
Hypoxia-Driven Responses in Chronic Kidney Disease

1
Institute of Experimental Medicine and Systems Biology, RWTH Aachen University Hospital, 52074 Aachen, Germany
2
Faculty of Science, Universidad Autónoma de Madrid, 28049 Madrid, Spain
*
Author to whom correspondence should be addressed.
Oxygen 2023, 3(3), 300-321; https://doi.org/10.3390/oxygen3030020
Submission received: 29 May 2023
/
Revised: 23 June 2023
/
Accepted: 6 July 2023
/
Published: 12 July 2023
Abstract
:Chronic kidney disease (CKD) affects 10% of the population. Fibrosis is the hallmark of CKD, which is marked by the deposit of extracellular matrix (ECM). This response is the final outcome of an unbalanced reaction to inflammation and wound healing and can be induced by a variety of insults, including hypoxia. Vascular damage results in an impaired tissue oxygen supply, inducing immune cell infiltration, tubule injury and the activation of ECM-secreting myofibroblasts. In turn, tubulointerstitial fibrosis development worsens oxygen diffusion. Hypoxia-inducible factor (HIF) is the primary transcriptional regulator of hypoxia-associated responses, such as oxidative stress and metabolic reprogramming, triggering a proinflammatory and profibrotic landscape. In this review, we discuss hypoxia-driven reprogramming in CKD as well as potential therapeutic approaches to target chronic hypoxia.
1. Chronic Kidney Disease
Chronic kidney disease (CKD) is a clinical setting with a gradual decline in renal function. It has high rates of morbidity and mortality and constitutes a significant medical, social and economic burden. It affects about 10% of the global population and affects roughly 30–40% of patients with high prevalent pathologies, such as diabetes mellitus and hypertension [1]. Acute kidney injury (AKI) is a risk factor for developing CKD, whereas CKD sensitizes patients to AKI [2]. While a lot has been learned about the mechanisms underlying these diseases, the causes predisposing AKI to CKD and vice versa remain largely elusive. High rates of cardiac diseases are reported in CKD patients with chronic inflammation, which may result in a reduced blood supply to the kidneys [3]. Dysregulation of the immune system following AKI could be a major factor contributing to increased risk of CKD based on the incomplete tissue relocation of immune cells after injury [4,5]. Genetic conditions such as polycystic kidney disease (PKD), immune glomerulopathies and exposure to environmental chemicals are less frequent causes of CKD. Within the next 20 years, CKD is ranked to be the fifth leading cause of death worldwide [6]. Although multiple interventions have been used to slow the progression of CKD, including strict control of blood pressure and glycaemia, correction of dyslipidemia, halting of potentially nephrotoxic drugs or smoking, blockade of the renin-angiotensin system to lower glomerular capillary pressure and immunosuppression in some rapidly progressing disorders, none of the current tactics have shown effectiveness to revert the progression to end-stage renal disease (ESRD). Sodium-glucose cotransporter-2 (SGLT2) inhibition showed promising in recent trials. However, most of CKD patients advance toward ESRD, which requires dialysis or kidney transplant [7].
Nephrons are the functional units of the kidney, formed by glomeruli and tubules. The glomerulus retains cells and big proteins while filtering the blood. The filtrate enters the tubule to produce the final urine by eliminating or adding substances to the tubular fluid. Kidneys keep the interior environment in a state of homeostasis by maintaining the proper balance of water, minerals, electrolytes and hydrogen ions as well as removing toxins. That is the case of uremic toxins, substances produced by protein metabolism that accumulate in patients with impaired renal function, contributing to CKD progression. Many of these protein-bound uremic toxins are gut-derived from the degradation of aromatic amino acids by intestinal bacteria. The most-studied ones are p-cresol, p-cresyl sulfate, p-cresyl glucuronide, indoxyl sulfate, indole-3-acetic acid, trimethylamine-N-oxide, phenylacetylglutamine and hippuric acid [8]. Regardless of etiology, CKD progression involves the development of tubulointerstitial and glomerular fibrosis. It involves the excessive accumulation of extracellular matrix proteins (ECM), such as fibronectin, proteoglycans and collagens, which replace the cellular living tissue. This is the result of an unbalanced response to inflammation and wound healing that activates ECM-secreting myofibroblasts [9].
Renal injury, caused by toxic compounds, hypoxia or proteinuria, promotes rarefaction of the peritubular microvasculature and damages the tubular epithelium, which leads to its subsequent dedifferentiation or cell death. Reduced number of functional nephrons results in hyperfiltration by the remnant kidney, increasing biomechanical forces that can determine cellular injury. The responses underlying tubular damage are complex, involving oxidative stress, metabolic alterations, cell cycle arrest, dedifferentiation, senescence, inflammatory mediators and epigenetic changes. After the first insult, the remaining cells go through a recovery phase during which regeneration mechanisms are activated to restore epithelial cell characteristics and functions. An incomplete repair promotes fibrosis and CKD progression. By secreting paracrine proinflammatory or profibrotic mediators, sublethal injured tubular epithelial cells (TECs) can act as fibrogenesis initiators [10]. The main profibrogenic cytokine is Transforming growth factor-β (TGF-β) [11]. In addition to a variety of cytokines, such as interleukin 1 (IL-1) and tumor necrosis factor (TNF), other key fibrogenic factors include platelet-derived growth factor (PDGF), connective tissue growth factor (CTGF), fibroblast growth factor 2 (FGF2), tumor necrosis factor-like weak inducer of apoptosis (TWEAK) and angiotensin II (Ang II) [12], triggering the activation of TGF-β, Notch, Wnt/β-Catenin, PDGF and Ang II/Reactive oxygen species (ROS)-signaling pathways. These proinflammatory/profibrotic factors are crucial for kidney fibrosis development by triggering ECM-producing myofibroblast activation, epithelial cell dedifferentiation/apoptosis and inflammatory cell recruitment/activation, perpetuating the fibrotic loop.
2. Hypoxia in the Kidney
Despite the significant blood flow to the kidney (up to 20% of the cardiac output in humans), it is an organ predisposed to suffer hypoxic damage. This is because its inefficient uptake of oxygen delivered by the renal artery (10% at most) due to an arterial-venous diffusional oxygen shunt. Indeed, the particular renal vasculature creates a cortico-medullary pO2 gradient from approximately 30 mmHg in the cortex to 10 mmHg in the medulla [13,14]. Furthermore, part of the delivered oxygen is consumed in mitochondrial respiration to produce large amounts of adenosine triphosphate (ATP). It is required for the active transport of the proximal tubule, which reabsorbs 80% of the filtrate that traverses the glomerulus. Therefore, both renal architecture and the limited ability of kidney tubules to generate energy under anaerobic conditions make the kidney a highly sensitive organ to changes in oxygen levels [15]. This particular setting facilitates erythropoietin-producing cells residing in the kidney to adjust the generation of erythropoietin (EPO) to variations in oxygen availability [16]. Of interest, the uremic toxin indoxyl sulfate can control the cell survival and differentiation of erythroid progenitors, altering erythropoiesis and contributing to anemia development in CKD patients [17].
To date, hypoxia along CKD progression is not conclusively demonstrated yet due to the lack of feasible methods. Techniques for assessing hypoxia in tissues include microelectrode-dependent measurements, pimonidazole staining, analysis of the HIF pathway, two-photon phosphorescence lifetime microscopy of oxygen in living animals, blood oxygenation level-dependent magnetic resonance imaging and positron emission tomography-computed tomography [18]. However, these methodologies have some drawbacks: limited accuracy and specificity, high operating costs and challenging implementation.
2.1. Factors Leading to Hypoxia
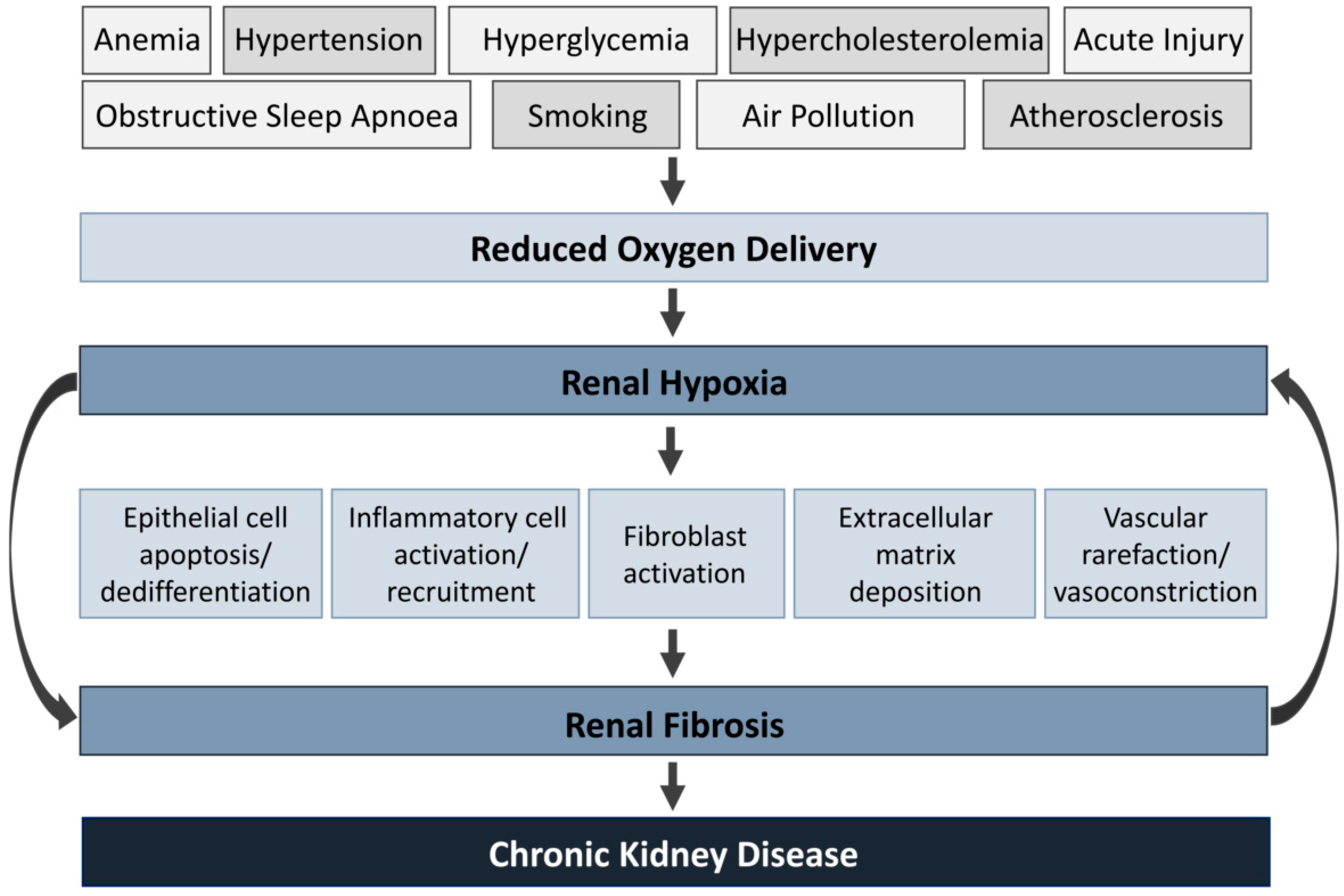
Systemic oxygenation is decreased by a combination of physiological, behavioral and environmental risk factors. They include anemia, hyperglycemia, hypertension, hypercholesterolemia, obstructive sleep apnea, smoking, air pollution and atherosclerosis [19,20,21,22,23]. Under prolonged hypoxia, pathological cellular processes are activated, including epithelial apoptosis/dedifferentiation, leukocyte activation/recruitment, fibroblast activation and ECM deposition [24,25,26,27,28]. Additionally, these processes result in renal fibrosis and vasoconstriction, worsening hypoxia by reducing erythrocyte access and limiting oxygen diffusion [29]. This loop acts as a vicious circle that will inevitably result in progressive CKD (Figure 1).
2.2. The Hypoxia-Inducible Factor System
The primary regulator of the cellular adaptive response to hypoxia is the hypoxia-inducible factor (HIF) pathway. HIFs are heterodimeric complexes formed by one of the three different oxygen-regulated α subunits (HIF-1α to -3 α) and one constitutional β subunit (HIF-b), which bind to the transcriptional coactivators C-AMP-Response Element-binding protein (CREB-binding protein)/binding protein p300 (CBP/p300) in the nucleus, increasing the expression of downstream genes (Figure 2) [30]. In the kidney, HIF-1α is mainly expressed in TECs under hypoxic conditions, while HIF-2α is limited to the endothelial and interstitial cells [31]. HIF has >1000 different target genes, including EPO, transferrin, vascular endothelial growth factor (VEGF) and glycolytic enzymes [32]. Genetic ablation of the HIF pathway, either HIF-1α or HIF-2α, aggravates renal injury, while global HIF-1α overexpression is protective [33,34,35].
The oxygen-dependent control of the HIF pathway is based on prolyl-4-hydroxylase domain (PHD1–3) enzymes and the asparagine hydroxylase factor inhibiting HIF (FIH) [36]. In normoxia, PHDs hydroxylate specific proline residues on HIF-α, which is recognized by von Hippel–Lindau tumor suppressor protein (VHL), triggering its proteasomal degradation via polyubiquitination. FIH-dependent asparagine hydroxylation of HIF-α prevents its interaction with CBP/p300, reducing HIF transactivation activity [37]. HIF-PHDs can be inhibited by structural analogs of 2-oxoglutarate, the intermediates of the Krebs cycle succinate and fumarate, ROS and nitric oxide, which results in HIF stabilization in normoxic conditions [38,39,40].
Accumulating evidence suggests that HIF-α can also be regulated by oxygen-independent degradation mechanisms. Thus, the heat-shock protein (HSP) 90, which stabilizes HIF-1, competes with the receptor of activated protein kinase C 1 (RACK1) to bind to HIF-1 and elongin C, triggering the ubiquitination of HIF-1 [41]. Post-translational modifications of HIF-1α and HIF-2α at specific residues through phosphorylation, acetylation, S-nitrosylation and sumoylation can also affect HIF protein stability and activity [42,43,44,45]. Additionally, it has been shown that HIF-α is subject to control at transcriptional and translational levels through the nuclear factor-κB (NF-κB) or the phosphatidylinositol 3-kinase (PI3K)/AKT/mammalian target of rapamycin (mTOR) pathways [46,47]. There is also evidence of HIF-independent gene regulation under hypoxia through more than 20 transcription factors [48,49,50].
HIF accumulation takes place at certain stages of CKD and is expected to protect from hypoxia. Nevertheless, its activation may be suboptimal [51]. Indoxyl sulfate also upregulates CBP/p300-interacting transactivator with Glu/Asp-rich carboxy-terminal domain 2 (CITED2) via post-transcriptional mRNA stabilization, which in turn can inhibit the interaction of p300 with HIF-1α C-terminal transactivation domain, suppressing HIF-1 activity [52].
