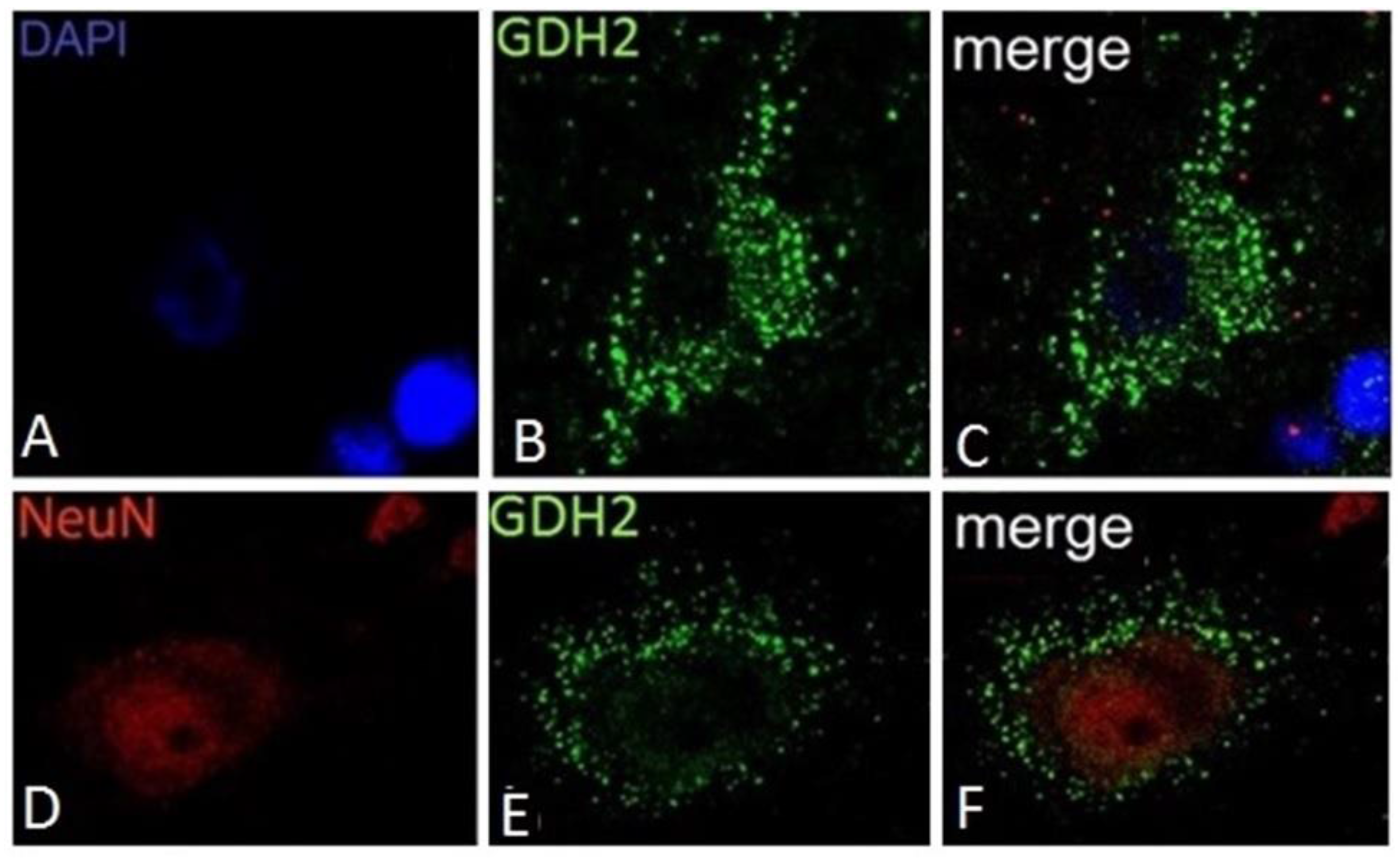
10. Expression of hGDH1 and hGDH2 in Human Cerebral Cortex
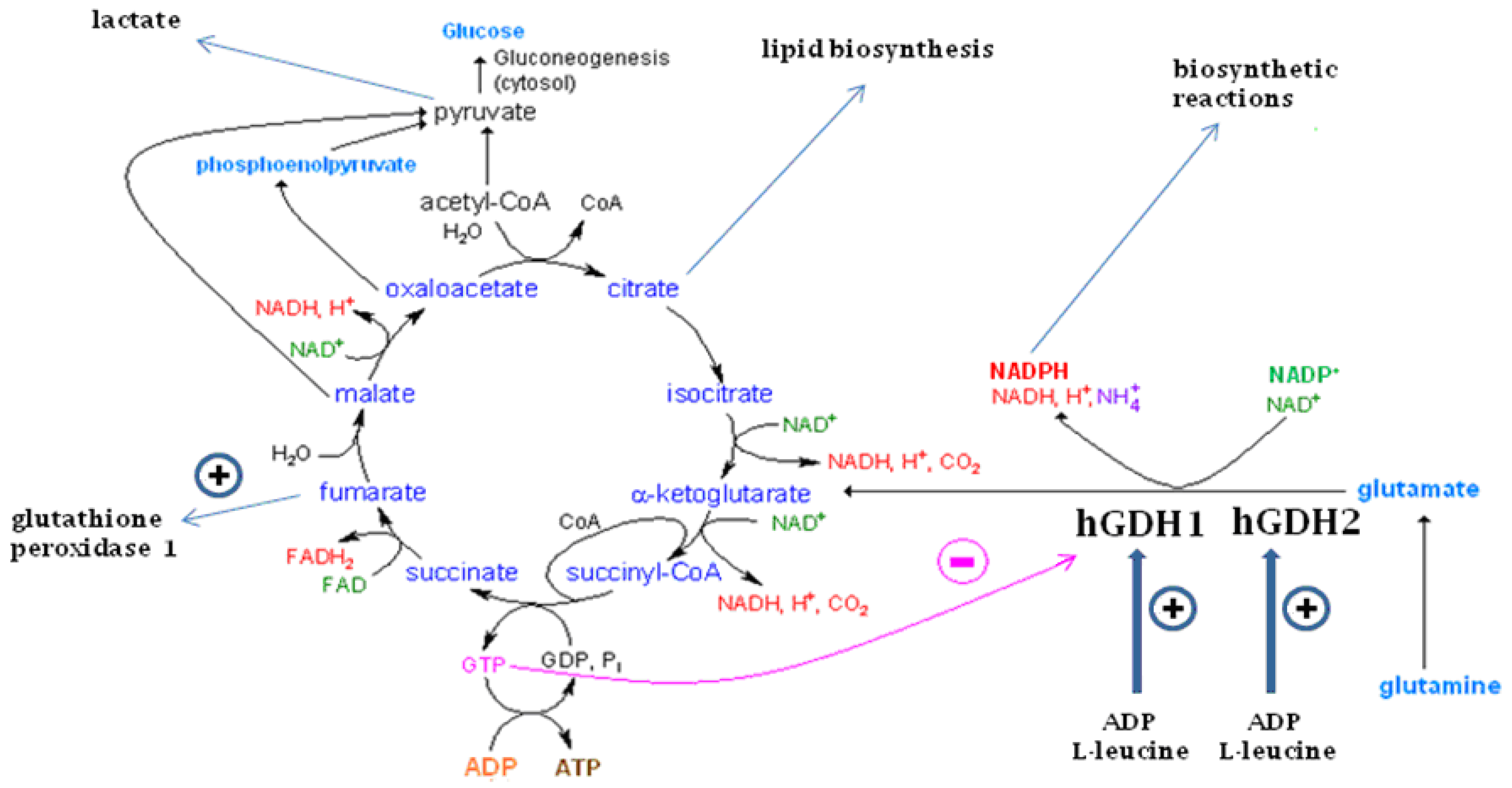
11. Functional Roles of the GDH Pathway in Various Tissues
12. Involvement of the GDH Pathway in Human Disorders
13. Involvement of hGDH1 and hGDH2 in the Biology of Glioma and Other Neoplasias
14. Conclusions
Acknowledgments
Conflicts of Interest
References
- Smith, E.L.; Austin, B.M.; Blumenthal, K.M.; Nyc, J.F. The Enzymes; Boyer, P.D., Ed.; Academic Press: New York, NY, USA, 1975; pp. 293–367. [Google Scholar]
- Duran, R.V.; Oppliger, W.; Robitaille, A.M.; Heiserich, L.; Skendaj, R.; Gottlieb, E.; Hall, M.N. Glutaminolysis activates Rag-mTORC1 signaling. Mol. Cell 2012, 47, 349–358. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Li, D.; Alesi, G.N.; Fan, J.; Kang, H.B.; Lu, Z.; Boggon, T.J.; Jin, P.; Yi, H.; Wright, E.R.; et al. Glutamate dehydrogenase 1 signals through antioxidant glutathione peroxidase 1 to regulate redox homeostasis and tumor growth. Cancer Cell 2015, 27, 257–270. [Google Scholar] [CrossRef] [PubMed]
- Engel, P.C. Glutamate Dehydrogenases: The Why and How of Coenzyme Specificity. Neurochem. Res. 2014, 39, 426–432. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.M.; Magasanik, B. Role of NAD-linked glutamate dehydrogenase in nitrogen metabolism in Saccharomyces cerevisiae. J. Bacteriol. 1990, 172, 4927–4935. [Google Scholar] [CrossRef] [PubMed]
- Avendaño, A.; Deluna, A.; Olivera, H.; Valenzuela, L.; Gonzalez, A. GDH3 encodes a glutamate dehydrogenase isozyme, a previously unrecognized route for glutamate biosynthesis in Saccharomyces cerevisiae. J. Bacteriol. 1997, 179, 5594–5597. [Google Scholar] [CrossRef] [PubMed]
- Plaitakis, A.; Zaganas, I.; Spanaki, C. Deregulation of glutamate dehydrogenase in human neurologic disorders. J. Neurosci Res. 2013, 91, 1007–1017. [Google Scholar] [CrossRef] [PubMed]
- Fontaine, J.X.; Tercé-Laforgue, T.; Armengaud, P.; Clément, G.; Renou, J.P.; Pelletier, S.; Catterou, M.; Azzopardi, M.; Gibon, Y.; Lea, P.J.; et al. Characterization of a NADH-dependent glutamate dehydrogenase mutant of Arabidopsis demonstrates the key role of this enzyme in root carbon and nitrogen metabolism. Plant Cell 2012, 24, 4044–4065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glevarec, G.; Bouton, S.; Jaspard, E.; Riou, M.T.; Cliquet, J.B.; Suzuki, A.; Limami, A.M. Respective roles of the glutamine synthetase/glutamate synthase cycle and glutamate dehydrogenase in ammonium and amino acid metabolism during germination and post-germinative growth in the model legume Medicago truncatula. Planta 2004, 219, 286–297. [Google Scholar] [CrossRef] [PubMed]
- Labboun, S.; Tercé-Laforgue, T.; Roscher, A.; Bedu, M.; Restivo, F.M.; Velanis, C.N.; Skopelitis, D.S.; Moschou, P.N.; Roubelakis-Angelakis, K.A.; Suzuki, A.; et al. Resolving the role of plant glutamate dehydrogenase. I. In vivo real time nuclear magnetic resonance spectroscopy experiments. Plant Cell Physiol. 2009, 50, 1761–1773. [Google Scholar] [CrossRef] [PubMed]
- Masclaux-Daubresse, C.; Reisdorf-Cren, M.; Pageau, K.; Lelandais, M.; Grandjean, O.; Kronenberger, J.; Valadier, M.H.; Feraud, M.; Jouglet, T.; Suzuki, A. Glutamine synthetase-glutamate synthase pathway and glutamate dehydrogenase play distinct roles in the sink-source nitrogen cycle in tobacco. Plant Physiol. 2006, 140, 444–456. [Google Scholar] [CrossRef] [PubMed]
- Dubois, F.; Tercé-Laforgue, T.; Gonzalez-Moro, M.B.; Estavillo, M.B.; Sangwan, R.; Gallais, A.; Hirel, B. Glutamate dehydrogenase in plants; is there a new story for an old enzyme? Plant Physiol. Biochem. 2003, 41, 565–576. [Google Scholar] [CrossRef]
- Fontaine, J.X.; Saladino, F.; Agrimonti, C.; Bedu, M.; Tercé-Laforgue, T.; Tétu, T.; Hirel, B.; Restivo, F.M.; Dubois, F. Control of the synthesis and subcellular targeting of the two GDH genes products in leaves and stems of Nicotiana plumbaginifolia and Arabidopsis thaliana. Plant Cell Physiol. 2006, 47, 410–418. [Google Scholar] [CrossRef] [PubMed]
- Tercé-Laforgue, T.; Dubois, F.; Ferrario-Mery, S.; Pou De Crecenzo, M.A.; Sangwan, R.; Hirel, B. Glutamate dehydrogenase of tobacco is mainly induced in the cytosol of phloem companion cells when ammonia is provided either externally or released during photorespiration. Plant Physiol. 2004, 136, 4308–4317. [Google Scholar] [CrossRef] [PubMed]
- Qiu, X.; Xie, W.; Lian, X.; Zhang, Q. Molecular analyses of the rice glutamate dehydrogenase gene family and their response to nitrogen and phosphorous deprivation. Plant Cell Rep. 2009, 28, 1115–1126. [Google Scholar] [CrossRef] [PubMed]
