

RONS and Oxidative Stress: An Overview of Basic Concepts
by
 , , , and
, , , and
Ana Karina Aranda-Rivera
1,2,†,
Alfredo Cruz-Gregorio
1,†,
Yalith Lyzet Arancibia-Hernández
1 ,
,
Estefani Yaquelin Hernández-Cruz
1,2 and
and
José Pedraza-Chaverri
1,*
1
Laboratory F-315, Department of Biology, Faculty of Chemistry, National Autonomous University of Mexico, Mexico City 04510, Mexico
2
Postgraduate in Biological Sciences, National Autonomous University of Mexico, Mexico City 04510, Mexico
*
Author to whom correspondence should be addressed.
†
These authors contributed equally to this work.
Oxygen 2022, 2(4), 437-478; https://doi.org/10.3390/oxygen2040030
Submission received: 4 September 2022
/
Revised: 1 October 2022
/
Accepted: 5 October 2022
/
Published: 10 October 2022
(This article belongs to the Special Issue Review Papers in Oxygen)
{kind=link}
{kind=link}
{kind=link}
Abstract
:Oxidative stress (OS) has greatly interested the research community in understanding damaging processes occurring in cells. OS is triggered by an imbalance between reactive oxygen species (ROS) production and their elimination by the antioxidant system; however, ROS function as second messengers under physiological conditions. ROS are produced from endogenous and exogenous sources. Endogenous sources involve mitochondria, nicotinamide adenine dinucleotide phosphate hydrogen (NADPH), oxidases (NOXs), endoplasmic reticulum (ER), xanthine oxidases (XO), endothelial nitric oxide synthase (eNOs), and others. In contrast, exogenous ROS might be generated through ultraviolet (UV) light, ionizing radiation (IR), contaminants, and heavy metals, among others. It can damage DNA, lipids, and proteins if OS is not controlled. To avoid oxidative damage, antioxidant systems are activated. In the present review, we focus on the basic concepts of OS, highlighting the production of reactive oxygen and nitrogen species (RONS) derived from internal and external sources and the last elimination. Moreover, we include the cellular antioxidant system regulation and their ability to decrease OS. External antioxidants are also proposed as alternatives to ameliorate OS. Finally, we review diseases involving OS and their mechanisms.
1. Introduction: Reactive Oxygen Species (ROS) and Reactive Nitrogen Species (RNS)
Reactive oxygen species (ROS) and reactive nitrogen species (RNS) are defined as unstable species containing oxygen (O2) and nitrogen that react quickly with other molecules in the cells [1]. This review considers ROS and RNS as reactive oxygen and nitrogen species (RONS) for practical purposes. The RONS containing one or more unpaired electrons are known as free radicals, while RONS without unpaired electrons are called non-free radicals. Free radical species include superoxide anion radical (O2•−), hydroxyl radical (•OH), alkoxyl (●OR), nitric oxide (NO•), and peroxyl radicals (●OOR); non-free radicals comprise hydrogen peroxide (H2O2), nitrogen dioxide (NO2), and peroxynitrite (ONOO−), among others [2]. O2•− is the principal precursor of RONS produced by the cells, and an increase in this radical is related to oxidative stress and cellular damage [3,4]. Thus, antioxidants are a powerful tool to control the overproduction of ROS and avoid oxidative damage.
2. Signaling and Physiological Functions of RONS
RONS are products of aerobic metabolism that regulate cellular processes such as survival, growth, proliferation, apoptosis, and others. Although RONS can cause damage to DNA, lipids, and proteins, they function as signaling molecules at low levels, participating in signaling pathways as second messengers [2]. RONS induce posttranslational modifications (PTM) in redox-sensitive residues such as cysteine (Cys), methionine (Met), lysine, arginine, proline, histidine, threonine, and tyrosine, Cys and Met being the most physiologically relevant [7,8,9]. Cys and Met residues contain sulfur groups on the chain sides, which makes them most prone to oxidation by RONS. The oxidation state depends on the microenvironment and pH because they modify the side chain of the protein pKa. For instance, thiol groups (SH) of Cys exposed to cytosol are deprotonated to thiolated (S-) to physiological pH, making them more sensitive to oxidation by RONS, which depend on pKa [10]. The principal RONS participating in cellular signaling are O2•− and H2O2, H2O2 being a more stable reactive species than O2•− [11]. Because H2O2 is highly diffusible, it is more suitable than O2•− for biological signaling [12].