2.3. Effects of Hypoxia
2.3.1. Inflammation
Hypoxia is associated with inflammation in models of progressive kidney disease. Immune cells can contribute not only to O2 deprivation at the site of inflammation, but also alter many of their responses to hypoxia, either promoting or repressing inflammation [5]. Renal inflammation, characterized by the infiltration of macrophages, neutrophils and lymphocytes, is present in kidney disease animal models and human renal biopsies (Figure 2). Inflammatory cells initiate phagocytosis and secrete inflammatory and profibrotic cytokines [53]. TGF-β, mainly released by macrophages and injured tubules, promotes renal wound healing by stimulating myofibroblast proliferation and reducing the production of extracellular matrix-degrading proteinases [54]. Renal cell death occurs by apoptosis or necrosis in response to renal insults, such ischemia or toxic agents. Damage-associated molecular patterns (DAMPs) generated by necrotic TECs stimulate Toll-like receptors on renal parenchymal cells and local immune cells to produce proinflammatory cytokines and chemokines [55]. The immunomodulatory medication Thalidomide attenuated macrophage infiltration and reduced kidney interstitial fibrosis [56]. KIM-1 expressed by injured tubules mediates small extracellular vesicle uptake by recognizing phosphatidylserine, a ligand of KIM-1, amplificating tubule inflammation induced by hypoxia [57]. Specific deletion of TGF-βRII in macrophages prevents from interstitial fibrosis following renal ischemia–reperfusion injury (IRI) [58]. Interleukin-1 receptor-associated kinase-M is linked to the transformation of macrophages from a M1 inflammatory to an M2 prorepair phenotype. Genetic ablation of this kinase led to a persistent M1 activation and severe fibrosis following IRI [59].
The transcription factor CCAAT/enhancer-binding protein δ (CEBPD) links hypoxia and inflammation in TECs in both acute and chronic hypoxic settings. Even under normoxic conditions, inflammatory stimuli like interleukin-1 can induce CEBPD, increasing HIF-1 expression, which in turn stimulates infiltration/activation of inflammatory cells. Importantly, hypoxia can induce CEBPD expression through NF-κB-dependent pathways [60]. The effect of HIF seems to largely depend on the target cell type. HIF-1 and HIF-2 activation has particular roles based on the macrophage phenotype, as HIF-1 is more related to M1 and HIF-2 to M2 [61]. In the mouse unilateral ureteral obstruction (UUO) model, HIF activation via myeloid-specific VHL-deletion not only decreased inflammation but had no discernible impact on renal fibrosis [62]. In contrast, global or conditional knockout of HIF in myeloid cells (driven by Lysozyme M promoter which targets macrophages and neutrophils) exhibited more severe inflammation [63]. Under ischemic conditions, neutrophils migrate into the renal interstitium, where they can harm TECs and peritubular capillary endothelial cells by releasing cytokines, chemokines, proteases and ROS. Adenosine 2A receptor agonists lessen neutrophil transmigration and improve renal function [64]. Blocking neutrophil integrin activation prevents neutrophil recruitment and reduces IRI [65]. Hypoxia induces neutrophil viability through HIF-1α-dependent NF-κB activity. HIF-1 boosts the expression of antimicrobial molecules in neutrophils [66]. Under hypoxic conditions, it has been demonstrated that the neutrophil-activating and survival factor MIP-1 (macrophage inflammatory protein-1) is induced, acting as an alternative mediator of neutrophil survival [67]. Furthermore, neutrophils also shape the tissue microenvironment by depleting local molecular oxygen [68]. In endothelial cells, inactivation of HIF-2α, but not HIF-1α, enhanced inflammatory cell infiltration. This was reversed by neutralizing vascular cell adhesion molecule-1 (VCAM1) and very late antigen-4 (VLA4) using specific antibodies [69]. CD8+ T cells immune response is also strengthened by the HIF-1α induced polarization of Th1 cells to Th2, together with regulatory T cell proliferation [70].
2.3.2. Oxidative Stress
Oxidative stress is a condition where the level of ROS or reactive nitrogen species (RNS) surpasses the cellular counteracting antioxidant capacity to scavenge them. Electrophiles include the superoxide radical, hydrogen peroxide (H2O2) and the hydroxyl radical, while RNS, such as peroxynitrite (ONOO−), nitrogen dioxide (•NO2) and dinitrogen trioxide (N2O3), are formed by reactions involving superoxide and nitric oxide (•NO) [71]. The •NO radical is produced by three L-arginine-dependent nitric oxide synthase (NOS) isoforms, all expressed in the kidney. It plays a role in controlling hypertension and acting as a powerful vasodilator. These reactive species can react with cellular macromolecules, such as lipids, proteins or DNA, altering their function. They can trigger the oxidation or nitration of amino acids like tyrosine, cysteine, methionine and tryptophan.
Mitochondria electron-transport chain (ETC) leakage, membrane-bound NADPH oxidases (NOXs) and endoplasmic reticulum (ER) are the primary sources of the majority of intracellular ROS [72]. The most abundant endogenous antioxidant is Glutathione (GSH) [73]. Other non-enzymatic antioxidants involve the synthesis of nucleophiles such as ascorbate, tocopherols and retinol. Nuclear factor erythroid 2-related factor 2 (Nrf2)-induced antioxidant protection is the main antioxidative response. NRF2 is sequestered in the cytosol by KEAP1. When ROS interact with certain cysteine residues in KEAP1, NRF2 is able to go into the nucleus and activate cytoprotective genes by attaching to antioxidant response elements (ARE). Other antioxidant enzymes include the NOX:quinone oxidoreductase1 (NQO1), glutathione S-transferase (GSTs), glutathione peroxidase (GPx), catalase and peroxiredoxin (PRx)/thioredoxin (TRx) [74].
Oxidative stress is involved in AKI to CKD progression (Figure 2). Oberg et al. compared the levels of three oxidative stress indicators (plasma protein carbonyl group content, plasma-free F2-isoprostane content and plasma protein reduced thiol content) in patients with stage 3–5 CKD and found that they had greater levels of oxidative stress [75]. The kidney is particularly susceptible to damage from oxidative stress because of its high levels of oxidation inside the mitochondria [76]. The substantial immunological response that occurs in sepsis-induced AKI promotes the activation of inducible NOS synthase and the overproduction of NO, which leads to endothelial damage, localized hypoxia and ROS [77]. Cisplatin-induced AKI is characterized by the generation of mitochondrial ROS (mtROS), reduced mitochondrial membrane potential and mitochondrial swelling [78]. While it is generally accepted that ROS increases during reoxygenation following hypoxia, it is more controversial whether ROS are elevated during hypoxia. Such increased mtROS and mitochondrial calcium content cause MPTP opening and the activation of different cell death pathways [79]. Additionally, mtROS also stimulates renal inflammation and NLRP3 activation [80]. Consistently, hyperactivation of Nrf2 in tubules halts the progression of tubular damage by suppressing IRI-mediated ROS during the early progressive phase in the IRI model [81]. Studies have reported reduced NO bioactivity in CKD patients [82]. This is due to compromised NO generation, increased metabolism or increased levels of NOS inhibitors. An imbalance of vasoactive substances triggers aberrant intrarenal vasoconstriction, which can also cause chronic hypoxia in the kidney. In addition to constrict glomerular arterioles, Ang II hampers the efficient utilization of oxygen in TECs through the production of oxidative stress by stimulating NADPH oxidase activity. It results in decreased NO bioavailability through peroxynitrite formation [83]. Because NO inhibits mitochondrial respiration, depletion of NO may accelerate mitochondrial respiration and decouple it from chemical energy use, which would result in tissue hypoxia [84]. Kidney damage induced by indoxyl sulfate may also operate through this mechanism [85]. In the hypoxic kidney, the stress-responsive transcription factor FoxO3 is activated in TECs likely as an adaptation to counteract hypoxic insults, attenuating CKD. Hypoxia inhibits FoxO3 prolyl hydroxylation and subsequent degradation, promoting autophagy and reducing oxidative injury [86].
In addition, mitochondrial dysfunction interferes the communication between mitochondria and ER, leading to tubular inflammation and fibrosis. ER controls protein synthesis, folding and degradation through the unfolded protein response (UPR) pathway. It promotes cellular survival by restoring ER and mitochondrial homeostasis through different signaling networks, which include ATF6 p50, XBP1 and ATF4, but if impaired, the UPR induces inflammation and cell death [87].
2.3.3. Metabolic Reprogramming
TECs need a huge amount of energy to cope with their reabsorption function. Although TECs reabsorb a lot of glucose, they barely consume it but are highly dependent on fatty acid oxidation (FAO), which makes them extremely vulnerable to hypoxia [88]. In particular, the mature nephron’s inability to produce anaerobic energy in combination with its basal low pO2 point to the S3 region of the proximal tubule as the main site for hypoxia-based damage [89,90]. During AKI and CKD, TECs are prone to metabolic reprogramming (Figure 2). Although FAO is transiently activated in PTECs (marked by Perilipin 2 (PLIN2) expression, a marker of intracellular lipid droplets) at the earliest stages of IRI and is linked with effective repair [91], its failure is considered a potential pathogenic component in the onset of renal disease. It involves the shift from FAO to glycolysis due to hypoxia, mitochondrial malfunction and disordered nutrient-sensing pathways [92]. HIF-1α enhances glucose uptake by promoting the expression of glucose transporters. By upregulating pyruvate dehydrogenase kinase (PDK) expression, HIF-1 can also boost lactate dehydrogenase (LDH) expression to promote the conversion of pyruvate to lactic acid and prevent it from entering the TCA cycle. HIF-1 also activates PKM2, the key enzyme in the glycolytic metabolism [93]. These changes are a double-edged role. On the one hand, increased glycolysis acts as a compensatory mechanism for ATP synthesis to maintain the normal function of the surviving TECs, playing also a protective role against antioxidative stress [94]. The activity of hexokinase (HK), a crucial enzyme in glycolysis, was significantly elevated in a sepsis-induced AKI animal model. This increased the flow of the pentose phosphate pathway (PPP), which keeps glutathione in its reduced state, thereby protecting the kidney from peroxide damage [95,96]. On the other hand, long-term shutdown of FAO and enhanced glycolysis lead to a proinflammatory response, lipid accumulation and fibrosis, contributing to AKI to CKD transition [97]. To fulfill the rapid energy demand associated with the ECM synthesis phenotype, myofibroblasts also undergo metabolic reprogramming marked by enhanced aerobic glycolysis [98]. In addition to provide a rapid energy-generating mechanism compared to oxidative phosphorylation (OXPHOS), glycolysis yields by-products such as lactate that control fibrosis [99]. It has been discovered that chicken ovalbumin upstream promoter-transcription factor II (COUP-TFII), a transcription factor that directly regulates PGC-1α expression, causes a metabolic reprogramming in myofibroblast that favors glycolysis and the production of profibrotic mediators [100]. Aerobic glycolysis inhibition has evolved as an antifibrotic therapy by reducing myofibroblast activation [101,102,103].
The pathophysiology of AKI to CKD transition is significantly influenced by the dysregulation of nicotinamide adenine dinucleotide (NAD+) metabolism, most likely due to the combination between decreased NAD+ synthesis and increased NAD+ consumption. NAD+ participates in metabolic activities including glycolysis, tricarboxylic acid (TCA) cycle and OXPHOS by acting as an electron carrier and forming nicotinamide adenine dinucleotide (NADH) [104]. NAD+ is an essential substrate for a number of crucial enzymes, including sirtuins. It controls mitochondrial homeostasis, cell senescence and DNA repair [105]. Reduced renal NAD+ may enhance vulnerability to AKI due to an impaired de novo NAD+ production pathway linked to kidney damage or aging [106]. AKI syndrome triggers accelerated consumption of NAD+ by poly (ADP-ribose) polymerases (PARP) and CD38 enzymes, contributing to reduce NAD+ availability. Treatment with TES-1025, a selective inhibitor of α-amino-β-carbox-ymuconate-ε-semialdehyde decarboxylase (ACMSD), a limiting enzyme in de novo NAD+ synthesis, rescued renal NAD+ decline and attenuated kidney injury [107,108]. In AKI and CKD models, nicotinamide mononucleotide (NMN) supplementation promoted the NAD+ salvage synthesis pathway and resulted in antifibrosis, antiaging and anti-inflammatory benefits [109,110].
Gluconeogenesis is also impaired during AKI. In experimental and human AKI, renal glucose release and lactate uptake were reduced, a metabolic change linked to patient mortality. Approximately 40% of endogenous gluconeogenesis is produced by the kidneys, which is involved in normoglycaemia during fasting and stress conditions. The generation of glucose from lactates is an energy-consuming process [111]. In AKI, severe lack of renal ATP supply is accompanied by NAD+ depletion [112]. Therefore, shifts from FAO to glycolysis, with concomitant impaired of NAD+ metabolism and gluconeogenesis seem to be cellular adaptations to the pathological environment.
In AKI to CKD transition, TECs display imbalance in mitochondrial quality control, characterized by altered mitochondrial biosynthesis, increased mitochondrial fission, decreased mitochondrial fusion and decreased mitophagy [113,114,115]. Another significant component that promotes this metabolism reprogramming is maladjustment of the nutrient-sensing mTOR/AMP-activated protein kinase (AMPK) signaling pathways, involved in maintaining renal energy homeostasis. mTOR complex 1 (mTORC1) increases HIF-1α translation, leading to an increased expression of the glycolytic enzymes HK and PKM2 along CKD progression [116]. When the ATP/AMP ration drops, AMPK increases ATP production by enhancing mitochondrial biosynthesis, FAO and OXPHOS. AMPK also inhibits the mTOR signaling pathway. In the cisplatin-induced AKI model, AMPK activation was reduced, which caused the overactivation of the mTOR signaling pathway and autophagy suppression. By contrast, AMPK activation shielded TECs and protected from fibrosis development in several AKI models [117,118].
Importantly, substantial changes in cell metabolism also accompany the phenotypic and functional alterations during transitions of other cell types involved in CKD. Thus, proinflammatory M1 macrophages primarily rely on glycolysis and have two breaks on the TCA cycle. By contrast, macrophages acquiring a M2 phenotype, with an important role in wound healing and resolving inflammation, are more dependent on OXPHOS. Although the activation of M1-like proinflammatory macrophages may be favored by ROS generation also contributing to their increased glycolysis and TCA blockage via HIF-1α stabilization, they are also important for M2 macrophage differentiation, suggesting a differential spatial and temporal role [119].
2.3.4. Fibrosis
Hypoxia triggers fibroblast activation which leads to ECM deposition. Fibrosis, in turn, exacerbates hypoxia by reducing the efficiency of oxygen diffusion by extending the distance between capillaries and TECs (Figure 2). Activated HIF binds to the promoters of profibrogenic genes and matrix-modifying factors including matrix metallopeptidase 2, tissue inhibitor of metalloproteinase 1 (TIMP-1), collagen I, plasminogen activator inhibitor-1 (PAI-1) and CTGF, resulting in the accumulation of ECM in the tubulointerstitium [24]. In this line, genetic deletion of HIF-1α in TECs reduced the number of S100A4-positive fibroblasts [120]. In the endothelium, persistent HIF-1α expression, induced by specific knockdown of PHD-2, caused endothelial–mesenchymal transition (EndoMT) and renal fibrosis [121]. Hypoxia stimulated endothelial PDGF-β production, triggering the proliferation of mesangial cells [122].
Besides the direct transcription activation of fibrogenic factors, HIF can interact with diverse profibrotic signaling pathways, including TGF-β, Notch, NF-κB and PI3K/Akt pathways, further regulating renal fibrosis [123]. HIF-1α can form a complex with Smad family member 3 (SMAD3), enhancing the expression of COL1A2 [124]. Conversely, the interaction between mTORC1 and SMAD3 under TGF-β treatment enhances HIF-1 induction and downstream collagen expression [125]. One possible mechanism is that TGF-β1-induced SMAD2/3 activation inhibits PHD2 expression [126]. Of importance, translational HIF regulation by TGF-β1 is cell type-specific. In TECs, HIF-1α expression is stimulated by TGF-β1/SMAD3 signaling, while HIF-2 induction is more prevalent in mesangial cells [127,128]. HIF accumulation not only induces TGF-β1, but also markedly increases Notch3 transcription [121]. Additionally, in the Ang II infusion model of hypertensive CKD, HIF-1α was demonstrated to be crucial to initiate glomerular injury and renal fibrosis by inducing the expression of vasoactive proteins [129]. Hypoxia causes renal fibrosis by activating the HIF-1α/HE4/NF-κB signaling, upregulating tissue inhibitor metalloproteinases 1, which may prevent ECM degradation [130]. Furthermore, HIF can also interact with PI3K/Akt to control fibrosis. HIF-1 induction in TECs activates the transcription of Bmi-1, which modulates PI3K/Akt signaling and promotes Snail-mediated TEC dedifferentiation [131].