- Spanaki, C.; Kotzamani, D.; Plaitakis, A. Widening Spectrum of Cellular and Subcellular Expression of Human GLUD1 and GLUD2 Glutamate Dehydrogenases Suggests Novel Functions. Neurochem Res. 2016, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Mastorodemos, V.; Zaganas, I.; Spanaki, C.; Bessa, M.; Plaitakis, A. Molecular basis of human glutamate dehydrogenase regulation under changing energy demands. J. Neurosci. Res. 2005, 79, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Peterson, P.E.; Smith, T.J. The structure of bovine glutamate dehydrogenase provides insights into the mechanism of allostery. Structure 1999, 7, 769–782. [Google Scholar] [CrossRef]
- Smith, T.J.; Schmidt, T.; Fang, J.; Wu, J.; Siuzdak, G.; Stanley, C.A. The structure of apo human glutamate dehydrogenase details subunit communication and allostery. J. Mol. Biol. 2002, 318, 765–777. [Google Scholar] [CrossRef]
- Banerjee, S.; Schmidt, T.; Fang, J.; Stanley, C.A.; Smith, T.J. Structural studies on ADP activation of mammalian glutamate dehydrogenase and the evolution of regulation. Biochemistry 2003, 42, 3446–3456. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Li, C.; Allen, A.; Stanley, C.A.; Smith, T.J. The structure and allosteric regulation of mammalian glutamate dehydrogenase. Arch. Biochem. Biophys. 2012, 519, 69–80. [Google Scholar] [CrossRef] [PubMed]
- Plaitakis, A.; Metaxari, M.; Shashidharan, P. Nerve tissue-specific (GLUD2) and housekeeping (GLUD1) human glutamate dehydrogenases are regulated by distinct allosteric mechanisms: Implications for biologic function. J. Neurochem. 2000, 75, 1862–1869. [Google Scholar] [CrossRef] [PubMed]
- Shashidharan, P.; Michaelidis, T.M.; Robakis, N.K.; Kresovali, A.; Papamatheakis, J.; Plaitakis, A. Novel human glutamate dehydrogenase expressed in neural and testicular tissues and encoded by an X-linked intronless gene. J. Biol. Chem. 1994, 269, 16971–16976. [Google Scholar] [PubMed]
- Shashidharan, P.; Clarke, D.D.; Ahmed, N.; Moschonas, N.; Plaitakis, A. Nerve tissue-specific human glutamate dehydrogenase that is thermolabile and highly regulated by ADP. J. Neurochem. 1997, 68, 1804–1811. [Google Scholar] [CrossRef] [PubMed]
- Burki, F.; Kaessmann, H. Birth and adaptive evolution of a hominoid gene that supports high neurotransmitter flux. Nat. Genet. 2004, 36, 1061–1063. [Google Scholar] [CrossRef] [PubMed]
- Zaganas, I.; Plaitakis, A. Single amino acid substitution (G456A) in the vicinity of the GTP binding domain of human housekeeping glutamate dehydrogenase markedly attenuates GTP inhibition and abolishes the cooperative behavior of the enzyme. J. Biol. Chem. 2002, 277, 26422–26428. [Google Scholar] [CrossRef] [PubMed]
- Kanavouras, K.; Mastorodemos, V.; Borompokas, N.; Spanaki, C.; Plaitakis, A. Properties and molecular evolution of human GLUD2 (neural and testicular tissue-specific) glutamate dehydrogenase. J. Neurosci. Res. 2007, 85, 3398–3406. [Google Scholar] [CrossRef] [PubMed]
- Zaganas, I.; Spanaki, C.; Karpusas, M.; Plaitakis, A. Substitution of Ser for Arg-443 in the regulatory domain of human housekeeping (GLUD1) glutamate dehydrogenase virtually abolishes basal activity and markedly alters the activation of the enzyme by ADP and L-leucine. J. Biol. Chem. 2002, 277, 46552–46558. [Google Scholar] [CrossRef] [PubMed]
- Mavrothalassitis, G.; Tzimagiorgis, G.; Mitsialis, A.; Zannis, V.; Plaitakis, A.; Papamatheakis, J.; Moschonas, N. Isolation and characterization of cDNA clones encoding human liver glutamate dehydrogenase: evidence for a small gene family. Proc. Natl. Acad. Sci. USA 1998, 85, 3494–3498. [Google Scholar] [CrossRef]
- Li, M.; Li, C.; Allen, A.; Stanley, C.A.; Smith, T.J. Glutamate dehydrogenase: Structure, allosteric regulation, and role in insulin homeostasis. Neurochem. Res. 2014, 39, 433–445. [Google Scholar] [CrossRef] [PubMed]
- Rice, D.W.; Baker, P.J.; Farrants, G.W.; Hornby, D.P. The crystal structure of glutamate dehydrogenase from Clostridium symbiosum at 0.6 nm resolution. Biochem. J. 1987, 242, 789–795. [Google Scholar] [CrossRef] [PubMed]
- Stillman, T.J.; Baker, P.J.; Britton, K.L.; Rice, D.W. Conformational flexibility in glutamate dehydrogenase. Role of water in substrate recognition and catalysis. J. Mol. Biol. 1993, 234, 1131–1139. [Google Scholar] [CrossRef] [PubMed]
- Stanley, C.A.; Lieu, Y.K.; Hsu, B.Y.; Burlina, A.B.; Greenberg, C.R.; Hopwood, N.J.; Perlman, K.; Rich, B.H.; Zammarchi, E.; Poncz, M. Hyperinsulinism and hyperammonemia in infants with regulatory mutations of the glutamate dehydrogenase gene. N. Engl. J. Med. 1998, 338, 1352–1357. [Google Scholar] [CrossRef] [PubMed]
- Bailey, J.; Bell, E.T.; Bell, J.E. Regulation of bovine glutamate dehydrogenase. The effects of pH and ADP. J. Biol. Chem. 1982, 257, 5579–5583. [Google Scholar] [PubMed]
- Zaganas, I.; Pajęcka, K.; Wendel Nielsen, C.; Schousboe, A.; Waagepetersen, H.S.; Plaitakis, A. The effect of pH and ADP on ammonia affinity for human glutamate dehydrogenases. Metab. Brain Dis. 2013, 28, 127–131. [Google Scholar] [CrossRef] [PubMed]