H2O2 is generated by different sources, including growth factors, chemical stressors, or chemokines. It is maintained under low concentrations to a nanomolecular range of 1–100 nm, being the central molecule implicated in cell signaling by regulating redox reactions; however, the final concentration of H2O2 depends on removal systems [13]. H2O2 easily crosses membranes by aquaporins and holds redox signaling through Cys residues. H2O2 oxidizes Cys residues in proteins producing sulfenic acid (R-SOH). If oxidation continues, R-SH is converted to sulfinic acid (R-SO2H), and later, if oxidation continues, to sulfonic acid (R-SO3H) [14]. The oxidation by H2O2-producing R-SO3H is irreversible, deactivating the protein suffering this modification. In cellular signaling, the inactivation of proteins is relevant for kinases and phosphatases because their inactivation avoids phosphorylation or the elimination of phosphate groups.
O2•− commonly does not cross membranes, except through the voltage-dependent anion channel (VDAC), and is maintained to 10−11 M [15]. O2•− is involved in activating the mitogen-activated protein kinase (MAPK) pathway through the Ras/Rac/Raf-1-MAPK pathway, which implies the generation of O2•− by endothelial cells via angiotensin II (Ang II). Moreover, Ang II induces the generation of O2•− through the nervous system, which increases vasopressin secretion and sympathetic outflow [16]. Another example of O2•− cellular signaling function involves the activation of the extracellular signal-regulated kinase (ERK) pathway, which is also a member of the MAPK family. Mechanistically, O2•− is generated through quinone 2,3-dimethoxy-1,4-naphthoquinone (DMNQ) and activates to peroxisome proliferator-activated receptor-γ (PPAR-γ), which stimulates ERK signaling [17].
Other cellular effects of O2•− are found in cancer cells. Cancer cells commonly support high oxidative stress levels, and RONS are utilized by these cells to turn on or turn off signaling pathways to grow, proliferate, and evade apoptosis. For instance, Yang et al. [18] found that the overexpression of v-Ha-ras augments O2•− production, inducing human keratinocyte (HaCaT) growth, suggesting that ras induces transformation in cancer cells. Therefore, O2•−-induced cellular signaling is required by living cells for the activation and deactivation of signaling pathways and their regulation.
3. Oxidative Stress
Oxidative stress involves an imbalance between the production of RONS and their elimination by the antioxidant system (described later). Redox balance also requires the regulation of master-switch signaling pathways such as nuclear factor kappa-light-chain-enhancer of activated B cells (NF-kB)/NF-κB inhibitor (IκB), nuclear factor erythroid-2 related factor 2/Kelch-like ECH-associated protein 1 (Nrf2/Keap-1), and Akt/Forkhead Box O3 (FOXO3), among others [19]. For instance, Nrf2 moves to the nucleus when Keap-1 suffers conformational changes due to the oxidation of residues Cys 226, Cys 613, and Cys 624 by RONS [20,21]. Nrf2 in the nucleus induces the expression of antioxidant enzymes, such as glutamate-cysteine ligase modifier (GCLM) subunits, heme oxygenase (HO-1), NADPH quinone oxidoreductase 1 (NQO1), glutathione S-transferase (GST), and glutathione peroxidase (GPx), among others [20,22,23].