2.4. Influence of Renal Hypoxia in AKI to CKD Progression
AKI consists in the sudden and persistent impairment of renal function that can be reversible if treated promptly and effectively. Clinical and animal research have demonstrated that AKI can eventually lead to CKD, even if followed by a full recovery of renal function. Of note, AKI severity and the frequency of AKI episodes predict the subsequent onset of CKD [132].
Historically, AKI models have been divided into two categories: ischemic and non-ischemic models [133]. However, hypoxia and HIF expression are not limited to the acute stage of ischemic AKI. HIF staining disappears 24 h after AKI induction. It is interesting to note that HIF reappeared between days 3 and 7 after AKI induction in the IRI model [33,134]. Human kidney transplant recipients have also reported similar delays in HIF activation and up to 5 weeks after an AKI episode [135]. These findings might potentially point to hypoxia as a side effect of tissue regeneration following kidney transplantation.
In response to nitric oxide synthase inhibition, folic acid nephropathy and IRI, mouse kidneys show a decrease in microvascular density. Although it is likely that HIF increases angiogenic factors like VEGF, which restore capillary density, this adaptation process typically fails because injured TECs are unable to produce sufficient VEGF [136]. Of note, prolonged hypoxia leads to the renal downregulation of the proangiogenic isoform 164 and the upregulation of the dys-angiogenic isoforms 120 and 188 of VEGF-A [137]. The inflammatory environment may contribute to the suppression of VEGF expression. Antiangiogenic factors (e.g., thrombospondin 1 and endostatin) are increased in several kidney disorders [138,139]. Furthermore, the incapability of endothelial progenitor cells potentially underlies poor capillary repair [140]. Complementarily, Kramann et al. proposed that the mechanism underlying capillary rarefaction is pericyte detachment from nearby capillary endothelial cells [141].
This persistent hypoxia induces several pathological processes including proinflammatory adhesion molecules and chemokines, which result in the accumulation of immune cells in the kidney [142]. Hypoxia induces leukocyte adherence to endothelial cells by activating 2-integrin [143]. Palm et al. demonstrated that dinitrophenol-induced renal hypoxia led macrophages to gather in the kidney [144]. Further, Polichnowski et al. suggested that capillary loss causes localized hypoxia that limits tubule regeneration [145]. Many in vitro studies have also shown that hypoxic injury causes apoptosis and TEC dedifferentiation. In AKI, recruited inflammatory cells and dedifferentiated TECs produce profibrotic cytokines, such as TGF-β, contributing to activate fibroblasts [146]. Under hypoxic conditions, these cells produced more collagen and tissue inhibitor of metalloproteinase 1 [24]. In turn, tubulointerstitial fibrosis exacerbates hypoxia because it widens the distance between capillaries and TECs, decreasing the effectiveness of oxygen transport [130]. CKD progresses as a result of the vicious cycle of tubulointerstitial fibrosis and hypoxia (Figure 1).
Increasing evidence has shown that proinflammatory and profibrotic gene expression is also regulated by hypoxia-induced epigenetic alterations. HIF-1 binds to the promoters of histone demethylases JMJD1A and JMJD2B, inducing their expression. In turn, it increases the expression of downstream genes by reducing promoter histone methylation [147,148]. In particular, chromosomal conformation capture tests demonstrated that the interaction between HIF-1 and lysine-specific demethylase 3A may cause a significant shift in chromatin conformation, leading to the erasure of suppressive histone marks [149]. Three weeks after IRI, proinflammatory and profibrotic genes, such as monocyte chemoattractant protein-1, TGF-β1 and collagen III, showed increased expression along with enhanced level of gene-activating histone modification (e.g., H3K4m3 or H3K9/14ac). Promoters of those genes also showed increased binding of chromatin-remodeling enzymes, such as Brahma-related gene 1 [150]. Zager et al. showed that IRI triggers endothelin-1 (ET-1) expression, which leads to gene-activating histone modifications close to the transcriptional start site of the ET-1 gene. This may exacerbate renal hypoxia by inducing vasoconstriction [151].
3. Hypoxia as a Therapeutic Target in CKD
Given that renal hypoxia is a final common path in CKD development and that the genetic suppression of the HIF pathway aggravates renal injury, HIF activation/stabilization is suggested as a potential therapy for CKD (Table A1) [152]. Dimethyloxalylglycine (DMOG), a competitive inhibitor of HIF-alpha prolyl hydroxylase (HIF-PH), improved proteinuria, mitochondrial function and cell survival in the rat subtotal nephrectomy model [153]. In hypertensive rats, DMOG also reduced glomerulosclerosis, fibrosis and proteinuria [154]. This effect was not observed in DMOG-treated animals lacking endothelial HIF-2 [69]. In the streptozotocin (STZ)-induced diabetic nephropathy rat model, cobalt chloride (CoCl2), another inhibitor of HIF hydroxylation, improved GFR, albuminuria and tubulointerstitial damage [155]. Additionally, CoCl2 had a protective effect in the Thy1 nephritis rat model, in the subtotal nephrectomy rat model and in Type 2 diabetic nephropathy [156,157]. Beside the beneficial effects of cobalt in experimental animals, long-term administration to humans has adverse effects. In this regard, the new HIF-PHD inhibitors (HIF-PHIs) roxadustat, daprodustat, vadadustat and enarodustat improved renal anemia and exerted a positive effect on the hemoglobin rate in CKD patients [158,159,160,161,162,163,164]. Enoradustat also reduced albuminuria and alleviated renal damage in the diabetic nephropathy ob/ob model [165]. Enoradustat was similarly protective in the rat subtotal nephrectomy model, decreasing fibrosis and inflammation [166].
While the pre-stimulation of the HIF pathway both genetically and pharmacologically reduces AKI episodes, its beneficial role in CKD progression is still controversial and mainly relies on pre-ischemic activation [167]. Schley et al. found that HIF-PHIs cause renal mononuclear phagocytes to adopt an anti-inflammatory phenotype [168]. By contrast, some other CKD preclinical studies showed that HIF-signaling activation in the kidney appears to be deleterious, as evidenced the stabilization of HIF-1 by genetic deletion of VHL in a 5/6 renal ablation model and the administration of an anti-HIF-1α drug in the UUO model [169]. Genetic endothelial-specific knockout of PHD2 (leading to constitutive HIF-1α and HIF-2α stabilization) also induced fibrosis and worsened kidney damage [121]. Similarly, genetic HIF-2a overexpression in the tubules exacerbated renal fibrosis [170]. Therefore, the role of HIF activation is likely to depend on the pathological context, cell type and timing. Additional investigation is necessary to clarify the implications and mechanisms of HIF-PHIs on CKD progression.
Strategies directed towards the preservation of peritubular capillaries early in the AKI process lead to maintaining renal oxygen levels and exert a protecting role in AKI to CKD transition. Thus, treatment with the angiogenic factors VEGF-121 or angiopoietin-1 administered early during injury suppressed AKI to CKD transition in the IRI model, by attenuating the loss of peritubular capillaries and subsequent tubulointerstitial fibrosis [171,172,173]. Protein kinase C comprises a superfamily of serine-threonine kinases with diverse functions in signal transduction and cellular regulation. Inactivation of PKC isoforms were shown to alleviate many diabetes- or hyperglycemia-associated vascular dysfunctions in the kidney [174]. Blocking PKCα signaling with the chemical inhibitor Go6976 also inhibited fibroblast activation and renal fibrosis [175]. Targeting fibrosis has been mainly addressed by blocking the TGF-β signaling pathway. It includes several strategies including Smad agonists/inhibitors and neutralizing antibodies against receptors/cytokines. However, the pleotropic nature of this pathway has limited their application due to potential side effects [176].
SGLT2 inhibitors (SGLT2is), a new class of antihyperglycemic drugs which act by targeting glucose reabsorption, also lessen principal kidney and cardiovascular problems in CKD patients [177]. Studies have suggested that SGLT2i may reduce cortical oxygen consumption. Reduced oxygen pressure could activate the HIF-signaling pathway and imitate systemic hypoxia [178,179]. In the IRI model, SGLT2i was also discovered to prevent renal capillary rarefaction and subsequent hypoxia and fibrosis. However, further research is required to decipher the renoprotective mechanisms of SGLT2 inhibition associated with tissue oxygenation [180].
Multiple substances directed to target mitochondria and correct imbalanced bioenergetics seem to prevent CKD progression. The PPARα agonists fenofibrate and clofibrate attenuated acute renal tubule injury by increasing FAO rate and inhibiting nuclear factor kappa-B (NF-κB) activity, oxidative stress, cellular apoptosis and fibrosis [181,182]. Additionally, promoting mitochondrial biosynthesis is a promising approach to block AKI to CKD progression [183]. In several AKI models, resveratrol (an SIRT1 agonist) and AICAR (an AMPK agonist) improved mitochondrial fitness and protected from renal fibrosis [184,185]. Mdivi-1, an inhibitor of mitochondrial fission protein DRP1, exerts a beneficial effect in several kidney diseases, attenuating tubular cell apoptosis and maintaining mitochondrial structure [186]. Enhancing mitophagy in TECs is also an effective strategy to treat AKI to CKD progression [115]. Berberine (BBR), a quaternary ammonium isoquinoline alkaloid, reduced cisplatin-induced TEC cytotoxicity by inducing mitophagy through the PINK1/Parkin signaling pathway [187]. Finally, a novel mitochondrial protectant, SS-31 (D-Arg-dimethylTyr-Lys-Phe-NH2), specifically binds to cardiolipin in the mitochondrial inner membrane to stabilize the mitochondrial structure, reducing ROS production. It was shown to prevent tubular apoptosis, interstitial fibrosis and glomerulosclerosis [188,189]. Although early restoration of TEC metabolism offers a promising therapeutic approach for AKI, the translation of drugs that target mitochondria and bioenergetics into the clinic is limited due to adverse off-target effects [190]. In addition, available studies have focused only on TECs, so whether other renal cell types undergo metabolic changes needs to be further explored.
Targeting hypoxia-associated oxidative stress in CKD using antioxidants has also emerged as a potential therapeutic strategy. Nrf2–ARE axis activation triggers strong antioxidative effects [191]. Therefore, the pharmacological stimulation of this system may be a promising target. Bardoxolone methyl, a Nrf2 activator that inhibits Keap1 through modifying Keap1-cysteine-151, was reported to increase glomerular filtration rate (GFR) in diabetic CKD patients on different clinical trials [192]. Inhibitors of xanthine oxidase, an enzyme involved in purine catabolism producing ROS, ameliorate renal damage and reduce uric acid levels, proinflammatory mediators and ROS [193]. That is the case of allopurinol, which slows down renal disease progression in CKD patients [194]. The inhibition of NOX enzymes has emerged as a promising approach to target ROS. Although NOX2 is also expressed, NOX4 is the most prevalent type in the kidney. [195]. Oral administration of APX-115, a pan-inhibitor of NOX enzymes, reduced fibrosis in the murine model of STZ-induced kidney disease [196]. Setanaxib (GKT137831), a first-in-class, dual inhibitor of NOX1/4, also showed renoprotective effects in this model [197]. Treatment of db/db mice with GKT136901, a NOX1/NOX4 inhibitor, blocked renal NOX4-dependent fibrotic signaling after exposure to high glucose [198]. Of note, H2S donors, such as sodium thiosulfate, maintain redox homeostasis by ROS scavenging and modifying cysteine residues on key signaling molecules, which exerts renal protection [199,200]. Vitamin E reduced cardiovascular disease and myocardial infarction events in hemodialysis patients [201]. A moderate dose of Vitamin C might be beneficial for CKD patients with iron deficiency [202]. CoQ10 supplementation restored the metabolic profile of CKD patients [203]. In renal IRI and Ang II-infused mouse models, pre-administration of MitoQ reduced oxidative damage and apoptosis by reducing aberrant mitochondrial fission and restoring mitophagy by activating the NRF2 pathway [204,205]. Plant-derived polyphenols, such as quercetin, curcumin and resveratrol, have also emerged as promising antioxidant agents with renoprotective effects [206]. Rassaf et al. discovered that supplementation with cocoa flavanol resulted in a reduction in endothelial dysfunction in ESRD patients [207]. However, well-powered clinical studies are still needed to obtain a meaningful evaluation of the effects of Nrf2 system-targeting compounds.
4. Conclusions
Renal hypoxia has emerged as a key mediator of CKD progression. Impaired oxygen supply leads to capillary rarefaction, tubular dedifferentiation, inflammatory reactions and fibrosis that in turn, contribute to worsen hypoxic conditions [208]. HIF is a master transcriptional regulator of cellular adaptation to hypoxia. Given experimental data suggesting suboptimal HIF activation in CKD, its stabilization is a novel and promising therapeutic target. Nevertheless, the majority of approaches are preventive, in which HIF is activated before the onset of AKI or CKD. Therefore, therapeutic strategies, in which HIF is activated after AKI occurs would be more clinically relevant to target AKI to CKD progression [209]. Due to discrepancies from such strategies, numerous challenging questions have emerged: what is the mechanism that links hypoxia and renal inflammation in the kidney? What is the cell type-specific spatiotemporal responsive pattern to hypoxia in the kidney? Is it possible to predict the outcome of kidney damage in response to hypoxia by developing early biomarkers and better resolution methods to monitor hypoxia? And, what is the long-term impact of the novel class of medications targeting HIF-PHIs on the development of AKI-CKD, considering its effectiveness in treating anemia?
Author Contributions
V.M. and A.R. wrote the manuscript and generated original figures. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Acknowledgments
V.M. is supported by a FEBS Long-Term Fellowship.
Conflicts of Interest
The authors declare no conflict of interest.
Appendix A

{kind=link}
{kind=link}
Table A1.
Pharmacological agents modulating the HIF pathway in rodent models of CKD.
| Treatment | Mecanism of Action | Effects | Experimental Model | Reference |
|---|---|---|---|---|
| CoCl2 | Transcriptional HIF-1α upregulator |
| Subtotal nephrectomy in rats | [210,211] |
| Hypertensive type 2 diabetic rats (SHR/NDmcr-cp) | [158] | ||
| Streptozotocin-induced diabetic nephropathy rats | [156] | ||
| Uninephrectomized Thy1 nephritis rats | [157] | ||
| DMOG | Prolyl hydroxylase inhibitor |
| DOCA-salt hypertensive rats | [155] |
| Dahl salt-sensitive rats | [212] | ||
| Deferoxamine | Iron chelator, Transcriptional HIF-1α upregulator |
| Unilateral ureteral obstruction | [213] |
| Enarodustat | Prolyl hydroxylase inhibitor |
| BTBR ob/ob mice | [166] |
| Subtotal nephrectomy in rats | [167] | ||
| Roxadustat | Prolyl hydroxylase inhibitor |
| Adenine-induced nephropathy | [169] |
| YC-1 | HIF-1β inhibitor |
| Type 1 diabetic mouse model OVE26 | [214] |
| Zinc | Blocks nuclear translocation of HIF-1β |
| Streptozotocin-induced diabetic nephropathy rats | [215] |
The studies showing a protective role of HIF activation are highlighted in green and those with a detrimental role are highlighted in red. DMOG: Dimethyloxaloylglycine, DOCA: Deoxycorticosterone acetate, ECM: Extracellular matrix, HIFα: Hypoxia Inducible Factor Subunit Alpha, HIFβ: Hypoxia Inducible Factor Subunit Beta, YC-1: 3-(5′-hydroxymethyl-2′-furyl)-1-benzyl indazole.