- Azarias, G.; Perreten, H.; Lengacher, S.; Poburko, D.; Demaurex, N.; Magistretti, P.J.; Chatton, J.Y. Glutamate transport decreases mitochondrial pH and modulates oxidative metabolism in astrocytes. J. Neurosci. 2011, 31, 3550–3559. [Google Scholar] [CrossRef] [PubMed]
- Van De Poll, M.C.G.; Soeters, P.B.; Deutz, N.E.P.; Fearon, K.C.H.; Dejong, C.H.C. Renal metabolism of amino acids: its role in interorgan amino acid exchange. Am. J. Clin. Nutr. 2004, 79, 185–197. [Google Scholar] [PubMed]
- Fahien, L.A.; Teller, J.K.; Macdonald, M.J.; Fahien, C.M. Regulation of glutamate dehydrogenase by Mg2+ and magnification of leucine activation by Mg2+. Mol. Pharmacol. 1990, 37, 943–949. [Google Scholar] [PubMed]
- Dimovasili, C.; Aschner, M.; Plaitakis, A.; Zaganas, I. Differential interaction of hGDH1 and hGDH2 with manganese: Implications for metabolism and toxicity. Neurochem. Int. 2015, 88, 60–65. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.S.; Vigneron, D.B.; Murphy-Boesch, J.; Nelson, S.J.; Kessler, H.B.; Coia, L.; Curran, W.; Brown, T.R. Free magnesium levels in normal human brain and brain tumors: 31P chemical-shift imaging measurements at 1.5 T. Proc. Natl. Acad. Sci. USA 1991, 88, 6810–6814. [Google Scholar] [CrossRef] [PubMed]
- Mastorodemos, V.; Kanavouras, K.; Sundaram, S.; Providaki, M.; Petraki, Z.; Kokkinidis, M.; Zaganas, I.; Logothetis, D.E.; Plaitakis, A. Side-chain interactions in the regulatory domain of human glutamate dehydrogenase determine basal activity and regulation. J. Neurochem. 2015, 133, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Dieter, H.; Koberstein, R.; Sund, H. Studies of glutamate dehydrogenase. The interaction of ADP [adenosine diphosphate], GTP [guanosin-5-triphosphate], and NADPH [nicotinoid adenine dinucleotide phosphate] in complex with glutamate dehydrogenase [beef liver]. Eur. J. Biochem. 1981, 115, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Sener, A.; Malaisse, W.J. L-leucine and a nonmetabolized analogue activate pancreatic islet glutamate dehydrogenase. Nature 1980, 288, 187–189. [Google Scholar] [CrossRef] [PubMed]
- Fahien, L.A.; Kmiotek, E. Regulation of glutamate dehydrogenase by palmitoyl-coenzyme A. Arch. Biochem. Biophys. 1981, 212, 247–253. [Google Scholar] [CrossRef]
- Jarzyna, R.; Lietz, T.; Bryła, J. Effect of polyamines on glutamate dehydrogenase within permeabilized kidney-cortex mitochondria and isolated renal tubules of rabbit. Biochem. Pharmacol. 1994, 47, 1387–1393. [Google Scholar] [CrossRef]
- Spanaki, C.; Zaganas, I.; Kounoupa, Z.; Plaitakis, A. The complex regulation of human GLUD1 and GLUD2 glutamate dehydrogenases and its implications in nerve tissue biology. Neurochem. Int. 2012, 61, 470–481. [Google Scholar] [CrossRef] [PubMed]
- Yielding, K.L.; Tomkins, G.M. Structural alterations in crystalline glutamic dehydrogenase induced by steroid hormones. Proc. Natl. Acad. Sci. USA 1960, 46, 1483–1488. [Google Scholar] [CrossRef] [PubMed]
- Tomkins, G.M.; Yielding, K.L.; Curran, J.F. The influence of diethylstilbestrol and adenosine diphosphate on pyridine nucleotide coenzyme binding by glutamic dehydrogenase. J. Biol. Chem. 1962, 237, 1704–1708. [Google Scholar] [PubMed]
- Fahien, L.A.; Shemisa, O. Effects of chlorpromazine on glutamate dehydrogenase. Mol. Pharmacol. 1970, 6, 156–163. [Google Scholar] [PubMed]
- Couée, I.; Tipton, K.F. The inhibition of glutamate dehydrogenase by some antipsychotic drugs. Biochem. Pharmacol. 1990, 39, 827–832. [Google Scholar] [CrossRef]
- Plaitakis, A.; Latsoudis, H.; Spanaki, C. The human GLUD2 glutamate dehydrogenase and its regulation in health and disease. Neurochem. Int. 2011, 59, 495–509. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Allen, A.; Kwagh, J.; Doliba, N.M.; Qin, W.; Najafi, H.; Collins, H.W.; Matschinsky, F.M.; Stanley, C.A.; Smith, T.J. Green tea polyphenols modulate insulin secretion by inhibiting glutamate dehydrogenase. J. Biol. Chem. 2006, 281, 10214–10221. [Google Scholar] [CrossRef] [PubMed]
- George, A.; Bell, J.E. Effects of adenosine 5'-diphosphate on bovine glutamate dehydrogenase: Diethyl pyrocarbonate modification. Biochemistry 1980, 19, 6057–6061. [Google Scholar] [CrossRef] [PubMed]
- Smith, T.J.; Stanley, C.A. Untangling the glutamate dehydrogenase allosteric nightmare. Trends Biochem. Sci. 2008, 33, 557–564. [Google Scholar] [CrossRef] [PubMed]
- Kanavouras, K.; Borompokas, N.; Latsoudis, H.; Stagourakis, A.; Zaganas, I.; Plaitakis, A. Mutations in human GLUD2 glutamate dehydrogenase affecting basal activity and regulation. J. Neurochem. 2009, 109, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Borompokas, N.; Papachatzaki, M.M.; Kanavouras, K.; Mastorodemos, V.; Zaganas, I.; Spanaki, C.; Plaitakis, A. Estrogen modification of human glutamate dehydrogenases is linked to enzyme activation state. J. Biol. Chem. 2010, 285, 31380–31387. [Google Scholar] [CrossRef] [PubMed]