The phosphatidylinositol 3-kinase (PI3K)/AKT signaling pathway regulates oxidative stress by inducing the activation of the transcription factor FOXO3, promoting the transcription of catalase (CAT) and SOD2 enzymes [24]. FOXO3 also regulates oxidative stress in mitochondria by interacting with peroxisome proliferator-activated receptor coactivator-1α (PGC-1α), the master biogenesis regulator, to augment the transcription of antioxidant enzymes [25]. PI3K can also regulate the activity of NF-κB/IκB via ROS. Mechanistically, the activation of PI3K activates the small GTPase RAC1, which stimulates NOXs, inducing ROS production. ROS could deactivate and send proteasome degradation to the NF-κB inhibitor, IκB [26]. The latter is because IκB contains Cys residues, which can be oxidized, releasing NF-κB to induce the expression of antioxidant response [27].
Therefore, oxidative stress is highly controlled in cells. However, disturbances in cells, such as the reduction in antioxidant enzymes or the elevation of RONS, can lead to a state of oxidative damage.
3.1. Sources of Oxidative Stress
RONS can be provided from extracellular or intracellular sources. Extracellular sources include ultraviolet (UV) light, ionizing radiation, heavy or transition metals, and others (revised later). In contrast, intracellular sources comprise mitochondria, peroxisomes, ER, and nicotinamide adenine dinucleotide phosphate hydrogen (NADPH) oxidases (NOXs) [28], which will be reviewed below.
3.1.1. Endogenous Sources
In human cells, more than 50 enzymes produce O2•− and H2O2, with mitochondria and NADPH oxidases being the principal sources for making them [29]. In the following sections, we reviewed the involvement of ROS sources and their contribution to redox signaling and oxidative stress.
Mitochondria
Mitochondria produce 1–3% ROS during the generation of ATP using the mitochondrial membrane potential through ATP synthase [30]; however, this percentage is commonly maintained at low levels because of the presence of an antioxidant system in mitochondria. Several processes in this cellular compartment are redox-dependent. Some examples include the reactions of enzyme members of the tricarboxylic acid (TCA) cycle such as pyruvate dehydrogenase (PDH), isocitrate dehydrogenase 2 (IDH2), and α-ketoglutarate dehydrogenase [31,32,33]; the reactions occur in complex I (CI) and III (CIII) because of contained Fe-S clusters and Cys groups. Fe-S is also contained in the mitochondrial enzyme aconitase 2 (Aco2), whose activity is inhibited under oxidative stress, releasing Fe2+ and subsequently deactivating Aco2 [6]. Another TCA cycle enzyme affected by ROS is citrate synthase, supported by the fact that antioxidants prevent its deactivation [34]. Furthermore, CI and CIII generate O2•− by having semiquinone (Q) and flavin (F) groups [35,36]. NOX4 is also present in the mitochondria and principally causes H2O2 in a mechanism explained in the following sections (Figure 1).
Indeed, the presence of NOX4 in mitochondria has been of great relevance, for example, in kidney diseases and cancer contexts [37,38]. In kidney disease, NOX4 interacts with mitochondria to augment ROS overproduction, which is related to the overactivation of redox signaling [37,39], whereas, in cancer cells, NOX4 functions as an energy sensor, associated with chemoresistance, and its inhibition sensitizes cancer cells to cell death [38]. Thus, in both cancer and renal diseases, the overactivation of NOX4 is deleterious. Another ROS source in the mitochondria is the electron transport system (ETS), which consists of CI-CIV, where the flow of electrons couples with proton gradient generation and crosses to the inner mitochondria membrane (IMM) to produce ATP through ATP synthase. Furthermore, ETS has two carriers, quinone and cytochrome c (cyt c), localized between CI and CII and CIII and CIV, respectively [40].