References
- Djudjaj, S.; Boor, P. Cellular and molecular mechanisms of kidney fibrosis. Mol. Asp. Med. 2019, 65, 16–36. [Google Scholar] [CrossRef] [PubMed]
- Kazancioglu, R. Risk factors for chronic kidney disease: An update. Kidney Int. Suppl. 2013, 3, 368–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, L.; Wei, Q.; Liu, J.; Yi, M.; Liu, Y.; Liu, H.; Sun, L.; Peng, Y.; Liu, F.; Venkatachalam, M.A.; et al. AKI on CKD: Heightened injury, suppressed repair, and the underlying mechanisms. Kidney Int. 2017, 92, 1071–1083. [Google Scholar] [CrossRef] [PubMed]
- Cheung, M.D.; Erman, E.N.; Moore, K.H.; Lever, J.M.; Li, Z.; LaFontaine, J.R.; Ghajar-Rahimi, G.; Liu, S.; Yang, Z.; Karim, R.; et al. Resident macrophage subpopulations occupy distinct microenvironments in the kidney. JCI Insight 2022, 7, e161078. [Google Scholar] [CrossRef] [PubMed]
- Lin, N.; Simon, M.C. Hypoxia-inducible factors: Key regulators of myeloid cells during inflammation. J. Clin. Investig. 2016, 126, 3661–3671. [Google Scholar] [CrossRef]
- AIRG-E, EKPF, ALCER, FRIAT, REDINREN, RICORS2040, SENEFRO; SET, ONT. CKD: The burden of disease invisible to research funders. Nefrol. Engl. Ed. 2022, 42, 65–84. [Google Scholar] [CrossRef]
- GBD Chronic Kidney Disease Collaboration. Global, regional, and national burden of chronic kidney disease, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2020, 395, 709–733. [Google Scholar] [CrossRef] [Green Version]
- Chao, C.T.; Chiang, C.K. Uremic toxins, oxidative stress, and renal fibrosis: An interwined complex. J. Ren. Nutr. 2015, 25, 155–159. [Google Scholar] [CrossRef]
- Humphreys, B.D. Mechanisms of Renal Fibrosis. Annu. Rev. Physiol. 2018, 80, 309–326. [Google Scholar] [CrossRef]
- Yu, S.M.; Bonventre, J.V. Acute kidney injury and maladaptive tubular repair leading to renal fibrosis. Curr. Opin. Nephrol. Hypertens. 2020, 29, 310–318. [Google Scholar] [CrossRef]
- Huebener, P.; Schwabe, R.F. Regulation of wound healing and organ fibrosis by toll-like receptors. Biochim. Biophys. Acta 2013, 1832, 1005–1017. [Google Scholar] [CrossRef] [Green Version]
- Duffield, J.S. Cellular and molecular mechanisms in kidney fibrosis. J. Clin. Investig. 2014, 124, 2299–2306. [Google Scholar] [CrossRef] [Green Version]
- Brezis, M.; Heyman, S.N.; Epstein, F.H. Determinants of intrarenal oxygenation. II. Hemodynamic effects. Am. J. Physiol. 1994, 267, F1063–F1068. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Edwards, A. Oxygen transport across vasa recta in the renal medulla. Am. J. Physiol. Heart Circ. Physiol. 2002, 283, H1042–H1055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thaysen, J.H.; Lassen, N.A.; Munck, O. Sodium transport and oxygen consumption in the mammalian kidney. Nature 1961, 190, 919–921. [Google Scholar] [CrossRef]
- Maxwell, P.H.; Osmond, M.K.; Pugh, C.W.; Heryet, A.; Nicholls, L.G.; Tan, C.C.; Doe, B.G.; Ferguson, D.J.; Johnson, M.H.; Ratcliffe, P.J. Identification of the renal erythropoietin-producing cells using transgenic mice. Kidney Int. 1993, 44, 1149–1162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamza, E.; Vallejo-Mudarra, M.; Ouled-Haddou, H.; Garcia-Caballero, C.; Guerrero-Hue, M.; Santier, L.; Rayego-Mateos, S.; Larabi, I.A.; Alvarez, J.C.; Garcon, L.; et al. Indoxyl sulfate impairs erythropoiesis at BFU-E stage in chronic kidney disease. Cell Signal. 2023, 104, 110583. [Google Scholar] [CrossRef] [PubMed]
- Hirakawa, Y.; Tanaka, T.; Nangaku, M. Renal Hypoxia in CKD; Pathophysiology and Detecting Methods. Front. Physiol. 2017, 8, 99. [Google Scholar] [CrossRef] [Green Version]
- Kanbay, A.; Buyukoglan, H.; Ozdogan, N.; Kaya, E.; Oymak, F.S.; Gulmez, I.; Demir, R.; Kokturk, O.; Covic, A. Obstructive sleep apnea syndrome is related to the progression of chronic kidney disease. Int. Urol. Nephrol. 2012, 44, 535–539. [Google Scholar] [CrossRef] [PubMed]
- Welch, W.J. Intrarenal oxygen and hypertension. Clin. Exp. Pharmacol. Physiol. 2006, 33, 1002–1005. [Google Scholar] [CrossRef]
- Iseki, K.; Kohagura, K. Anemia as a risk factor for chronic kidney disease. Kidney Int. Suppl. 2007, 72, S4–S9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yacoub, R.; Habib, H.; Lahdo, A.; Al Ali, R.; Varjabedian, L.; Atalla, G.; Kassis Akl, N.; Aldakheel, S.; Alahdab, S.; Albitar, S. Association between smoking and chronic kidney disease: A case control study. BMC Public Health 2010, 10, 731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zager, R.A.; Johnson, A.; Anderson, K.; Wright, S. Cholesterol ester accumulation: An immediate consequence of acute in vivo ischemic renal injury. Kidney Int. 2001, 59, 1750–1761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Norman, J.T.; Clark, I.M.; Garcia, P.L. Hypoxia promotes fibrogenesis in human renal fibroblasts. Kidney Int 2000, 58, 2351–2366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falanga, V.; Zhou, L.; Yufit, T. Low oxygen tension stimulates collagen synthesis and COL1A1 transcription through the action of TGF-beta1. J. Cell Physiol. 2002, 191, 42–50. [Google Scholar] [CrossRef]
- Albina, J.E.; Henry, W.L., Jr.; Mastrofrancesco, B.; Martin, B.A.; Reichner, J.S. Macrophage activation by culture in an anoxic environment. J. Immunol. 1995, 155, 4391–4396. [Google Scholar] [CrossRef]
- Luscinskas, F.W.; Ma, S.; Nusrat, A.; Parkos, C.A.; Shaw, S.K. Leukocyte transendothelial migration: A junctional affair. Semin. Immunol. 2002, 14, 105–113. [Google Scholar] [CrossRef]
- Khan, S.; Cleveland, R.P.; Koch, C.J.; Schelling, J.R. Hypoxia induces renal tubular epithelial cell apoptosis in chronic renal disease. Lab. Investig. 1999, 79, 1089–1099. [Google Scholar]
- Mimura, I.; Nangaku, M. The suffocating kidney: Tubulointerstitial hypoxia in end-stage renal disease. Nat. Rev. Nephrol. 2010, 6, 667–678. [Google Scholar] [CrossRef]
- Semenza, G.L. Oxygen sensing, hypoxia-inducible factors, and disease pathophysiology. Annu. Rev. Pathol. 2014, 9, 47–71. [Google Scholar] [CrossRef]
- Haase, V.H. Hypoxia-inducible factors in the kidney. Am. J. Physiol. Ren. Physiol. 2006, 291, F271–F281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nangaku, M.; Eckardt, K.U. Hypoxia and the HIF system in kidney disease. J. Mol. Med. 2007, 85, 1325–1330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conde, E.; Alegre, L.; Blanco-Sanchez, I.; Saenz-Morales, D.; Aguado-Fraile, E.; Ponte, B.; Ramos, E.; Saiz, A.; Jimenez, C.; Ordonez, A.; et al. Hypoxia inducible factor 1-alpha (HIF-1 alpha) is induced during reperfusion after renal ischemia and is critical for proximal tubule cell survival. PLoS ONE 2012, 7, e33258. [Google Scholar] [CrossRef]
- Kojima, I.; Tanaka, T.; Inagi, R.; Kato, H.; Yamashita, T.; Sakiyama, A.; Ohneda, O.; Takeda, N.; Sata, M.; Miyata, T.; et al. Protective role of hypoxia-inducible factor-2alpha against ischemic damage and oxidative stress in the kidney. J. Am. Soc. Nephrol. 2007, 18, 1218–1226. [Google Scholar] [CrossRef] [Green Version]
- Fahling, M.; Mathia, S.; Paliege, A.; Koesters, R.; Mrowka, R.; Peters, H.; Persson, P.B.; Neumayer, H.H.; Bachmann, S.; Rosenberger, C. Tubular von Hippel-Lindau knockout protects against rhabdomyolysis-induced AKI. J. Am. Soc. Nephrol. 2013, 24, 1806–1819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schofield, C.J.; Ratcliffe, P.J. Oxygen sensing by HIF hydroxylases. Nat. Rev. Mol. Cell Biol. 2004, 5, 343–354. [Google Scholar] [CrossRef] [PubMed]
- Gunter, J.; Ruiz-Serrano, A.; Pickel, C.; Wenger, R.H.; Scholz, C.C. The functional interplay between the HIF pathway and the ubiquitin system—More than a one-way road. Exp. Cell Res. 2017, 356, 152–159. [Google Scholar] [CrossRef] [Green Version]
- Frost, J.; Frost, M.; Batie, M.; Jiang, H.; Rocha, S. Roles of HIF and 2-Oxoglutarate-Dependent Dioxygenases in Controlling Gene Expression in Hypoxia. Cancers 2021, 13, 350. [Google Scholar] [CrossRef]
- Laukka, T.; Mariani, C.J.; Ihantola, T.; Cao, J.Z.; Hokkanen, J.; Kaelin, W.G., Jr.; Godley, L.A.; Koivunen, P. Fumarate and Succinate Regulate Expression of Hypoxia-inducible Genes via TET Enzymes. J. Biol. Chem. 2016, 291, 4256–4265. [Google Scholar] [CrossRef] [Green Version]
- Berchner-Pfannschmidt, U.; Tug, S.; Kirsch, M.; Fandrey, J. Oxygen-sensing under the influence of nitric oxide. Cell Signal. 2010, 22, 349–356. [Google Scholar] [CrossRef]
- Liu, Y.V.; Baek, J.H.; Zhang, H.; Diez, R.; Cole, R.N.; Semenza, G.L. RACK1 competes with HSP90 for binding to HIF-1alpha and is required for O(2)-independent and HSP90 inhibitor-induced degradation of HIF-1alpha. Mol. Cell 2007, 25, 207–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richard, D.E.; Berra, E.; Gothie, E.; Roux, D.; Pouyssegur, J. p42/p44 mitogen-activated protein kinases phosphorylate hypoxia-inducible factor 1alpha (HIF-1alpha) and enhance the transcriptional activity of HIF-1. J. Biol. Chem. 1999, 274, 32631–32637. [Google Scholar] [CrossRef] [Green Version]
- Geng, H.; Liu, Q.; Xue, C.; David, L.L.; Beer, T.M.; Thomas, G.V.; Dai, M.S.; Qian, D.Z. HIF1alpha protein stability is increased by acetylation at lysine 709. J. Biol. Chem. 2012, 287, 35496–35505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yasinska, I.M.; Sumbayev, V.V. S-nitrosation of Cys-800 of HIF-1alpha protein activates its interaction with p300 and stimulates its transcriptional activity. FEBS Lett. 2003, 549, 105–109. [Google Scholar] [CrossRef] [Green Version]
- Berta, M.A.; Mazure, N.; Hattab, M.; Pouyssegur, J.; Brahimi-Horn, M.C. SUMOylation of hypoxia-inducible factor-1alpha reduces its transcriptional activity. Biochem. Biophys. Res. Commun. 2007, 360, 646–652. [Google Scholar] [CrossRef] [PubMed]
- Rius, J.; Guma, M.; Schachtrup, C.; Akassoglou, K.; Zinkernagel, A.S.; Nizet, V.; Johnson, R.S.; Haddad, G.G.; Karin, M. NF-kappaB links innate immunity to the hypoxic response through transcriptional regulation of HIF-1alpha. Nature 2008, 453, 807–811. [Google Scholar] [CrossRef] [Green Version]
- Hudson, C.C.; Liu, M.; Chiang, G.G.; Otterness, D.M.; Loomis, D.C.; Kaper, F.; Giaccia, A.J.; Abraham, R.T. Regulation of hypoxia-inducible factor 1alpha expression and function by the mammalian target of rapamycin. Mol. Cell. Biol. 2002, 22, 7004–7014. [Google Scholar] [CrossRef] [Green Version]
- Yan, S.F.; Zou, Y.S.; Mendelsohn, M.; Gao, Y.; Naka, Y.; Du Yan, S.; Pinsky, D.; Stern, D. Nuclear factor interleukin 6 motifs mediate tissue-specific gene transcription in hypoxia. J. Biol. Chem. 1997, 272, 4287–4294. [Google Scholar] [CrossRef] [Green Version]
- Cummins, E.P.; Berra, E.; Comerford, K.M.; Ginouves, A.; Fitzgerald, K.T.; Seeballuck, F.; Godson, C.; Nielsen, J.E.; Moynagh, P.; Pouyssegur, J.; et al. Prolyl hydroxylase-1 negatively regulates IkappaB kinase-beta, giving insight into hypoxia-induced NFkappaB activity. Proc. Natl. Acad. Sci. USA 2006, 103, 18154–18159. [Google Scholar] [CrossRef]
- Batie, M.; Frost, J.; Frost, M.; Wilson, J.W.; Schofield, P.; Rocha, S. Hypoxia induces rapid changes to histone methylation and reprograms chromatin. Science 2019, 363, 1222–1226. [Google Scholar] [CrossRef]
- Shu, S.; Wang, Y.; Zheng, M.; Liu, Z.; Cai, J.; Tang, C.; Dong, Z. Hypoxia and Hypoxia-Inducible Factors in Kidney Injury and Repair. Cells 2019, 8, 207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, T.; Yamaguchi, J.; Higashijima, Y.; Nangaku, M. Indoxyl sulfate signals for rapid mRNA stabilization of Cbp/p300-interacting transactivator with Glu/Asp-rich carboxy-terminal domain 2 (CITED2) and suppresses the expression of hypoxia-inducible genes in experimental CKD and uremia. FASEB J. 2013, 27, 4059–4075. [Google Scholar] [CrossRef]