- Spanaki, C.; Kotzamani, D.; Petraki, Z.; Drakos, E.; Plaitakis, A. Expression of human GLUD1 and GLUD2 glutamate dehydrogenases in steroid producing tissues. Mol. Cell Endocrinol. 2015, 415, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Herrero-Yraola, A.; Bakhit, S.M.; Franke, P.; Weise, C.; Schweiger, M.; Jorcke, D.; Ziegler, M. Regulation of glutamate dehydrogenase by reversible ADP-ribosylation in mitochondria. EMBO J. 2001, 20, 2404–2412. [Google Scholar] [CrossRef] [PubMed]
- Haigis, M.C.; Mostoslavsky, R.; Haigis, K.M.; Fahie, K.; Christodoulou, D.C.; Murphy, A.J.; Valenzuela, D.M.; Yancopoulos, G.D.; Karow, M.; Blander, G.; et al. SIRT4 inhibits glutamate dehydrogenase and opposes the effects of calorie restriction in pancreatic beta cells. Cell 2006, 126, 941–954. [Google Scholar] [CrossRef] [PubMed]
- Molven, A.; Matre, G.E.; Duran, M.; Wanders, R.J.; Rishaug, U.; Njølstad, P.R.; Jellum, E.; Søvik, O. Familial hyperinsulinemic hypoglycemia caused by a defect in the SCHAD enzyme of mitochondrial fatty acid oxidation. Diabetes 2004, 53, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Chen, P.; Palladino, A.; Narayan, S.; Russell, L.K.; Sayed, S.; Xiong, G.; Chen, J.; Stokes, D.; Butt, Y.M.; et al. Mechanism of hyperinsulinism in short-chain 3-hydroxyacyl-CoA dehydrogenase deficiency involves activation of glutamate dehydrogenase. J. Biol. Chem. 2010, 285, 31806–31818. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, E.; Schmidt, F.W. Distribution pattern of several enzymes in human liver and its variations during cell damage.III. On the methodology of enzyme determination in human organ extracts and serum. Enzymol. Biol. Clin. 1963, 35, 73–79. [Google Scholar]
- Salganicoff, L.; De Robertis, E. Subcellular distribution of the enzymes of the glutamic accidm glutamine and gamma-aminobutyric acid cycles in rat brain. J. Neurochem. 1965, 12, 287–309. [Google Scholar] [CrossRef] [PubMed]
- Fahien, L.A.; Strmecki, M.; Smith, S. Studies of gluconeogenic mitochondrial enzymes. I. A new method of preparing bovine liver glutamate dehydrogenase and effects of purification methods on properties of the enzyme. Arch. Biochem. Biophys. 1969, 130, 449–455. [Google Scholar] [CrossRef]
- Aoki, C.; Milner, T.A.; Berger, S.B.; Sheu, K.F.; Blass, J.P.; Pickel, V.M. Glial glutamate dehydrogenase: ultrastructural localization and regional distribution in relation to the mitochondrial enzyme, cytochrome oxidase. J. Neurosci. Res. 1987, 18, 305–318. [Google Scholar] [CrossRef] [PubMed]
- Rothe, F.; Brosz, M.; Storm-Mathisen, J. Quantitative ultrastructural localization of glutamate dehydrogenase in the rat cerebellar cortex. Neuroscience 1994, 62, 1133–1146. [Google Scholar] [CrossRef]
- Colon, A.D.; Plaitakis, A.; Perakis, A.; Berl, S.; Clarke, D.D. Purification and characterization of a soluble and a particulate glutamate dehydrogenase from rat brain. J. Neurochem. 1986, 46, 1811–1819. [Google Scholar] [CrossRef] [PubMed]
- Rajas, F.; Rousset, B. A membrane-bound form of glutamate dehydrogenase possesses an ATP-dependent high-affinity microtubule-binding activity. Biochem. J. 1993, 295, 447–455. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.K.; Shin, S.; Cho, S.S.; Park, J.S. Purification and characterization of glutamate dehydrogenase as another isoprotein binding to the membrane of rough endoplasmic reticulum. J. Cell Biochem. 1999, 76, 244–253. [Google Scholar] [CrossRef]
- Di Prisco, G.; Banay-Schwartz, M.; Strecker, H.J. Glutamate dehydrogenase in nuclear and mitochondrial fractions of rat liver. Biochem. Biophys. Res. Commun. 1968, 33, 606–612. [Google Scholar] [CrossRef]
- Lai, J.C.; Sheu, K.F.; Kim, Y.T.; Clarke, D.D.; Blass, J.P. The subcellular localization of glutamate dehydrogenase (GDH): is GDH a marker for mitochondria in brain? Neurochem. Res. 1986, 11, 733–744. [Google Scholar] [CrossRef] [PubMed]
- Panda, P.; Suar, M.; Singh, D.; Pandey, S.M.; Chaturvedi, M.M.; Purohit, J.S. Characterization of nuclear glutamate dehydrogenase of chicken liver and brain. Protein Pept. Lett. 2011, 18, 1194–1203. [Google Scholar] [CrossRef] [PubMed]
- Spanaki, C.; Kotzamani, D.; Kleopa, K.; Plaitakis, A. Evolution of GLUD2 Glutamate Dehydrogenase Allows Expression in Human Cortical Neurons. Mol. Neurobiol. 2016, 53, 5140–5148. [Google Scholar] [CrossRef] [PubMed]
- Camardella, L.; Di Prisco, G.; Garofano, F.; Guerrini, A.M. Nuclear and cytoplasmic glutamate dehydrogenases (NADP-dependent) in Saccharomyces cerevisiae. Biochem. Biophys. Res. Commun. 1975, 64, 773–777. [Google Scholar] [CrossRef]
- Tiwari, A.K.; Panda, P.; Purohit, J.S. Evaluation of sub-cellular distribution of glutamate dehydrogenase (GDH) in Drosophila melanogaster larvae. Acta Histochem. 2014, 116, 297–303. [Google Scholar] [CrossRef] [PubMed]