The electron donors fade ETS CI and CII NADH and FADH2, which provide the TCA cycle or β-oxidation, beginning the electron transport. Then, electrons are transferred to ubiquinone (CoQ), and four electrons are pumped from the mitochondria matrix to the intermembrane space (IMS). CII does not pump electrons and is part of the TCA cycle enzyme, the succinate dehydrogenase (SDH), but this complex transfers two electrons to CoQ. Both CI and CII reduce COQ to ubiquinol (QH2). CIII transfers two electrons from QH2 to cyt c and pumps four protons to IMS. Finally, CIV captures four electrons from cyt c, transferred to oxygen to produce H2O. Four protons are translocated to IMS. The proton flow induces a pH and electrochemical gradient, which is used by ATP synthase to produce ATP through oxidative phosphorylation, known as OXPHOS.
Because electron leak is common during OXPHOS, several proteins of ETS suffer redox modifications that function as protective mechanisms to avoid the deactivation of mitochondrial proteins. As mentioned before, CI and CII are the primary producers of ROS, although CII also generate them to a lesser extent. This way, CI and CII release O2•− and H2O2 to the mitochondrial matrix, while CIII releases them towards the cristae lumen and intermembrane space [41]. O2•− does not readily pass through cell membranes and is relatively short-lived. An increase in proton motive force slows electron flow in the ETS, augmenting ROS generation because electrons can interact with molecules such as molecular oxygen, which generates ROS. In the mitochondria, O2•− is produced in the mitochondrial electron transport chain (ETC).
NADPH Oxidases (NOXs)
NOXs comprise seven members, including NOX1, NOX2, NOX3, NOX4, NOX5, DUOX1, and DUOX2, present in cellular membranes and essentially produce O2•−. The role of NOXs was discovered in neutrophils by a respiratory burst realized by NOX2, which produces large ROS quantities for bacterial elimination. Other NOX functions include gene expression regulation, proliferation, hormone synthesis, and cell differentiation [3,42]. NOXs comprise almost six subunits: the membranes-bound subunits gp91phox and p22phox and the cytoplasmic regulatory subunits p40phox, p47phox, and p67phox, and the GTPase Rac1 [43,44]. The subcellular localization of NOXs depends on cellular types.
NOXs produce O2•−, attributed to the catalytic subunit gp91phox (or NOX2) activated when cytoplasmatic subunits are assembled. Mechanistically, the binding of growth factor to growth factor receptor (GFR) activates PI3K, which activates the catalytic activity of GTPase Rac1. The latter induces the assembly of cytosolic subunits to stimulate gp91phox. Then, electrons of NADPH are transferred to O2 to produce O2•−, which is converted to H2O2. Although low quantities of H2O2 can freely diffuse through the membrane, it is easily transported by aquaporins, which activate redox-signaling pathways (Figure 1).
NOX4 is localized in mitochondria and does not contain cytosolic subunits, producing H2O2 as the principal ROS. NOX4-induced H2O2 has been reported to cause the opening of the mitochondrial adenosine triphosphate (ATP)-sensitive potassium K channel (mt-KATP), which decreases mitochondrial membrane potential (Figure 1) [45]. The latter induces mtROS overproduction, inducing mitochondrial dysfunction. The interplay between mitochondria and NOX4 has been associated with cardiovascular and kidney diseases [39].
Peroxisomes
Peroxisomes induce the catabolism of fatty acids (FAs), producing H2O2 as the principal subproduct, generated through oxidases such as fatty acyl-CoA and D-amino acid oxidases. Because peroxisomes contain high levels of CAT and peroxiredoxin 5, H2O2 is rapidly degraded to H2O (Figure 1). One of the functional consequences of peroxisomal H2O2 is the ability to deactivate phosphatidylinositol-3,4,5-trisphosphate 3-phosphatase (PTEN) and/or activate FOXO3 [46]. Moreover, it has been reported that peroxisome-induced ROS might regulate autophagy, a process involving the degradation and recycling of cellular components. This regulation was attributed to the presence of tuberous sclerosis complex (TSC), regulating mTORC1 response to ROS into peroxisomes, which deactivates mTORC1 to begin the first steps of the autophagy mechanism [47].