- Eltzschig, H.K.; Carmeliet, P. Hypoxia and inflammation. N. Engl. J. Med. 2011, 364, 656–665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sureshbabu, A.; Muhsin, S.A.; Choi, M.E. TGF-beta signaling in the kidney: Profibrotic and protective effects. Am. J. Physiol. Ren. Physiol. 2016, 310, F596–F606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ludes, P.O.; de Roquetaillade, C.; Chousterman, B.G.; Pottecher, J.; Mebazaa, A. Role of Damage-Associated Molecular Patterns in Septic Acute Kidney Injury, From Injury to Recovery. Front. Immunol. 2021, 12, 606622. [Google Scholar] [CrossRef]
- Santana, A.C.; Degaspari, S.; Catanozi, S.; Delle, H.; de Sa Lima, L.; Silva, C.; Blanco, P.; Solez, K.; Scavone, C.; Noronha, I.L. Thalidomide suppresses inflammation in adenine-induced CKD with uraemia in mice. Nephrol. Dial. Transplant. 2013, 28, 1140–1149. [Google Scholar] [CrossRef]
- Chen, J.; Tang, T.T.; Cao, J.Y.; Li, Z.L.; Zhong, X.; Wen, Y.; Shen, A.R.; Liu, B.C.; Lv, L.L. KIM-1 augments hypoxia-induced tubulointerstitial inflammation through uptake of small extracellular vesicles by tubular epithelial cells. Mol. Ther. 2022, 31, 1437–1450. [Google Scholar] [CrossRef]
- Chung, S.; Overstreet, J.M.; Li, Y.; Wang, Y.; Niu, A.; Wang, S.; Fan, X.; Sasaki, K.; Jin, G.N.; Khodo, S.N.; et al. TGF-beta promotes fibrosis after severe acute kidney injury by enhancing renal macrophage infiltration. JCI Insight 2018, 3, e123563. [Google Scholar] [CrossRef]
- Lech, M.; Grobmayr, R.; Ryu, M.; Lorenz, G.; Hartter, I.; Mulay, S.R.; Susanti, H.E.; Kobayashi, K.S.; Flavell, R.A.; Anders, H.J. Macrophage phenotype controls long-term AKI outcomes--kidney regeneration versus atrophy. J. Am. Soc. Nephrol. 2014, 25, 292–304. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi, J.; Tanaka, T.; Eto, N.; Nangaku, M. Inflammation and hypoxia linked to renal injury by CCAAT/enhancer-binding protein delta. Kidney Int. 2015, 88, 262–275. [Google Scholar] [CrossRef] [Green Version]
- Takeda, N.; O’Dea, E.L.; Doedens, A.; Kim, J.W.; Weidemann, A.; Stockmann, C.; Asagiri, M.; Simon, M.C.; Hoffmann, A.; Johnson, R.S. Differential activation and antagonistic function of HIF-alpha isoforms in macrophages are essential for NO homeostasis. Genes Dev. 2010, 24, 491–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, H.; Gilbert, V.; Liu, Q.; Kapitsinou, P.P.; Unger, T.L.; Rha, J.; Rivella, S.; Schlondorff, D.; Haase, V.H. Myeloid cell-derived hypoxia-inducible factor attenuates inflammation in unilateral ureteral obstruction-induced kidney injury. J. Immunol. 2012, 188, 5106–5115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tateishi, Y.; Osada-Oka, M.; Tanaka, M.; Shiota, M.; Izumi, Y.; Ishimura, E.; Motoyama, K.; Inaba, M.; Miura, K. Myeloid HIF-1 attenuates the progression of renal fibrosis in murine obstructive nephropathy. J. Pharmacol. Sci. 2015, 127, 181–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Awad, A.S.; Rouse, M.; Huang, L.; Vergis, A.L.; Reutershan, J.; Cathro, H.P.; Linden, J.; Okusa, M.D. Compartmentalization of neutrophils in the kidney and lung following acute ischemic kidney injury. Kidney Int. 2009, 75, 689–698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yago, T.; Petrich, B.G.; Zhang, N.; Liu, Z.; Shao, B.; Ginsberg, M.H.; McEver, R.P. Blocking neutrophil integrin activation prevents ischemia-reperfusion injury. J. Exp. Med. 2015, 212, 1267–1281. [Google Scholar] [CrossRef] [PubMed]
- Peyssonnaux, C.; Datta, V.; Cramer, T.; Doedens, A.; Theodorakis, E.A.; Gallo, R.L.; Hurtado-Ziola, N.; Nizet, V.; Johnson, R.S. HIF-1alpha expression regulates the bactericidal capacity of phagocytes. J. Clin. Investig. 2005, 115, 1806–1815. [Google Scholar] [CrossRef] [Green Version]
- Walmsley, S.R.; Print, C.; Farahi, N.; Peyssonnaux, C.; Johnson, R.S.; Cramer, T.; Sobolewski, A.; Condliffe, A.M.; Cowburn, A.S.; Johnson, N.; et al. Hypoxia-induced neutrophil survival is mediated by HIF-1alpha-dependent NF-kappaB activity. J. Exp. Med. 2005, 201, 105–115. [Google Scholar] [CrossRef] [Green Version]
- Campbell, E.L.; Bruyninckx, W.J.; Kelly, C.J.; Glover, L.E.; McNamee, E.N.; Bowers, B.E.; Bayless, A.J.; Scully, M.; Saeedi, B.J.; Golden-Mason, L.; et al. Transmigrating neutrophils shape the mucosal microenvironment through localized oxygen depletion to influence resolution of inflammation. Immunity 2014, 40, 66–77. [Google Scholar] [CrossRef] [Green Version]
- Kapitsinou, P.P.; Sano, H.; Michael, M.; Kobayashi, H.; Davidoff, O.; Bian, A.; Yao, B.; Zhang, M.Z.; Harris, R.C.; Duffy, K.J.; et al. Endothelial HIF-2 mediates protection and recovery from ischemic kidney injury. J. Clin. Investig. 2014, 124, 2396–2409. [Google Scholar] [CrossRef] [Green Version]
- Doedens, A.L.; Phan, A.T.; Stradner, M.H.; Fujimoto, J.K.; Nguyen, J.V.; Yang, E.; Johnson, R.S.; Goldrath, A.W. Hypoxia-inducible factors enhance the effector responses of CD8(+) T cells to persistent antigen. Nat. Immunol. 2013, 14, 1173–1182. [Google Scholar] [CrossRef] [Green Version]
- Moldogazieva, N.T.; Mokhosoev, I.M.; Feldman, N.B.; Lutsenko, S.V. ROS and RNS signalling: Adaptive redox switches through oxidative/nitrosative protein modifications. Free Radic. Res. 2018, 52, 507–543. [Google Scholar] [CrossRef]
- Laurindo, F.R.; Araujo, T.L.; Abrahao, T.B. Nox NADPH oxidases and the endoplasmic reticulum. Antioxid. Redox Signal. 2014, 20, 2755–2775. [Google Scholar] [CrossRef] [Green Version]
- Espinosa-Diez, C.; Miguel, V.; Mennerich, D.; Kietzmann, T.; Sanchez-Perez, P.; Cadenas, S.; Lamas, S. Antioxidant responses and cellular adjustments to oxidative stress. Redox Biol. 2015, 6, 183–197. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, T.; Nioi, P.; Pickett, C.B. The Nrf2-antioxidant response element signaling pathway and its activation by oxidative stress. J. Biol. Chem. 2009, 284, 13291–13295. [Google Scholar] [CrossRef] [Green Version]
- Oberg, B.P.; McMenamin, E.; Lucas, F.L.; McMonagle, E.; Morrow, J.; Ikizler, T.A.; Himmelfarb, J. Increased prevalence of oxidant stress and inflammation in patients with moderate to severe chronic kidney disease. Kidney Int. 2004, 65, 1009–1016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, H.J.; Shirakawa, H. Oxidative Stress and Mitochondrial Dysfunction in Chronic Kidney Disease. Cells 2022, 12, 88. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, F.; Assreuy, J.; Sordi, R. The role of nitric oxide in sepsis-associated kidney injury. Biosci. Rep. 2022, 42, BSR20220093. [Google Scholar] [CrossRef] [PubMed]
- Mapuskar, K.A.; Wen, H.; Holanda, D.G.; Rastogi, P.; Steinbach, E.; Han, R.; Coleman, M.C.; Attanasio, M.; Riley, D.P.; Spitz, D.R.; et al. Persistent increase in mitochondrial superoxide mediates cisplatin-induced chronic kidney disease. Redox Biol. 2019, 20, 98–106. [Google Scholar] [CrossRef]
- Briston, T.; Roberts, M.; Lewis, S.; Powney, B.; Staddon, J.M.; Szabadkai, G.; Duchen, M.R. Mitochondrial permeability transition pore: Sensitivity to opening and mechanistic dependence on substrate availability. Sci. Rep. 2017, 7, 10492. [Google Scholar] [CrossRef] [Green Version]
- Abais, J.M.; Zhang, C.; Xia, M.; Liu, Q.; Gehr, T.W.; Boini, K.M.; Li, P.L. NADPH oxidase-mediated triggering of inflammasome activation in mouse podocytes and glomeruli during hyperhomocysteinemia. Antioxid. Redox Signal. 2013, 18, 1537–1548. [Google Scholar] [CrossRef] [Green Version]
- Nezu, M.; Souma, T.; Yu, L.; Suzuki, T.; Saigusa, D.; Ito, S.; Suzuki, N.; Yamamoto, M. Transcription factor Nrf2 hyperactivation in early-phase renal ischemia-reperfusion injury prevents tubular damage progression. Kidney Int. 2017, 91, 387–401. [Google Scholar] [CrossRef]
- Wever, R.; Boer, P.; Hijmering, M.; Stroes, E.; Verhaar, M.; Kastelein, J.; Versluis, K.; Lagerwerf, F.; van Rijn, H.; Koomans, H.; et al. Nitric oxide production is reduced in patients with chronic renal failure. Arter. Thromb. Vasc. Biol. 1999, 19, 1168–1172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Godin, N.; Liu, F.; Lau, G.J.; Brezniceanu, M.L.; Chenier, I.; Filep, J.G.; Ingelfinger, J.R.; Zhang, S.L.; Chan, J.S. Catalase overexpression prevents hypertension and tubular apoptosis in angiotensinogen transgenic mice. Kidney Int. 2010, 77, 1086–1097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carlstrom, M. Nitric oxide signalling in kidney regulation and cardiometabolic health. Nat. Rev. Nephrol. 2021, 17, 575–590. [Google Scholar] [CrossRef] [PubMed]
- Tumur, Z.; Niwa, T. Indoxyl sulfate inhibits nitric oxide production and cell viability by inducing oxidative stress in vascular endothelial cells. Am. J. Nephrol. 2009, 29, 551–557. [Google Scholar] [CrossRef]
- Li, L.; Kang, H.; Zhang, Q.; D’Agati, V.D.; Al-Awqati, Q.; Lin, F. FoxO3 activation in hypoxic tubules prevents chronic kidney disease. J. Clin. Investig. 2019, 129, 2374–2389. [Google Scholar] [CrossRef] [Green Version]
- Bartoszewska, S.; Collawn, J.F. Unfolded protein response (UPR) integrated signaling networks determine cell fate during hypoxia. Cell. Mol. Biol. Lett. 2020, 25, 18. [Google Scholar] [CrossRef] [Green Version]
- Bhargava, P.; Schnellmann, R.G. Mitochondrial energetics in the kidney. Nat. Rev. Nephrol. 2017, 13, 629–646. [Google Scholar] [CrossRef]
- Hansen, J.; Sealfon, R.; Menon, R.; Eadon, M.T.; Lake, B.B.; Steck, B.; Anjani, K.; Parikh, S.; Sigdel, T.K.; Zhang, G.; et al. A reference tissue atlas for the human kidney. Sci. Adv. 2022, 8, eabn4965. [Google Scholar] [CrossRef]
- Scholz, H.; Boivin, F.J.; Schmidt-Ott, K.M.; Bachmann, S.; Eckardt, K.U.; Scholl, U.I.; Persson, P.B. Kidney physiology and susceptibility to acute kidney injury: Implications for renoprotection. Nat. Rev. Nephrol. 2021, 17, 335–349. [Google Scholar] [CrossRef]
- Li, H.; Dixon, E.E.; Wu, H.; Humphreys, B.D. Comprehensive single-cell transcriptional profiling defines shared and unique epithelial injury responses during kidney fibrosis. Cell Metab. 2022, 34, 1977–1998. [Google Scholar] [CrossRef] [PubMed]
- Lan, R.; Geng, H.; Singha, P.K.; Saikumar, P.; Bottinger, E.P.; Weinberg, J.M.; Venkatachalam, M.A. Mitochondrial Pathology and Glycolytic Shift during Proximal Tubule Atrophy after Ischemic AKI. J. Am. Soc. Nephrol. 2016, 27, 3356–3367. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.W.; Tchernyshyov, I.; Semenza, G.L.; Dang, C.V. HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006, 3, 177–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faivre, A.; Verissimo, T.; Auwerx, H.; Legouis, D.; de Seigneux, S. Tubular Cell Glucose Metabolism Shift During Acute and Chronic Injuries. Front. Med. 2021, 8, 742072. [Google Scholar] [CrossRef]
- Smith, J.A.; Stallons, L.J.; Schnellmann, R.G. Renal cortical hexokinase and pentose phosphate pathway activation through the EGFR/Akt signaling pathway in endotoxin-induced acute kidney injury. Am. J. Physiol. Ren. Physiol. 2014, 307, F435–F444. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.L.; Zhang, R.; Anand, P.; Stomberski, C.T.; Qian, Z.; Hausladen, A.; Wang, L.; Rhee, E.P.; Parikh, S.M.; Karumanchi, S.A.; et al. Metabolic reprogramming by the S-nitroso-CoA reductase system protects against kidney injury. Nature 2019, 565, 96–100. [Google Scholar] [CrossRef]
- Kang, H.M.; Ahn, S.H.; Choi, P.; Ko, Y.A.; Han, S.H.; Chinga, F.; Park, A.S.; Tao, J.; Sharma, K.; Pullman, J.; et al. Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nat. Med. 2015, 21, 37–46. [Google Scholar] [CrossRef]
- Henderson, N.C.; Rieder, F.; Wynn, T.A. Fibrosis: From mechanisms to medicines. Nature 2020, 587, 555–566. [Google Scholar] [CrossRef]