- Mastorodemos, V.; Kotzamani, D.; Zaganas, I.; Arianoglou, G.; Latsoudis, H.; Plaitakis, A. Human GLUD1 and GLUD2 glutamate dehydrogenase localize to mitochondria and endoplasmic reticulum. Biochem. Cell Biol. 2009, 87, 505–516. [Google Scholar] [CrossRef] [PubMed]
- Rosso, L.; Marques, A.C.; Reichert, A.S.; Kaessmann, H. Mitochondrial targeting adaptation of the hominoid-specific glutamate dehydrogenase driven by positive Darwinian selection. PLoS Genet. 2008, 4, e1000150. [Google Scholar] [CrossRef] [PubMed]
- Cormack, B.P.; Valdivia, R.H.; Falkow, S. FACS-optimized mutants of the green fluorescent protein (GFP). Gene. 1996, 173, 33–38. [Google Scholar] [CrossRef]
- Kotzamani, D.; and Plaitakis, A. Alpha helical structures in the leader sequence of human GLUD2 glutamate dehydrogenase responsible for mitochondrial import. Neurochem. Int. 2012, 61, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Kalef-Ezra, E.; Kotzamani, D.; Zaganas, I.; Katrakili, N.; Plaitakis, A.; Tokatlidis, K. Import of a major mitochondrial enzyme depends on synergy between two distinct helices of its presequence. Biochem. J. 2016, 473, 2813–2829. [Google Scholar] [CrossRef] [PubMed]
- Kachroo, A.H.; Laurent, J.M.; Yellman, C.M.; Meyer, A.G.; Wilke, C.O.; Marcotte, E.M. Systematic humanization of yeast genes reveals conserved functions and genetic modularity. Science 2015, 348, 921–925. [Google Scholar] [CrossRef] [PubMed]
- Altmann, K.; Dürr, M.; Westermann, B. Saccharomyces cerevisiae as a model organism to study mitochondrial biology: general considerations and basic procedures. Methods Mol. Biol. 2007, 372, 81–90. [Google Scholar] [PubMed]
- Roise, D.; Schatz, G. Mitochondrial presequences. J. Biol. Chem. 1988, 263, 4509–4511. [Google Scholar] [PubMed]
- Chacinska, A.; Koehler, C.M.; Milenkovic, D.; Lithgow, T.; Pfanner, N. Importing mitochondrial proteins: Machineries and mechanisms. Cell 2009, 138, 628–644. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Patel, N.D.; Hui, D.; Pal, R.; Hafez, M.M.; Sayed-Ahmed, M.M.; Al-Yahya, A.A.; Michaelis, E.K. Gene expression patterns in the hippocampus during the development and aging of Glud1 (Glutamate Dehydrogenase 1) transgenic and wild type mice. BMC Neurosci. 2014, 15, 37–53. [Google Scholar] [CrossRef] [PubMed]
- Yano, M.; Kanazawa, M.; Terada, K.; Takeya, M.; Hoogenraad, N.; Mori, M. Functional analysis of human mitochondrial receptor Tom20 for protein import into mitochondria. J. Biol. Chem. 1998, 273, 26844–26851. [Google Scholar] [CrossRef] [PubMed]
- Hachiya, N.; Mihara, K.; Suda, K.; Horst, M.; Schatz, G.; Lithgow, T. Reconstitution of the initial steps of mitochondrial protein import. Nature 1995, 376, 705–709. [Google Scholar] [CrossRef] [PubMed]
- Botman, D.; Tigchelaar, W.; Van Noorden, C.J. Determination of glutamate dehydrogenase activity and its kinetics in mouse tissues using metabolic mapping (quantitative enzyme histochemistry). J. Histochem. Cytochem. 2014, 62, 802–812. [Google Scholar] [CrossRef] [PubMed]
- Treberg, J.R.; Banh, S.; Pandey, U.; Weihrauch, D. Intertissue differences for the role of glutamate dehydrogenase in metabolism. Neurochem. Res. 2014, 39, 516–526. [Google Scholar] [CrossRef] [PubMed]
- Spanaki, C.; Zaganas, I.; Kleopa, K.A.; Plaitakis, A. Human GLUD2 glutamate dehydrogenase is expressed in neural and testicular supporting cells. J. Biol. Chem. 2010, 285, 16748–16756. [Google Scholar] [CrossRef] [PubMed]
- Spanaki, C.; Kotzamani, D.; Petraki, Z.; Drakos, E.; Plaitakis, A. Heterogeneous cellular distribution of glutamate dehydrogenase in brain and in non-neural tissues. Neurochem. Res. 2014, 39, 500–515. [Google Scholar] [CrossRef] [PubMed]
- Zaganas, I.; Spanaki, C.; Plaitakis, A. Expression of human GLUD2 glutamate dehydrogenase in human tissues: Functional implications. Neurochem. Int. 2012, 61, 455–462. [Google Scholar] [CrossRef] [PubMed]
- Leong, S.F.; Clark, J.B. Regional development of glutamate dehydrogenase in the rat brain. J. Neurochem. 1984, 43, 106–111. [Google Scholar] [CrossRef] [PubMed]
- Rothe, F.; Schmidt, W.; Wolf, G. Postnatal changes in the activity of glutamate dehydrogenase and aspartate aminotransferase in the rat nervous system with special reference to the glutamate transmitter metabolism. Brain Res. 1983, 313, 67–74. [Google Scholar] [CrossRef]
- Zaganas, I.; Waagepetersen, H.S.; Georgopoulos, P.; Sonnewald, U.; Plaitakis, A.; Schousboe, A. Differential expression of glutamate dehydrogenase in cultured neurons and astrocytes from mouse cerebellum and cerebral cortex. J. Neurosci. Res. 2001, 66, 909–913. [Google Scholar] [CrossRef] [PubMed]
- Aoki, C.; Milner, T.A.; Sheu, K.F.; Blass, J.P.; Pickel, V.M. Regional distribution of astrocytes with intense immunoreactivity for glutamate dehydrogenase in rat brain: implications for neuron-glia interactions in glutamate transmission. J. Neurosci. 1987, 7, 2214–2231. [Google Scholar] [PubMed]