Endoplasmic Reticulum (ER)
Another critical source of ROS is ER, which generates principally H2O2 during the folding of proteins. The formation of H2O2 is attributed to disulfide isomerases, which generate one oxidizing equivalent for every disulfide formed. Thereby, H2O2 is the resulting subproduct of protein folding.
ER and mitochondria maintain contact through mitochondrial membranes, known as MAMs, functioning as a regulator of lipid transport, Ca2+ signaling, and cell death; thus, any disturbance in ER affects mitochondria [48,49]. This is the case for ER stress triggered by hypoxia, oxidative stress, saturated FA deposition, and others, causing the accumulation of unfolded proteins in the ER lumen. The latter induces the release of glucose-related protein 78 kDa (GRP78) and the posterior activation of unfolded protein response (UPR). UPR comprises three signaling pathways: the protein kinase activated by double-stranded RNA(PKR)-like ER kinase (PERK), the activating transcription factor 6 (ATF6), and the inositol-requiring transmembrane kinase/endonuclease alpha (IRE1α) [50]. The principal objective of UPR is to reestablish homeostasis and restore the functions of ER, including protein folding, synthesis, and transport (Figure 1) [51].
PERK is a transmembrane protein dimerized and autophosphorylated to activate the regulatory subunit of the translation initiation component (eIF2 complex), eukaryotic initiation factor 2 alpha (eIF-2α), which decreases protein synthesis. The phosphorylated eIF-2α (peIF-2α) augments the transcription of ATF4 to trigger the transcription of chaperones, antioxidant response, amino acids, and autophagy. However, the sustained activation of this pathway can lead to apoptosis mediated by C/EBP homologous protein (CHOP). Another pathway involves the Ser/Thr IRE1α, which is activated and autophosphorylated to induce the splicing of the noncoding sequence of the X box-binding protein 1 (XBP1) mRNA. This splicing leads to the formation of XBP1s (the active form) that induces the transcription of UPR-targeted genes [52]. Under chronic ER stress, IRE1α forms a complex with the tumor necrosis factor receptor-associated factor 2 (TRAF2) to promote the activation of cell death. Finally, ATF6 dissociates from GRP78 and translocates to the Golgi apparatus to induce ATF6 processing in its N-terminal domain, allowing its translocation to the nucleus.
Xanthine Oxidases (XOs)
XOs are enzymes localized in the cytosol involved in the catabolism of purine nucleotides. XO functions by oxidizing hypoxanthine and xanthine bases to uric acid, the final product of purine catabolism, reactions where H2O2 is produced. Thus, H2O2 is the principal product of this enzyme [53]. The importance of XO is highlighted for its contribution to inflammasome activation in macrophages [54]. The XO-induced inflammasome activation by releasing H2O2 promotes the activation of PI3K/AKT, the signaling pathway that induces NLRP3 activation through the generation of mitochondrial ROS (mtROS) [54]. In organs such as the heart and kidney, hypoxanthine and xanthine substrates are also produced by the ATP breaking during ischemic episodes, inducing H2O2 and O2•− production following reperfusion [2]. Deleterious consequences have been suggested regarding XO overactivation in cardiovascular diseases because H2O2 and O2•− are generated, which decrease nitric oxide production [55]. Furthermore, XO-released H2O2 and O2•− can cause the oxidation of lipids or proteins, which alters homeostasis by affecting signaling pathways or disrupting cellular membrane integrity [56]. Therefore, XO is required for the immune response, but its overactivation causes damage.
Myeloperoxidase (MPO)
MPO is a peroxidase enzyme containing a heme group. This is mainly expressed in neutrophils and monocytes and has microbicide activity against pathogens by producing hypochlorous acid (HOCl) and its intermediates [57]. In addition to the protective role of MPO against pathogens, this has an essential role as a local mediator in tissue damage and inflammation in inflammatory diseases, released into phagolysosomes and extracellular compartments. Thus, MPO is required for immune response.