- Kottmann, R.M.; Kulkarni, A.A.; Smolnycki, K.A.; Lyda, E.; Dahanayake, T.; Salibi, R.; Honnons, S.; Jones, C.; Isern, N.G.; Hu, J.Z.; et al. Lactic Acid Is Elevated in Idiopathic Pulmonary Fibrosis and Induces Myofibroblast Differentiation via pH-Dependent Activation of Transforming Growth Factor-β. Am. J. Respir. Crit. Care Med. 2012, 186, 740–751. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Galichon, P.; Xiao, X.; Figueroa-Ramirez, A.C.; Tamayo, D.; Lee, J.J.; Kalocsay, M.; Gonzalez-Sanchez, D.; Chancay, M.S.; McCracken, K.W.; et al. Orphan nuclear receptor COUP-TFII enhances myofibroblast glycolysis leading to kidney fibrosis. EMBO Rep. 2021, 22, e51169. [Google Scholar] [CrossRef]
- Wei, Q.; Su, J.; Dong, G.; Zhang, M.; Huo, Y.; Dong, Z. Glycolysis inhibitors suppress renal interstitial fibrosis via divergent effects on fibroblasts and tubular cells. Am. J. Physiol. Ren. Physiol. 2019, 316, F1162–F1172. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.; Jiang, L.; Xu, J.; Bai, F.; Zhou, Y.; Yuan, Q.; Luo, J.; Zen, K.; Yang, J. Inhibiting aerobic glycolysis suppresses renal interstitial fibroblast activation and renal fibrosis. Am. J. Physiol. Ren. Physiol. 2017, 313, F561–F575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, H.; Zhu, J.; Chang, L.; Liang, C.; Li, X.; Wang, W. 3-Bromopyruvate decreased kidney fibrosis and fibroblast activation by suppressing aerobic glycolysis in unilateral ureteral obstruction mice model. Life Sci. 2021, 272, 119206. [Google Scholar] [CrossRef]
- Chini, C.C.S.; Zeidler, J.D.; Kashyap, S.; Warner, G.; Chini, E.N. Evolving concepts in NAD(+) metabolism. Cell Metab. 2021, 33, 1076–1087. [Google Scholar] [CrossRef] [PubMed]
- Covarrubias, A.J.; Perrone, R.; Grozio, A.; Verdin, E. NAD(+) metabolism and its roles in cellular processes during ageing. Nat. Rev. Mol. Cell Biol. 2021, 22, 119–141. [Google Scholar] [CrossRef]
- Ralto, K.M.; Rhee, E.P.; Parikh, S.M. NAD(+) homeostasis in renal health and disease. Nat. Rev. Nephrol. 2020, 16, 99–111. [Google Scholar] [CrossRef]
- Pellicciari, R.; Liscio, P.; Giacche, N.; De Franco, F.; Carotti, A.; Robertson, J.; Cialabrini, L.; Katsyuba, E.; Raffaelli, N.; Auwerx, J. alpha-Amino-beta-carboxymuconate-epsilon-semialdehyde Decarboxylase (ACMSD) Inhibitors as Novel Modulators of De Novo Nicotinamide Adenine Dinucleotide (NAD(+)) Biosynthesis. J. Med. Chem. 2018, 61, 745–759. [Google Scholar] [CrossRef]
- Katsyuba, E.; Mottis, A.; Zietak, M.; De Franco, F.; van der Velpen, V.; Gariani, K.; Ryu, D.; Cialabrini, L.; Matilainen, O.; Liscio, P.; et al. De novo NAD(+) synthesis enhances mitochondrial function and improves health. Nature 2018, 563, 354–359. [Google Scholar] [CrossRef]
- Guan, Y.; Wang, S.R.; Huang, X.Z.; Xie, Q.H.; Xu, Y.Y.; Shang, D.; Hao, C.M. Nicotinamide Mononucleotide, an NAD(+) Precursor, Rescues Age-Associated Susceptibility to AKI in a Sirtuin 1-Dependent Manner. J. Am. Soc. Nephrol. 2017, 28, 2337–2352. [Google Scholar] [CrossRef] [Green Version]
- Zheng, M.; Cai, J.; Liu, Z.; Shu, S.; Wang, Y.; Tang, C.; Dong, Z. Nicotinamide reduces renal interstitial fibrosis by suppressing tubular injury and inflammation. J. Cell. Mol. Med. 2019, 23, 3995–4004. [Google Scholar] [CrossRef] [Green Version]
- Legouis, D.; Faivre, A.; Cippa, P.E.; de Seigneux, S. Renal gluconeogenesis: An underestimated role of the kidney in systemic glucose metabolism. Nephrol. Dial. Transplant. 2022, 37, 1417–1425. [Google Scholar] [CrossRef]
- Legouis, D.; Ricksten, S.E.; Faivre, A.; Verissimo, T.; Gariani, K.; Verney, C.; Galichon, P.; Berchtold, L.; Feraille, E.; Fernandez, M.; et al. Altered proximal tubular cell glucose metabolism during acute kidney injury is associated with mortality. Nat. Metab. 2020, 2, 732–743. [Google Scholar] [CrossRef]
- Brooks, C.; Wei, Q.; Cho, S.G.; Dong, Z. Regulation of mitochondrial dynamics in acute kidney injury in cell culture and rodent models. J. Clin. Investig. 2009, 119, 1275–1285. [Google Scholar] [CrossRef]
- Xiao, X.; Hu, Y.; Quiros, P.M.; Wei, Q.; Lopez-Otin, C.; Dong, Z. OMA1 mediates OPA1 proteolysis and mitochondrial fragmentation in experimental models of ischemic kidney injury. Am. J. Physiol. Ren. Physiol. 2014, 306, F1318–F1326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuo, Z.; Jing, K.; Wu, H.; Wang, S.; Ye, L.; Li, Z.; Yang, C.; Pan, Q.; Liu, W.J.; Liu, H.F. Mechanisms and Functions of Mitophagy and Potential Roles in Renal Disease. Front. Physiol. 2020, 11, 935. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.C.; Quintin, J.; Cramer, R.A.; Shepardson, K.M.; Saeed, S.; Kumar, V.; Giamarellos-Bourboulis, E.J.; Martens, J.H.; Rao, N.A.; Aghajanirefah, A.; et al. mTOR- and HIF-1alpha-mediated aerobic glycolysis as metabolic basis for trained immunity. Science 2014, 345, 1250684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bao, H.; Zhang, Q.; Liu, X.; Song, Y.; Li, X.; Wang, Z.; Li, C.; Peng, A.; Gong, R. Lithium targeting of AMPK protects against cisplatin-induced acute kidney injury by enhancing autophagy in renal proximal tubular epithelial cells. FASEB J. 2019, 33, 14370–14381. [Google Scholar] [CrossRef] [Green Version]
- Jin, K.; Ma, Y.; Manrique-Caballero, C.L.; Li, H.; Emlet, D.R.; Li, S.; Baty, C.J.; Wen, X.; Kim-Campbell, N.; Frank, A.; et al. Activation of AMP-activated protein kinase during sepsis/inflammation improves survival by preserving cellular metabolic fitness. FASEB J. 2020, 34, 7036–7057. [Google Scholar] [CrossRef] [Green Version]
- Wculek, S.K.; Dunphy, G.; Heras-Murillo, I.; Mastrangelo, A.; Sancho, D. Metabolism of tissue macrophages in homeostasis and pathology. Cell. Mol. Immunol. 2022, 19, 384–408. [Google Scholar] [CrossRef]
- Higgins, D.F.; Kimura, K.; Bernhardt, W.M.; Shrimanker, N.; Akai, Y.; Hohenstein, B.; Saito, Y.; Johnson, R.S.; Kretzler, M.; Cohen, C.D.; et al. Hypoxia promotes fibrogenesis in vivo via HIF-1 stimulation of epithelial-to-mesenchymal transition. J. Clin. Investig. 2007, 117, 3810–3820. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Zeng, H.; Chen, S.T.; Zhou, L.; Xie, X.J.; He, X.; Tao, Y.K.; Tuo, Q.H.; Deng, C.; Liao, D.F.; et al. Ablation of endothelial prolyl hydroxylase domain protein-2 promotes renal vascular remodelling and fibrosis in mice. J. Cell. Mol. Med. 2017, 21, 1967–1978. [Google Scholar] [CrossRef] [PubMed]
- Eng, E.; Holgren, C.; Hubchak, S.; Naaz, P.; Schnaper, H.W. Hypoxia regulates PDGF-B interactions between glomerular capillary endothelial and mesangial cells. Kidney Int. 2005, 68, 695–703. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Ning, X.; Li, R.; Yang, Z.; Yang, X.; Sun, S.; Qian, Q. Signalling pathways involved in hypoxia-induced renal fibrosis. J. Cell. Mol. Med. 2017, 21, 1248–1259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baumann, B.; Hayashida, T.; Liang, X.; Schnaper, H.W. Hypoxia-inducible factor-1alpha promotes glomerulosclerosis and regulates COL1A2 expression through interactions with Smad3. Kidney Int. 2016, 90, 797–808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rozen-Zvi, B.; Hayashida, T.; Hubchak, S.C.; Hanna, C.; Platanias, L.C.; Schnaper, H.W. TGF-beta/Smad3 activates mammalian target of rapamycin complex-1 to promote collagen production by increasing HIF-1alpha expression. Am. J. Physiol. Ren. Physiol. 2013, 305, F485–F494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, W.Q.; Zhu, Q.; Hu, J.; Li, P.L.; Zhang, F.; Li, N. Hypoxia-inducible factor prolyl-hydroxylase-2 mediates transforming growth factor beta 1-induced epithelial-mesenchymal transition in renal tubular cells. Biochim. Biophys. Acta 2013, 1833, 1454–1462. [Google Scholar] [CrossRef] [Green Version]
- Hanna, C.; Hubchak, S.C.; Liang, X.; Rozen-Zvi, B.; Schumacker, P.T.; Hayashida, T.; Schnaper, H.W. Hypoxia-inducible factor-2alpha and TGF-beta signaling interact to promote normoxic glomerular fibrogenesis. Am. J. Physiol. Ren. Physiol. 2013, 305, F1323–F1331. [Google Scholar] [CrossRef] [Green Version]
- Basu, R.K.; Hubchak, S.; Hayashida, T.; Runyan, C.E.; Schumacker, P.T.; Schnaper, H.W. Interdependence of HIF-1alpha and TGF-beta/Smad3 signaling in normoxic and hypoxic renal epithelial cell collagen expression. Am. J. Physiol. Ren. Physiol. 2011, 300, F898–F905. [Google Scholar] [CrossRef] [Green Version]
- Luo, R.; Zhang, W.; Zhao, C.; Zhang, Y.; Wu, H.; Jin, J.; Zhang, W.; Grenz, A.; Eltzschig, H.K.; Tao, L.; et al. Elevated Endothelial Hypoxia-Inducible Factor-1alpha Contributes to Glomerular Injury and Promotes Hypertensive Chronic Kidney Disease. Hypertension 2015, 66, 75–84. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Liu, L.; Bai, M.; Liu, M.; Wei, L.; Yang, Z.; Qian, Q.; Ning, X.; Sun, S. Hypoxia-induced HE4 in tubular epithelial cells promotes extracellular matrix accumulation and renal fibrosis via NF-kappaB. FASEB J. 2020, 34, 2554–2567. [Google Scholar] [CrossRef]
- Du, R.; Xia, L.; Ning, X.; Liu, L.; Sun, W.; Huang, C.; Wang, H.; Sun, S. Hypoxia-induced Bmi1 promotes renal tubular epithelial cell-mesenchymal transition and renal fibrosis via PI3K/Akt signal. Mol. Biol. Cell 2014, 25, 2650–2659. [Google Scholar] [CrossRef]
- Hsu, R.K.; Hsu, C.Y. The Role of Acute Kidney Injury in Chronic Kidney Disease. Semin. Nephrol. 2016, 36, 283–292. [Google Scholar] [CrossRef] [Green Version]
- Hukriede, N.A.; Soranno, D.E.; Sander, V.; Perreau, T.; Starr, M.C.; Yuen, P.S.T.; Siskind, L.J.; Hutchens, M.P.; Davidson, A.J.; Burmeister, D.M.; et al. Experimental models of acute kidney injury for translational research. Nat. Rev. Nephrol. 2022, 18, 277–293. [Google Scholar] [CrossRef]
- Rosenberger, C.; Heyman, S.N.; Rosen, S.; Shina, A.; Goldfarb, M.; Griethe, W.; Frei, U.; Reinke, P.; Bachmann, S.; Eckardt, K.U. Up-regulation of HIF in experimental acute renal failure: Evidence for a protective transcriptional response to hypoxia. Kidney Int. 2005, 67, 531–542. [Google Scholar] [CrossRef] [Green Version]
- Rosenberger, C.; Pratschke, J.; Rudolph, B.; Heyman, S.N.; Schindler, R.; Babel, N.; Eckardt, K.U.; Frei, U.; Rosen, S.; Reinke, P. Immunohistochemical detection of hypoxia-inducible factor-1alpha in human renal allograft biopsies. J. Am. Soc. Nephrol. 2007, 18, 343–351. [Google Scholar] [CrossRef] [Green Version]
- Mayer, G. Capillary rarefaction, hypoxia, VEGF and angiogenesis in chronic renal disease. Nephrol. Dial. Transplant. 2011, 26, 1132–1137. [Google Scholar] [CrossRef] [Green Version]
- Mattot, V.; Moons, L.; Lupu, F.; Chernavvsky, D.; Gomez, R.A.; Collen, D.; Carmeliet, P. Loss of the VEGF(164) and VEGF(188) isoforms impairs postnatal glomerular angiogenesis and renal arteriogenesis in mice. J. Am. Soc. Nephrol. 2002, 13, 1548–1560. [Google Scholar] [CrossRef] [Green Version]
- Kang, D.H.; Joly, A.H.; Oh, S.W.; Hugo, C.; Kerjaschki, D.; Gordon, K.L.; Mazzali, M.; Jefferson, J.A.; Hughes, J.; Madsen, K.M.; et al. Impaired angiogenesis in the remnant kidney model: I. Potential role of vascular endothelial growth factor and thrombospondin-1. J. Am. Soc. Nephrol. 2001, 12, 1434–1447. [Google Scholar] [CrossRef]
- Maciel, T.T.; Coutinho, E.L.; Soares, D.; Achar, E.; Schor, N.; Bellini, M.H. Endostatin, an antiangiogenic protein, is expressed in the unilateral ureteral obstruction mice model. J. Nephrol. 2008, 21, 753–760. [Google Scholar]
- Goligorsky, M.S.; Yasuda, K.; Ratliff, B. Dysfunctional endothelial progenitor cells in chronic kidney disease. J. Am. Soc. Nephrol. 2010, 21, 911–919. [Google Scholar] [CrossRef]
- Kramann, R.; Wongboonsin, J.; Chang-Panesso, M.; Machado, F.G.; Humphreys, B.D. Gli1(+) Pericyte Loss Induces Capillary Rarefaction and Proximal Tubular Injury. J. Am. Soc. Nephrol. 2017, 28, 776–784. [Google Scholar] [CrossRef]