- Wenthold, R.J.; Altschuler, R.A.; Skaggs, K.K.; Reeks, K.A. Immunocytochemical characterization of glutamate dehydrogenase in the cerebellum of the rat. J. Neurochem. 1987, 48, 636–643. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, A.; Kugler, P. Cellular and regional expression of glutamate dehydrogenase in the rat nervous system: non-radioactive in situ hybridization and comparative immunocytochemistry. Neuroscience 1999, 92, 293–308. [Google Scholar] [CrossRef]
- Nissim, I.; Brosnan, M.E.; Yudkoff, M.; Brosnan, J.T. Studies of hepatic glutamine metabolism in the perfused rat liver with (15) N-labeled glutamine. J. Biol. Chem. 1999, 274, 28958–28965. [Google Scholar] [CrossRef] [PubMed]
- Krebs, H.A.; Gascoyne, T. The redox state of the nicotinamide-adenine dinucleotides in rat liver homogenates. Biochem. J. 1968, 108, 513–520. [Google Scholar] [CrossRef] [PubMed]
- Brosnan, J.T.; Brosnan, M.E.; Charron, R.; Nissim, I. A mass isotopomer study of urea and glutamine synthesis from 15N-labeled ammonia in the perfused rat liver. J. Biol. Chem. 1996, 271, 16199–16207. [Google Scholar] [CrossRef] [PubMed]
- Boon, L.; Geerts, W.J.; Jonker, A.; Lamers, W.H.; Van Noorden, C.J. High protein diet induces pericentral glutamate dehydrogenase and ornithine aminotransferase to provide sufficient glutamate for pericentral detoxification of ammonia in rat liver lobules. Histochem. Cell Biol. 1999, 111, 445–452. [Google Scholar] [CrossRef] [PubMed]
- Cooper, A.J.; Nieves, E.; Coleman, A.E.; Filc-DeRicco, S.; Gelbard, A.S. Short-term metabolic fate of [13N] ammonia in rat liver in vivo. J. Biol. Chem. 1987, 262, 1073–1080. [Google Scholar] [PubMed]
- Cooper, A.J.; Nieves, E.; Rosenspire, K.C.; Filc-DeRicco, S.; Gelbard, A.S.; Brusilow, S.W. Short-term metabolic fate of 13N-labeled glutamate, alanine, and glutamine(amide) in rat liver. J. Biol. Chem. 1988, 263, 12268–12273. [Google Scholar] [PubMed]
- Carobbio, S.; Ishihara, H.; Fernandez-Pascual, S.; Bartley, C.; Martin-Del-Rio, R.; Maechler, P. Insulin secretion profiles are modified by overexpression of glutamate dehydrogenase in pancreatic islets. Diabetologia 2004, 47, 266–276. [Google Scholar] [CrossRef] [PubMed]
- Vetterli, L.; Carobbio, S.; Frigerio, F.; Karaca, M.; Maechler, P. The Amplifying Pathway of the β-Cell Contributes to Diet-induced Obesity. J. Biol. Chem. 2016, 291, 13063–13075. [Google Scholar] [CrossRef] [PubMed]
- Sonnewald, U.; Westergaard, N.; Petersen, S.B.; Unsgård, G.; Schousboe, A. Metabolism of [U-13C] glutamate in astrocytes studied by 13C NMR spectroscopy: Incorporation of more label into lactate than into glutamine demonstrates the importance of the tricarboxylic acid cycle. J. Neurochem. 1993, 61, 1179–1182. [Google Scholar] [CrossRef] [PubMed]
- Treberg, J.R.; Clow, K.A.; Greene, K.A.; Brosnan, M.E.; Brosnan, J.T. Systemic activation of glutamate dehydrogenase increases renal ammoniagenesis: implications for the hyperinsulinism/hyperammonemia syndrome. Am. J. Physiol. Endocrinol. Metab. 2010, 298, E1219–E1225. [Google Scholar] [CrossRef] [PubMed]
- Spanaki, C.; Plaitakis, A. The role of glutamate dehydrogenase in mammalian ammonia metabolism. Neurotox. Res. 2012, 21, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Cooper, A.J. 13N as a tracer for studying glutamate metabolism. Neurochem. Int. 2011, 59, 456–464. [Google Scholar] [CrossRef] [PubMed]
- Cooper, A.J.; McDonald, J.M.; Gelbard, A.S.; Gledhill, R.F.; Duffy, T.E. The metabolic fate of 13N-labeled ammonia in rat brain. J. Biol. Chem. 1979, 254, 4982–4992. [Google Scholar] [PubMed]
- Berl, S.; Takagaki, G.; Clarke, D.D.; Waelsch, H. Metabolic compartments in vivo. Ammonia and glutamic acid metabolism in brain and liver. J. Biol. Chem. 1962, 237, 2562–2569. [Google Scholar] [PubMed]
- Nadler, J.V.; White, W.F.; Vaca, K.W.; Perry, B.W.; Cotman, C.W. Biochemical correlates of transmission mediated by glutamate and aspartate. J. Neurochem. 1978, 31, 147–155. [Google Scholar] [CrossRef] [PubMed]
- Schousboe, A.; Scafidi, S.; Bak, L.K.; Waagepetersen, H.S.; McKenna, M.C. Glutamate metabolism in the brain focusing on astrocytes. Adv. Neurobiol. 2014, 11, 13–30. [Google Scholar] [PubMed]
- Yu, A.C.; Schousboe, A.; Hertz, L. Metabolic fate of 14C-labeled glutamate in astrocytes in primary cultures. J. Neurochem. 1982, 39, 954–960. [Google Scholar] [CrossRef] [PubMed]
- McKenna, M.C.; Sonnewald, U.; Huang, X.; Stevenson, J.; Zielke, H.R. Exogenous glutamate concentration regulates the metabolic fate of glutamate in astrocytes. J. Neurochem. 1996, 66, 386–393. [Google Scholar] [CrossRef] [PubMed]
- McKenna, M.C.; Stridh, M.H.; McNair, L.F.; Sonnewald, U.; Waagepetersen, H.S.; Schousboe, A. Gluatamte oxidation in astrocytes: roles of glutamate dehydrogenase and aminotransferases. J. Neurosci. Res. 2016, 94, 1561–1571. [Google Scholar] [CrossRef] [PubMed]