MPO activation is also strongly related to atherosclerosis and cardiovascular diseases since mice overexpressing MPO show accelerating atherosclerotic plaque development [58]. Moreover, MPO activation is present in the initiation and progression of cardiac diseases [59] and inflammatory events in kidney and pulmonary diseases [60,61].
Endothelial Nitric Oxide Synthase (eNOS)
NOS hydroxylates L-arginine to Nω-hydroxy-l-arginine, which later is oxidized to L-citrulline and NO• [62]. NO• is an essential molecule of cellular signaling that functions protectively in vascular endothelium, as eNOS is the most critical enzyme for NO• production [63]. In addition, NO• is synthesized by the isoform neuronal NOS (nNOS), expressed in neurons, and inducible NOS (iNOS), associated with inflammation.
eNOS is constitutively expressed, requiring the presence of increased intracellular calcium (Ca2+). The principal mechanism for eNOS activation consists of increased Ca2+-calmodulin binding, but posttranslational modifications such as phosphorylation, lipid attachment, or S-nitrosylation regulate eNOS activity. eNOS requires dimerization, the substrate L-arginine and the cofactor (6R)-5,6,7,8-tetrahydro-l-biopterin (BH4). NOS catalyzes the electron transfer mediated by flavin from C-terminal domain-bound NADPH to N-terminal heme. Calmodulin augments the electron transfer from NADPH to the heme center, where electrons reduce O2.
3.1.2. Exogenous and Environmental Sources
The primary source of reactive oxygen species (ROS) in the body is physiological metabolism; however, ROS can also be generated through exposure to external factors such as ionizing (IR) and ultraviolet (UV) radiation, biological organisms, pollutants, food, medicines, and drugs such as alcohol and tobacco (Figure 2).
UV Light
UV light is part of the solar emission spectrum that lies between the electromagnetic radiation spectrum of X-rays and visible light. It has wavelengths ranging from 100 nm to 400 nm [64]. UV light can be subdivided according to its wavelengths into several categories: UVA (320–400 nm), UVB (290–320 nm), and UVC (220–290 nm) [64]. UVA and UVB are essential for producing vitamin D; however, overexposure to these causes skin damage, sunburn, and even the initiation of carcinogenic processes [65,66]. In contrast, UVC rays are completely absorbed by atmospheric ozone and have little effect on human health. However, accidental human exposure to these rays, mainly from lamps that kill microbes, causes corneal burns and snow blindness [64,67].
Humans are exposed to UV radiation through sun exposure and artificial sources [68]. UV radiation generates ROS, including O2•−, singlet oxygen (1O2), •OH, and H2O2, through various mechanisms that directly affect cellular components or photosensitization [69]. These mechanisms include structural damage to the enzyme CAT [70] and increased nitric NOS synthesis [71]. UV radiation also decreases the expression of protein kinase C (PKC), causing an increase in the production of ROS [72]. In addition, UV rays have been shown to damage DNA due to the reported rise in 8-OHdG, a marker of oxidative DNA damage. This marker is usually associated with •OH; however, it has been proposed that the 8-OHdG generated by UV radiation is formed through a mechanism involving 1O2 [73]. UV radiation also damages mitochondrial DNA (mtDNA), deteriorating the ETC and, therefore, inducing ROS production. This can further damage mtDNA, creating a vicious cycle [74]. Likewise, UV radiation causes the activation of the enzyme NOX, the elimination of a proton and an electron from lipid molecules, which produces lipid radicals. It promotes the release of mediators of inflammation, generating oxidative stress [64,75].
It should be noted that the epidermis is the part most damaged by UV rays. Oxidative damage has been observed in cutaneous chromophores such as DNA, urocanic acid, aromatic amino acids, retinoids, carotenoids, bilirubin (BR), flavins, hemoglobin, melanin, and NAD(P)H. Chromophores can be directly damaged or act as photosensitizers, resulting in increased ROS [76,77].