- Ascon, M.; Ascon, D.B.; Liu, M.; Cheadle, C.; Sarkar, C.; Racusen, L.; Hassoun, H.T.; Rabb, H. Renal ischemia-reperfusion leads to long term infiltration of activated and effector-memory T lymphocytes. Kidney Int. 2009, 75, 526–535. [Google Scholar] [CrossRef] [Green Version]
- Kong, T.; Eltzschig, H.K.; Karhausen, J.; Colgan, S.P.; Shelley, C.S. Leukocyte adhesion during hypoxia is mediated by HIF-1-dependent induction of beta2 integrin gene expression. Proc. Natl. Acad. Sci. USA 2004, 101, 10440–10445. [Google Scholar] [CrossRef]
- Friederich-Persson, M.; Thorn, E.; Hansell, P.; Nangaku, M.; Levin, M.; Palm, F. Kidney hypoxia, attributable to increased oxygen consumption, induces nephropathy independently of hyperglycemia and oxidative stress. Hypertension 2013, 62, 914–919. [Google Scholar] [CrossRef] [Green Version]
- Polichnowski, A.J.; Lan, R.; Geng, H.; Griffin, K.A.; Venkatachalam, M.A.; Bidani, A.K. Severe renal mass reduction impairs recovery and promotes fibrosis after AKI. J. Am. Soc. Nephrol. 2014, 25, 1496–1507. [Google Scholar] [CrossRef] [Green Version]
- Ferenbach, D.A.; Bonventre, J.V. Mechanisms of maladaptive repair after AKI leading to accelerated kidney ageing and CKD. Nat. Rev. Nephrol. 2015, 11, 264–276. [Google Scholar] [CrossRef] [Green Version]
- Beyer, S.; Kristensen, M.M.; Jensen, K.S.; Johansen, J.V.; Staller, P. The histone demethylases JMJD1A and JMJD2B are transcriptional targets of hypoxia-inducible factor HIF. J. Biol. Chem. 2008, 283, 36542–36552. [Google Scholar] [CrossRef] [Green Version]
- Krieg, A.J.; Rankin, E.B.; Chan, D.; Razorenova, O.; Fernandez, S.; Giaccia, A.J. Regulation of the histone demethylase JMJD1A by hypoxia-inducible factor 1 alpha enhances hypoxic gene expression and tumor growth. Mol. Cell. Biol. 2010, 30, 344–353. [Google Scholar] [CrossRef] [Green Version]
- Mimura, I.; Nangaku, M.; Kanki, Y.; Tsutsumi, S.; Inoue, T.; Kohro, T.; Yamamoto, S.; Fujita, T.; Shimamura, T.; Suehiro, J.; et al. Dynamic change of chromatin conformation in response to hypoxia enhances the expression of GLUT3 (SLC2A3) by cooperative interaction of hypoxia-inducible factor 1 and KDM3A. Mol. Cell. Biol. 2012, 32, 3018–3032. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Romo, R.; Berman, N.; Gomez, A.; Bobadilla, N.A. Epigenetic regulation in the acute kidney injury to chronic kidney disease transition. Nephrology 2015, 20, 736–743. [Google Scholar] [CrossRef]
- Zager, R.A.; Johnson, A.C.; Andress, D.; Becker, K. Progressive endothelin-1 gene activation initiates chronic/end-stage renal disease following experimental ischemic/reperfusion injury. Kidney Int. 2013, 84, 703–712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hill, P.; Shukla, D.; Tran, M.G.; Aragones, J.; Cook, H.T.; Carmeliet, P.; Maxwell, P.H. Inhibition of hypoxia inducible factor hydroxylases protects against renal ischemia-reperfusion injury. J. Am. Soc. Nephrol. 2008, 19, 39–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, J.L.; Pham, H.; Li, Y.; Hall, E.; Perkins, G.A.; Ali, S.S.; Patel, H.H.; Singh, P. Hypoxia-inducible factor-1alpha activation improves renal oxygenation and mitochondrial function in early chronic kidney disease. Am. J. Physiol. Ren. Physiol. 2017, 313, F282–F290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kato, S.; Takahashi, T.; Miyata, N.; Roman, R.J. DMOG, a Prolyl Hydroxylase Inhibitor, Increases Hemoglobin Levels without Exacerbating Hypertension and Renal Injury in Salt-Sensitive Hypertensive Rats. J. Pharmacol. Exp. Ther. 2020, 372, 166–174. [Google Scholar] [CrossRef]
- Nordquist, L.; Friederich-Persson, M.; Fasching, A.; Liss, P.; Shoji, K.; Nangaku, M.; Hansell, P.; Palm, F. Activation of hypoxia-inducible factors prevents diabetic nephropathy. J. Am. Soc. Nephrol. 2015, 26, 328–338. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, T.; Matsumoto, M.; Inagi, R.; Miyata, T.; Kojima, I.; Ohse, T.; Fujita, T.; Nangaku, M. Induction of protective genes by cobalt ameliorates tubulointerstitial injury in the progressive Thy1 nephritis. Kidney Int. 2005, 68, 2714–2725. [Google Scholar] [CrossRef] [Green Version]
- Ohtomo, S.; Nangaku, M.; Izuhara, Y.; Takizawa, S.; Strihou, C.; Miyata, T. Cobalt ameliorates renal injury in an obese, hypertensive type 2 diabetes rat model. Nephrol. Dial. Transplant. 2008, 23, 1166–1172. [Google Scholar] [CrossRef] [Green Version]
- Akizawa, T.; Iwasaki, M.; Yamaguchi, Y.; Majikawa, Y.; Reusch, M. Phase 3, Randomized, Double-Blind, Active-Comparator (Darbepoetin Alfa) Study of Oral Roxadustat in CKD Patients with Anemia on Hemodialysis in Japan. J. Am. Soc. Nephrol. 2020, 31, 1628–1639. [Google Scholar] [CrossRef]
- Dhillon, S. Daprodustat: First Approval. Drugs 2020, 80, 1491–1497. [Google Scholar] [CrossRef]
- Markham, A. Vadadustat: First Approval. Drugs 2020, 80, 1365–1371. [Google Scholar] [CrossRef]
- Markham, A. Enarodustat: First Approval. Drugs 2021, 81, 169–174. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Lan, J.; Dong, F.; Duan, P. Effectiveness of hypoxia-induced factor prolyl hydroxylase inhibitor for managing anemia in chronic kidney disease: A systematic review and meta-analysis. Eur. J. Clin. Pharmacol. 2021, 77, 491–507. [Google Scholar] [CrossRef] [PubMed]
- Flight, M.H. Deal watch: AstraZeneca bets on FibroGen’s anaemia drug. Nat. Rev. Drug Discov. 2013, 12, 730. [Google Scholar] [CrossRef] [PubMed]
- Takkavatakarn, K.; Thammathiwat, T.; Phannajit, J.; Katavetin, P.; Praditpornsilpa, K.; Eiam-Ong, S.; Susantitaphong, P. The impacts of hypoxia-inducible factor stabilizers on laboratory parameters and clinical outcomes in chronic kidney disease patients with renal anemia: A systematic review and meta-analysis. Clin. Kidney J. 2023, 16, 845–858. [Google Scholar] [CrossRef]
- Sugahara, M.; Tanaka, S.; Tanaka, T.; Saito, H.; Ishimoto, Y.; Wakashima, T.; Ueda, M.; Fukui, K.; Shimizu, A.; Inagi, R.; et al. Prolyl Hydroxylase Domain Inhibitor Protects against Metabolic Disorders and Associated Kidney Disease in Obese Type 2 Diabetic Mice. J. Am. Soc. Nephrol. 2020, 31, 560–577. [Google Scholar] [CrossRef]
- Uchida, L.; Tanaka, T.; Saito, H.; Sugahara, M.; Wakashima, T.; Fukui, K.; Nangaku, M. Effects of a prolyl hydroxylase inhibitor on kidney and cardiovascular complications in a rat model of chronic kidney disease. Am. J. Physiol. Ren. Physiol. 2020, 318, F388–F401. [Google Scholar] [CrossRef]
- Yu, X.; Fang, Y.; Liu, H.; Zhu, J.; Zou, J.; Xu, X.; Jiang, S.; Ding, X. The balance of beneficial and deleterious effects of hypoxia-inducible factor activation by prolyl hydroxylase inhibitor in rat remnant kidney depends on the timing of administration. Nephrol. Dial. Transplant. 2012, 27, 3110–3119. [Google Scholar] [CrossRef] [Green Version]
- Schley, G.; Klanke, B.; Kalucka, J.; Schatz, V.; Daniel, C.; Mayer, M.; Goppelt-Struebe, M.; Herrmann, M.; Thorsteinsdottir, M.; Palsson, R.; et al. Mononuclear phagocytes orchestrate prolyl hydroxylase inhibition-mediated renoprotection in chronic tubulointerstitial nephritis. Kidney Int. 2019, 96, 378–396. [Google Scholar] [CrossRef]
- Kimura, K.; Iwano, M.; Higgins, D.F.; Yamaguchi, Y.; Nakatani, K.; Harada, K.; Kubo, A.; Akai, Y.; Rankin, E.B.; Neilson, E.G.; et al. Stable expression of HIF-1alpha in tubular epithelial cells promotes interstitial fibrosis. Am. J. Physiol. Ren. Physiol. 2008, 295, F1023–F1029. [Google Scholar] [CrossRef] [Green Version]
- Schietke, R.E.; Hackenbeck, T.; Tran, M.; Gunther, R.; Klanke, B.; Warnecke, C.L.; Knaup, K.X.; Shukla, D.; Rosenberger, C.; Koesters, R.; et al. Renal tubular HIF-2alpha expression requires VHL inactivation and causes fibrosis and cysts. PLoS ONE 2012, 7, e31034. [Google Scholar] [CrossRef] [Green Version]
- Leonard, E.C.; Friedrich, J.L.; Basile, D.P. VEGF-121 preserves renal microvessel structure and ameliorates secondary renal disease following acute kidney injury. Am. J. Physiol. Ren. Physiol. 2008, 295, F1648–F1657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, Y.J.; Kim, D.H.; Lee, A.S.; Lee, S.; Kang, K.P.; Lee, S.Y.; Jang, K.Y.; Sung, M.J.; Park, S.K.; Kim, W. Peritubular capillary preservation with COMP-angiopoietin-1 decreases ischemia-reperfusion-induced acute kidney injury. Am. J. Physiol. Ren. Physiol. 2009, 297, F952–F960. [Google Scholar] [CrossRef] [Green Version]
- Engel, J.E.; Williams, E.; Williams, M.L.; Bidwell, G.L., 3rd; Chade, A.R. Targeted VEGF (Vascular Endothelial Growth Factor) Therapy Induces Long-Term Renal Recovery in Chronic Kidney Disease via Macrophage Polarization. Hypertension 2019, 74, 1113–1123. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Gobe, G. Protein kinase C activation and its role in kidney disease. Nephrology 2006, 11, 428–434. [Google Scholar] [CrossRef] [PubMed]
- Xue, X.; Ren, J.; Sun, X.; Gui, Y.; Feng, Y.; Shu, B.; Wei, W.; Lu, Q.; Liang, Y.; He, W.; et al. Protein kinase Calpha drives fibroblast activation and kidney fibrosis by stimulating autophagic flux. J. Biol. Chem. 2018, 293, 11119–11130. [Google Scholar] [CrossRef] [Green Version]
- Park, C.H.; Yoo, T.H. TGF-beta Inhibitors for Therapeutic Management of Kidney Fibrosis. Pharmaceuticals 2022, 15, 1485. [Google Scholar] [CrossRef]
- Vallon, V.; Verma, S. Effects of SGLT2 Inhibitors on Kidney and Cardiovascular Function. Annu. Rev. Physiol. 2021, 83, 503–528. [Google Scholar] [CrossRef]
- Marathias, K.P.; Lambadiari, V.A.; Markakis, K.P.; Vlahakos, V.D.; Bacharaki, D.; Raptis, A.E.; Dimitriadis, G.D.; Vlahakos, D.V. Competing Effects of Renin Angiotensin System Blockade and Sodium-Glucose Cotransporter-2 Inhibitors on Erythropoietin Secretion in Diabetes. Am. J. Nephrol. 2020, 51, 349–356. [Google Scholar] [CrossRef]
- Layton, A.T.; Vallon, V.; Edwards, A. Predicted consequences of diabetes and SGLT inhibition on transport and oxygen consumption along a rat nephron. Am. J. Physiol. Ren. Physiol. 2016, 310, F1269–F1283. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Nakano, D.; Guan, Y.; Hitomi, H.; Uemura, A.; Masaki, T.; Kobara, H.; Sugaya, T.; Nishiyama, A. A sodium-glucose cotransporter 2 inhibitor attenuates renal capillary injury and fibrosis by a vascular endothelial growth factor-dependent pathway after renal injury in mice. Kidney Int. 2018, 94, 524–535. [Google Scholar] [CrossRef] [Green Version]
- Zuo, N.; Zheng, X.; Liu, H.; Ma, X. Fenofibrate, a PPARalpha agonist, protect proximal tubular cells from albumin-bound fatty acids induced apoptosis via the activation of NF-kB. Int. J. Clin. Exp. Pathol. 2015, 8, 10653–10661. [Google Scholar]
- Tanaka, Y.; Kume, S.; Araki, S.; Isshiki, K.; Chin-Kanasaki, M.; Sakaguchi, M.; Sugimoto, T.; Koya, D.; Haneda, M.; Kashiwagi, A.; et al. Fenofibrate, a PPARalpha agonist, has renoprotective effects in mice by enhancing renal lipolysis. Kidney Int. 2011, 79, 871–882. [Google Scholar] [CrossRef] [Green Version]
- Clark, A.J.; Parikh, S.M. Targeting energy pathways in kidney disease: The roles of sirtuins, AMPK, and PGC1alpha. Kidney Int. 2021, 99, 828–840. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Gao, Y.; Zhang, Q.; Wei, S.; Chen, Z.; Dai, X.; Zeng, Z.; Zhao, K.S. SIRT1/3 Activation by Resveratrol Attenuates Acute Kidney Injury in a Septic Rat Model. Oxidative Med. Cell. Longev. 2016, 2016, 7296092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, Y.A.; Bae, S.Y.; Ahn, S.Y.; Kim, J.; Kwon, Y.J.; Jung, W.Y.; Ko, G.J. Resveratrol Ameliorates Contrast Induced Nephropathy Through the Activation of SIRT1-PGC-1alpha-Foxo1 Signaling in Mice. Kidney Blood Press. Res. 2017, 42, 641–653. [Google Scholar] [CrossRef]