- Cooper, A.J.; Jeitner, T.M. Central role of glutamate metabolism in the maintenance of nitrogen homeostasis in normal and hyperammonemic brain. Biomolecules 2016, 6, 16. [Google Scholar] [CrossRef] [PubMed]
- Frigerio, F.; Karaca, M.; De Roo, M.; Mlynárik, V.; Skytt, D.M.; Carobbio, S.; Pajęcka, K.; Waagepetersen, H.S.; Gruetter, R.; Muller, D.; et al. Deletion of glutamate dehydrogenase 1 (Glud1) in the central nervous system affects glutamate handling without altering synaptic transmission. J. Neurochem. 2012, 123, 342–348. [Google Scholar] [CrossRef] [PubMed]
- Nissen, J.D.; Pajęcka, K.; Stridh, M.H.; Skytt, D.M.; Waagepetersen, H.S. Dysfunctional TCA-cycle metabolism in glutamate dehydrogenase deficient astrocytes. Glia 2015, 63, 2313–2326. [Google Scholar] [CrossRef] [PubMed]
- Karaca, M.; Frigerio, F.; Migrenne, S.; Martin-Levilain, J.; Skytt, D.M.; Pajecka, K.; Martin-del-Rio, R.; Gruetter, R.; Tamarit-Rodriguez, J.; Waagepetersen, H.S.; et al. GDH-Dependent glutamate oxidation in the brain dictates peripheral energy substrate distribution. Cell Rep. 2015, 13, 365–375. [Google Scholar] [CrossRef] [PubMed]
- Bao, X.; Pal, R.; Hascup, K.N.; Wang, Y.; Wang, W.T.; Xu, W.; Hui, D.; Agbas, A.; Wang, X.; Michaelis, M.L.; et al. Transgenic expression of Glud1 (glutamate dehydrogenase 1) in neurons: in vivo model of enhanced glutamate release, altered synaptic plasticity, and selective neuronal vulnerability. J. Neurosci. 2009, 29, 13929–13944. [Google Scholar] [CrossRef] [PubMed]
- Schaller, B.; Mekle, R.; Xin, L.; Kunz, N.; Gruetter, R. Net increase of lactate and glutamate concentration in activated human visual cortex detected with magnetic resonance spectroscopy at 7 tesla. J. Neurosci. Res. 2013, 91, 1076–1083. [Google Scholar] [CrossRef] [PubMed]
- Palaiologos, G.; Hertz, L.; Schousboe, A. Role of aspartate aminotransferase and mitochondrial dicarboxylate transport for release of endogenously and exogenously supplied neurotransmitter in glutamatergic neurons. Neurochem. Res. 1989, 14, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Guo, S.; Jiang, X.; Bryk, J.; Naumann, R.; Enard, W.; Tomita, M.; Sugimoto, M.; Khaitovich, P.; Pääbo, S. Mice carrying a human GLUD2 gene recapitulate aspects of human transcriptome and metabolome development. Proc. Natl. Acad Sci. USA 2016, 113, 5358–5363. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Nishimura, M.C.; Kharbanda, S.; Peale, F.; Deng, Y.; Daemen, A.; Forrest, W.F.; Kwong, M.; Hedehus, M.; Hatzivassiliou, G.; et al. Hominoid-specific enzyme GLUD2 promotes growth of IDH1R132H glioma. Proc. Natl. Acad. Sci. USA 2014, 111, 14217–14222. [Google Scholar] [CrossRef] [PubMed]
- Salminen, A.; Kauppinen, A.; Hiltunen, M.; Kaarniranta, K. Krebs cycle intermediates regulate DNA and histone methylation: epigenetic impact on the aging process. Ageing Res. Rev. 2014, 16, 45–65. [Google Scholar] [CrossRef] [PubMed]
- Carey, B.W.; Finley, L.W.; Cross, J.R.; Allis, C.D.; Thompson, C.B. Intracellular α-ketoglutarate maintains the pluripotency of embryonic stem cells. Nature 2015, 518, 413–416. [Google Scholar] [CrossRef] [PubMed]
- Raizen, D.M.; Brooks-Kayal, A.; Steinkrauss, L.; Tennekoon, G.I.; Stanley, C.A.; Kelly, A. Central nervous system hyperexcitability associated with glutamate dehydrogenase gain of function mutations. J. Pediatr. 2005, 146, 388–394. [Google Scholar] [CrossRef] [PubMed]
- Scharfman, H.E. Metabolic control of epilepsy. Science 2015, 347, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Malthankar-Phatak, G.H.; De Lanerolle, N.; Eid, T.; Spencer, D.D.; Behar, K.L.; Spencer, S.S.; Kim, J.H.; Lai, J.C. Differential glutamate dehydrogenase (GDH) activity profile in patients with temporal lobe epilepsy. Epilepsia 2006, 47, 1292–1299. [Google Scholar] [CrossRef] [PubMed]
- Sherwin, L.A. Neuroactive amino acids in focally epileptic human brain: a review. Neurochem. Res. 1999, 24, 1387–1395. [Google Scholar] [CrossRef] [PubMed]
- Vega Rasgado, L.A.; Reyes, G.C.; Díaz, F.V. Anticonvulsant drugs, brain glutamate dehydrogenase activity and oxygen consumption. ISRN Pharmacol. 2012, 2012. [Google Scholar] [CrossRef] [PubMed]
- Vega Rasgado, L.A.; Reyes, G.C.; Díaz, F.V. Effect of convulsant drugs in GDH activity and oxygen consumption in mouse brain. J. Med. Med. Sci. 2013, 4, 34–42. [Google Scholar]
- Vega Rasgado, L.A.; Reyes, G.C.; Díaz, F.V. Modulation of brain glutamate dehydrogenase as a tool for controlling seizures. Acta. Pharm. 2015, 65, 443–452. [Google Scholar] [CrossRef] [PubMed]
- Bahi-Buisson, N.; Roze, E.; Dionisi, C.; Escande, F.; Valayannopoulos, V.; Feillet, F.; Heinrichs, C.; Chadefaux-Vekemans, B.; Dan, B.; De Lonlay, P. Neurological aspects of hyperinsulinism-hyperammonaemia syndrome. Dev. Med. Child Neurol. 2008, 50, 945–949. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, R.; Goto, S.; Sako, W.; Miyashiro, A.; Kim, I.; Escande, F.; Harada, M.; Morigaki, R.; Asanuma, K.; Mizobuchi, Y.; et al. Generalized dystonia in a patient with a novel mutation in the GLUD1 gene. Mov. Disord. 2012, 27, 1198–1199. [Google Scholar] [CrossRef] [PubMed]