- Perry, H.M.; Huang, L.; Wilson, R.J.; Bajwa, A.; Sesaki, H.; Yan, Z.; Rosin, D.L.; Kashatus, D.F.; Okusa, M.D. Dynamin-Related Protein 1 Deficiency Promotes Recovery from AKI. J. Am. Soc. Nephrol. 2018, 29, 194–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, J.; Xue, Q.; Kuang, L.; Xie, L.; Luo, R.; Nie, X. Berberine alleviates cisplatin-induced acute kidney injury by regulating mitophagy via PINK 1/Parkin pathway. Transl. Androl. Urol. 2020, 9, 1712–1724. [Google Scholar] [CrossRef] [PubMed]
- Birk, A.V.; Liu, S.; Soong, Y.; Mills, W.; Singh, P.; Warren, J.D.; Seshan, S.V.; Pardee, J.D.; Szeto, H.H. The mitochondrial-targeted compound SS-31 re-energizes ischemic mitochondria by interacting with cardiolipin. J. Am. Soc. Nephrol. 2013, 24, 1250–1261. [Google Scholar] [CrossRef] [Green Version]
- Szeto, H.H.; Liu, S.; Soong, Y.; Wu, D.; Darrah, S.F.; Cheng, F.Y.; Zhao, Z.; Ganger, M.; Tow, C.Y.; Seshan, S.V. Mitochondria-targeted peptide accelerates ATP recovery and reduces ischemic kidney injury. J. Am. Soc. Nephrol. 2011, 22, 1041–1052. [Google Scholar] [CrossRef] [Green Version]
- Stoker, M.L.; Newport, E.; Hulit, J.C.; West, A.P.; Morten, K.J. Impact of pharmacological agents on mitochondrial function: A growing opportunity? Biochem. Soc. Trans. 2019, 47, 1757–1772. [Google Scholar] [CrossRef] [Green Version]
- Ngo, V.; Duennwald, M.L. Nrf2 and Oxidative Stress: A General Overview of Mechanisms and Implications in Human Disease. Antioxidants 2022, 11, 2345. [Google Scholar] [CrossRef] [PubMed]
- Kanda, H.; Yamawaki, K. Bardoxolone methyl: Drug development for diabetic kidney disease. Clin. Exp. Nephrol. 2020, 24, 857–864. [Google Scholar] [CrossRef] [PubMed]
- Pisano, A.; Cernaro, V.; Gembillo, G.; D’Arrigo, G.; Buemi, M.; Bolignano, D. Xanthine Oxidase Inhibitors for Improving Renal Function in Chronic Kidney Disease Patients: An Updated Systematic Review and Meta-Analysis. Int. J. Mol. Sci. 2017, 18, 2283. [Google Scholar] [CrossRef] [Green Version]
- Goicoechea, M.; de Vinuesa, S.G.; Verdalles, U.; Ruiz-Caro, C.; Ampuero, J.; Rincon, A.; Arroyo, D.; Luno, J. Effect of allopurinol in chronic kidney disease progression and cardiovascular risk. Clin. J. Am. Soc. Nephrol. 2010, 5, 1388–1393. [Google Scholar] [CrossRef] [Green Version]
- Sedeek, M.; Nasrallah, R.; Touyz, R.M.; Hebert, R.L. NADPH oxidases, reactive oxygen species, and the kidney: Friend and foe. J. Am. Soc. Nephrol. 2013, 24, 1512–1518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, G.; Uddin, M.J.; Lee, G.; Jiang, S.; Cho, A.; Lee, J.H.; Lee, S.R.; Bae, Y.S.; Moon, S.H.; Lee, S.J.; et al. A novel pan-Nox inhibitor, APX-115, protects kidney injury in streptozotocin-induced diabetic mice: Possible role of peroxisomal and mitochondrial biogenesis. Oncotarget 2017, 8, 74217–74232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jha, J.C.; Gray, S.P.; Barit, D.; Okabe, J.; El-Osta, A.; Namikoshi, T.; Thallas-Bonke, V.; Wingler, K.; Szyndralewiez, C.; Heitz, F.; et al. Genetic targeting or pharmacologic inhibition of NADPH oxidase nox4 provides renoprotection in long-term diabetic nephropathy. J. Am. Soc. Nephrol. 2014, 25, 1237–1254. [Google Scholar] [CrossRef] [Green Version]
- Sedeek, M.; Callera, G.; Montezano, A.; Gutsol, A.; Heitz, F.; Szyndralewiez, C.; Page, P.; Kennedy, C.R.; Burns, K.D.; Touyz, R.M.; et al. Critical role of Nox4-based NADPH oxidase in glucose-induced oxidative stress in the kidney: Implications in type 2 diabetic nephropathy. Am. J. Physiol. Ren. Physiol. 2010, 299, F1348–F1358. [Google Scholar] [CrossRef]
- Kurreck, A.; Gronau, F.; Alberto Vilchez, M.E.; Abels, W.; Enghard, P.; Brandl, A.; Francis, R.; Fohre, B.; Lojewski, C.; Pratschke, J.; et al. Sodium Thiosulfate Reduces Acute Kidney Injury in Patients Undergoing Cytoreductive Surgery Plus Hyperthermic Intraperitoneal Chemotherapy with Cisplatin: A Single-Center Observational Study. Ann. Surg. Oncol. 2022, 29, 152–162. [Google Scholar] [CrossRef]
- Bijarnia, R.K.; Bachtler, M.; Chandak, P.G.; van Goor, H.; Pasch, A. Sodium thiosulfate ameliorates oxidative stress and preserves renal function in hyperoxaluric rats. PLoS ONE 2015, 10, e0124881. [Google Scholar] [CrossRef]
- Rein, J. Vitamin E use in preventing coronary heart disease in patients undergoing dialysis. Mayo Clin. Proc. 2002, 77, 295. [Google Scholar] [CrossRef] [PubMed]
- Karahan, S.; Afsar, B.; Kanbay, M. Ascorbic acid: A promising agent in chronic kidney disease? Clin. Kidney J. 2018, 11, 530–531. [Google Scholar] [CrossRef] [PubMed]
- Bakhshayeshkaram, M.; Lankarani, K.B.; Mirhosseini, N.; Tabrizi, R.; Akbari, M.; Dabbaghmanesh, M.H.; Asemi, Z. The Effects of Coenzyme Q10 Supplementation on Metabolic Profiles of Patients with Chronic Kidney Disease: A Systematic Review and Meta-analysis of Randomized Controlled Trials. Curr. Pharm. Des. 2018, 24, 3710–3723. [Google Scholar] [CrossRef]
- Dare, A.J.; Bolton, E.A.; Pettigrew, G.J.; Bradley, J.A.; Saeb-Parsy, K.; Murphy, M.P. Protection against renal ischemia-reperfusion injury in vivo by the mitochondria targeted antioxidant MitoQ. Redox Biol. 2015, 5, 163–168. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Z.; Liang, W.; Chen, Z.; Hu, J.; Feng, J.; Cao, Y.; Ma, Y.; Ding, G. Mitoquinone Protects Podocytes from Angiotensin II-Induced Mitochondrial Dysfunction and Injury via the Keap1-Nrf2 Signaling Pathway. Oxidative Med. Cell. Longev. 2021, 2021, 1394486. [Google Scholar] [CrossRef] [PubMed]
- Ashkar, F.; Bhullar, K.S.; Wu, J. The Effect of Polyphenols on Kidney Disease: Targeting Mitochondria. Nutrients 2022, 14, 3115. [Google Scholar] [CrossRef]
- Rassaf, T.; Rammos, C.; Hendgen-Cotta, U.B.; Heiss, C.; Kleophas, W.; Dellanna, F.; Floege, J.; Hetzel, G.R.; Kelm, M. Vasculoprotective Effects of Dietary Cocoa Flavanols in Patients on Hemodialysis: A Double-Blind, Randomized, Placebo-Controlled Trial. Clin. J. Am. Soc. Nephrol. 2016, 11, 108–118. [Google Scholar] [CrossRef] [Green Version]
- Ullah, M.M.; Basile, D.P. Role of Renal Hypoxia in the Progression From Acute Kidney Injury to Chronic Kidney Disease. Semin. Nephrol. 2019, 39, 567–580. [Google Scholar] [CrossRef]
- Tanaka, S.; Tanaka, T.; Nangaku, M. Hypoxia and hypoxia-inducible factors in chronic kidney disease. Ren. Replace. Ther. 2016, 2, 25. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, T.; Kojima, I.; Ohse, T.; Ingelfinger, J.R.; Adler, S.; Fujita, T.; Nangaku, M. Cobalt promotes angiogenesis via hypoxia-inducible factor and protects tubulointerstitium in the remnant kidney model. Lab. Investig. 2005, 85, 1292–1307. [Google Scholar] [CrossRef] [Green Version]
- Deng, A.; Arndt, M.A.; Satriano, J.; Singh, P.; Rieg, T.; Thomson, S.; Tang, T.; Blantz, R.C. Renal protection in chronic kidney disease: Hypoxia-inducible factor activation vs. angiotensin II blockade. Am. J. Physiol. Ren. Physiol. 2010, 299, F1365–F1373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dallatu, M.K.; Nwokocha, E.; Agu, N.; Myung, C.; Newaz, M.A.; Garcia, G.; Truong, L.D.; Oyekan, A.O. The Role of Hypox-ia-Inducible Factor/Prolyl Hydroxylation Pathway in Deoxycorticosterone Acetate/Salt Hypertension in the Rat. J. Hypertens. 2014, 3, 184. [Google Scholar] [CrossRef]
- Ikeda, Y.; Ozono, I.; Tajima, S.; Imao, M.; Horinouchi, Y.; Izawa-Ishizawa, Y.; Kihira, Y.; Miyamoto, L.; Ishizawa, K.; Tsuchi-ya, K.; et al. Iron chelation by deferoxamine prevents renal interstitial fibrosis in mice with unilateral ureteral obstruction. PLoS ONE 2014, 9, e89355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nayak, B.K.; Shanmugasundaram, K.; Friedrichs, W.E.; Cavaglierii, R.C.; Patel, M.; Barnes, J.; Block, K. HIF-1 Mediates Renal Fibrosis in OVE26 Type 1 Diabetic Mice. Diabetes 2016, 65, 1387–1397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Liang, D.; Fan, J.; Lian, X.; Zhao, Y.; Wang, X.; Chi, Z.H.; Zhang, P. Zinc Attenuates Tubulointerstitial Fibrosis in Diabetic Nephropathy Via Inhibition of HIF Through PI-3K Signaling. Biol. Trace Elem. Res. 2016, 173, 372–383. [Google Scholar] [CrossRef]
Figure 1.
Renal hypoxia in the pathophysiology of CKD progression. Environmental, behavioral and physiologic risk factors reduce systemic oxygenation. It triggers a range of pathological cellular processes including epithelial cell apoptosis/dedifferentiation, inflammatory cell activation/recruitment, fibroblast activation, ECM deposition and vascular rarefaction/vasoconstriction. These processes cause tubulointerstitial fibrosis, further aggravating hypoxia by limiting oxygen diffusion. This vicious cycle results in CKD progression.
Figure 1.
Renal hypoxia in the pathophysiology of CKD progression. Environmental, behavioral and physiologic risk factors reduce systemic oxygenation. It triggers a range of pathological cellular processes including epithelial cell apoptosis/dedifferentiation, inflammatory cell activation/recruitment, fibroblast activation, ECM deposition and vascular rarefaction/vasoconstriction. These processes cause tubulointerstitial fibrosis, further aggravating hypoxia by limiting oxygen diffusion. This vicious cycle results in CKD progression.
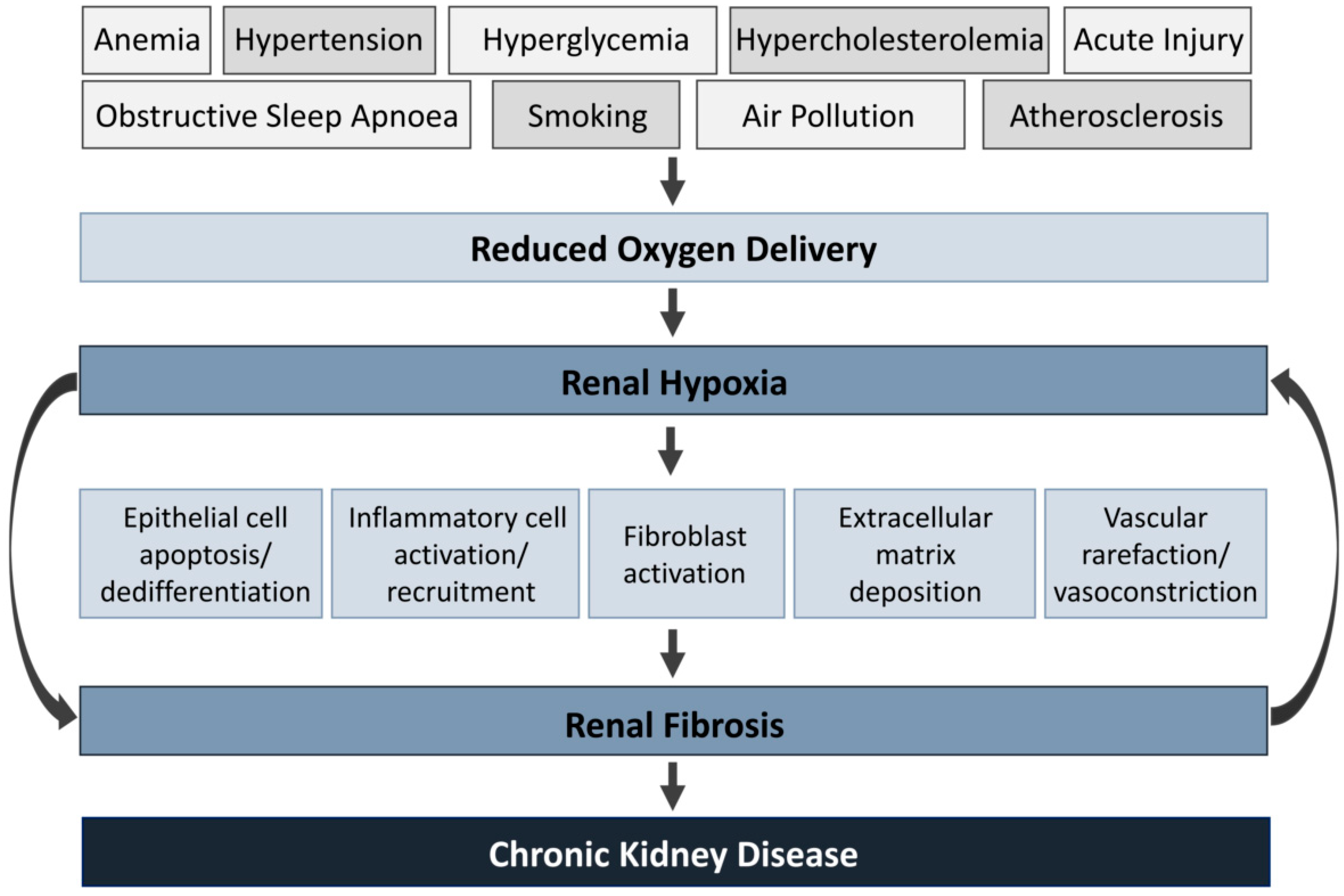
Figure 2.
Effects and mechanisms of hypoxia on the progression of CKD. Hypoxia activates the Hypoxia-inducible factor (HIF) system, which triggers a transcriptional adaptive response regulating inflammation, metabolism, oxidative stress and fibrosis. Black arrows and red blunt arrows represent positive and negative regulation, respectively. Green and red arrows represent enhanced and decreased cellular processes, respectively. ARE: Antioxidant response element, CBP/p300: C-AMP-Response Element-binding protein (CREB-binding protein)/binding protein p300, DC: Dendritic cell, ECM: Extracellular matrix, FAO: Fatty acid oxidation, HIFα: Hypoxia Inducible Factor Subunit Alpha, HIFβ: Hypoxia Inducible Factor Subunit Beta, HP: Hydroxyproline, HRE: Hypoxia response element, iNOS: Inducible nitric oxide synthase, M1: M1 macrophage type 1, M2: Macrophage type 2, mtROS: Mitochondrial reactive oxygen species, NAD+: Nicotinamide adenine dinucleotide, NF-κb: Nuclear factor kappa B, NRF2: Nuclear factor erythroid 2-related factor 2, PDH: Pyruvate dehydrogenase complex, VHL: Von Hippel–Lindau Tumor Suppressor, Ub: Ubiquitin.
Figure 2.
Effects and mechanisms of hypoxia on the progression of CKD. Hypoxia activates the Hypoxia-inducible factor (HIF) system, which triggers a transcriptional adaptive response regulating inflammation, metabolism, oxidative stress and fibrosis. Black arrows and red blunt arrows represent positive and negative regulation, respectively. Green and red arrows represent enhanced and decreased cellular processes, respectively. ARE: Antioxidant response element, CBP/p300: C-AMP-Response Element-binding protein (CREB-binding protein)/binding protein p300, DC: Dendritic cell, ECM: Extracellular matrix, FAO: Fatty acid oxidation, HIFα: Hypoxia Inducible Factor Subunit Alpha, HIFβ: Hypoxia Inducible Factor Subunit Beta, HP: Hydroxyproline, HRE: Hypoxia response element, iNOS: Inducible nitric oxide synthase, M1: M1 macrophage type 1, M2: Macrophage type 2, mtROS: Mitochondrial reactive oxygen species, NAD+: Nicotinamide adenine dinucleotide, NF-κb: Nuclear factor kappa B, NRF2: Nuclear factor erythroid 2-related factor 2, PDH: Pyruvate dehydrogenase complex, VHL: Von Hippel–Lindau Tumor Suppressor, Ub: Ubiquitin.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Miguel, V.; Rojo, A. Hypoxia-Driven Responses in Chronic Kidney Disease. Oxygen 2023, 3, 300-321. https://doi.org/10.3390/oxygen3030020
AMA Style
Miguel V, Rojo A. Hypoxia-Driven Responses in Chronic Kidney Disease. Oxygen. 2023; 3(3):300-321. https://doi.org/10.3390/oxygen3030020
Chicago/Turabian StyleMiguel, Verónica, and Alba Rojo. 2023. "Hypoxia-Driven Responses in Chronic Kidney Disease" Oxygen 3, no. 3: 300-321. https://doi.org/10.3390/oxygen3030020