- Plaitakis, A.; Latsoudis, H.; Kanavouras, K.; Ritz, B.; Bronstein, J.M.; Skoula, I.; Mastorodemos, V.; Papapetropoulos, S.; Borompokas, N.; Zaganas, I.; et al. Gain-of-function variant in GLUD2 glutamate dehydrogenase modifies Parkinson's disease onset. Eur. J. Hum. Genet. 2010, 18, 336–341. [Google Scholar] [CrossRef] [PubMed]
- Boroughs, L.K.; DeBerardinis, R.J. Metabolic pathways promoting cancer cell survival and growth. Nat. Cell Biol. 2015, 17, 351–359. [Google Scholar] [CrossRef] [PubMed]
- McGuirk, S.; Gravel, S.P.; Deblois, G.; Papadopoli, D.J.; Faubert, B.; Wegner, A.; Hiller, K.; Avizonis, D.; Akavia, U.D.; Jones, R.G.; et al. PGC-1α supports glutamine metabolism in breast cancer. Cancer Metab. 2013, 1, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Timmerman, L.A.; Holton, T.; Yuneva, M.; Louie, R.J.; Padró, M.; Daemen, A.; Hu, M.; Chan, D.A.; Ethier, S.P.; Van 't Veer, L.J.; et al. Glutamine sensitivity analysis identifies the xCT antiporter as a common triple-negative breast tumor therapeutic target. Cancer Cell. 2013, 24, 450–465. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Moss, T.; Mangala, L.S.; Marini, J.; Zhao, H.; Wahlig, S.; Armaiz-Pena, G.; Jiang, D.; Achreja, A.; Win, J.; et al. Metabolic shifts toward glutamine regulate tumor growth, invasion and bioenergetics in ovarian cancer. Mol. Syst. Biol. 2014, 10, 728–750. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J.; Cheng, T. Q’s next: The diverse functions of glutamine in metabolism, cell biology and cancer. Oncogene 2010, 29, 313–324. [Google Scholar] [CrossRef] [PubMed]
- Csibi, A.; Fendt, S.M.; Li, C.; Poulogiannis, G.; Choo, A.Y.; Chapski, D.J.; Jeong, S.M.; Dempsey, J.M.; Parkhitko, A.; Morrison, T.; et al. The mTORC1 pathway stimulates glutamine metabolism and cell proliferation by repressing SIRT4. Cell 2013, 153, 840–854. [Google Scholar] [CrossRef]
- Friday, E.; Oliver, R.; Welbourne, T.; Turturro, F. Glutaminolysis and glycolysis regulation by troglitazone in breast cancer cells: Relationship to mitochondrial membrane potential. J. Cell. Physiol. 2011, 226, 511–519. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Zhu, J.; Yu, M.; Cai, C.; Zhou, Y.; Yu, M.; Fu, Z.; Gong, Y.; Yang, B.; Li, Y.; et al. Glutamate dehydrogenase is a novel prognostic marker and predicts metastases in colorectal cancer patients. J. Transl. Med. 2015, 13, 144–153. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wang, G.; Mao, Q.; Li, S.; Xiong, W.; Lin, Y.; Ge, J. Glutamate dehydrogenase (GDH) regulates bioenergetics and redox homeostasis in human glioma. Oncotarget 2016. [Google Scholar] [CrossRef]
- Yang, C.; Sudderth, J.; Dang, T.; Bachoo, R.J.; MacDonald, J.G.; DeBernardinis, R.J. Glioblastoma cells require glutamate dehydrogenase to survive impairments of glucose metabolism or Akt signaling. Cancer Res. 2009, 69, 7986–7993. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Ye, D.; Guan, K.L.; Xiong, Y. IDH1 and IDH2 mutations in tumorigenesis: mechanistic insights and clinical perspectives. Clin. Cancer Res. 2012, 18, 5562–5571. [Google Scholar] [CrossRef] [PubMed]
- Parsons, D.W.; Jones, S.; Zhang, X.; Lin, J.C.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Siu, I.M.; Gallia, G.L.; et al. An integrated genomic analysis of human glioblastoma multiforme. Science 2008, 321, 1807–1812. [Google Scholar] [CrossRef] [PubMed]
- Waitkus, M.S.; Diplas, B.H.; Yan, H. Isocitrate dehydrogenase mutations in gliomas. Neuro. Oncol. 2016, 18, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Gross, S.; Cairns, R.A.; Minden, M.D.; Driggers, E.M.; Bittinger, M.A.; Jang, H.G.; Sasaki, M.; Jin, S.; Schenkein, D.P.; Su, S.M.; et al. Cancer-associated metabolite 2-hydroxyglutarate accumulates in acute myelogenous leukemia with isocitrate dehydrogenase 1 and 2 mutations. J. Exp. Med. 2010, 207, 339–344. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Plaitakis, A.; Kalef-Ezra, E.; Kotzamani, D.; Zaganas, I.; Spanaki, C. The Glutamate Dehydrogenase Pathway and Its Roles in Cell and Tissue Biology in Health and Disease. Biology 2017, 6, 11. https://doi.org/10.3390/biology6010011
Plaitakis A, Kalef-Ezra E, Kotzamani D, Zaganas I, Spanaki C. The Glutamate Dehydrogenase Pathway and Its Roles in Cell and Tissue Biology in Health and Disease. Biology. 2017; 6(1):11. https://doi.org/10.3390/biology6010011
Chicago/Turabian StylePlaitakis, Andreas, Ester Kalef-Ezra, Dimitra Kotzamani, Ioannis Zaganas, and Cleanthe Spanaki. 2017. "The Glutamate Dehydrogenase Pathway and Its Roles in Cell and Tissue Biology in Health and Disease" Biology 6, no. 1: 11. https://doi.org/10.3390/biology6010011