Finally, increased UV radiation-induced ROS activates different signaling pathways, especially in the pathophysiology of skin diseases. The principal signaling pathways activated for this mechanism are MAPKs, including ERK and c-Jun-N-terminal kinase (JNK), leading to the subsequent activation of the activator protein 1 (AP-1) [78]. UV radiation also causes the release of labile iron (Fe), which is involved in the activation of NF-kB [79,80]. AP-1 and NF-kB are essential in regulating various genes involved in processes such as the cell cycle, proliferation, and apoptosis [79].
Ionizing Radiation (IR)
IR is produced naturally in the environment by natural radioisotopes found in the ground and cosmic rays reaching the Earth’s surface [81,82]. IR is a form of energy transfer that can cause the ionization of matter [83]. This energy is transmitted by electromagnetic waves, including X-radiation, gamma radiation, and UV radiation [84]. Currently, IR has different uses both in medicine and outside of it. These uses include radiation therapy, cancer treatment, smoke detectors, the sterilization of medical equipment, and food irradiation to kill bacteria and pasteurize food [85]. However, IR in large quantities induces a broad spectrum of alterations, such as oxidative stress.
IR penetrates living organisms’ cells, which induces the ionization of organic and inorganic compounds [86]. IR causes the radiolysis of water molecules, which is the primary process for forming ROS [87,88]. ROS also occur because mitochondria are susceptible to IR, which causes mitochondrial dysfunction and therefore increases ROS production [89]. Einor et al. [90] performed a meta-analysis with 41 studies on various biological matrices and showed that IR generates ROS even at low doses. In addition, irradiation has been reported to increase concentrations of malondialdehyde (MDA), NO•, and calcium ions while decreasing SOD and CAT activity and glutathione (GSH) concentration in models both in vivo and in vitro [91,92,93].
On the other hand, IR also alters the nuclear and mitochondrial DNA molecule and the proteins responsible for stabilizing it, making the DNA more susceptible to damage by ROS, generating deletions, mutations, and other lethal genetic effects [94,95,96]. In addition, genes that encode antioxidant enzymes can suffer mutations. For example, in a clinical study conducted by 29 doctors from the radiology and radiotherapy departments of Chu Hedi Chaker Sfax, Tunisia, a close relationship between the low-activity GSTP1 genotypes, GSTT1 null, GSTM1 null, and stress biomarkers was found, especially with the MDA level and the activities of the SOD and CAT enzymes [97]. Likewise, in another clinical study conducted on American radiological technologists who became ill with breast cancer, it was shown that the mutation in the PTGS2 gene, which encodes the cyclooxygenase-2 enzyme (COX-2), can modify the risk of breast cancer due to occupational exposure to radiation [98]. Furthermore, a meta-analysis demonstrated increased mutations in more than 30 species exposed to radiation in the Chernobyl disaster 25 years later [99].
In addition to mutations, exposure to IR generates epigenetic modifications [100]. In workers exposed to radon in the uranium mines, significant hypermethylation of the DNA of tumor suppressor genes was detected [101]. Aberrating hypermethylation was also observed in a fraction of patients with renal cell carcinomas that lived in radio-contaminated areas after the Chernobyl accident [102]. Finally, Szumiel [103] proposes an interdependence between the oxidant stress induced by IR, the alterations in the mitochondrial DNA, and the epigenetic changes in the nuclear DNA, where the production of ROS induced by IR causes mutations in the mitochondrial DNA, which in turn contributes to the production of ROS, generating a vicious circle. Mitochondrial DNA mutations affect the epigenetic control mechanisms of the nuclear DNA by decreasing the activity of methyltransferase and, therefore, causing the global hypomethylation of DNA.
Contaminants
Accelerated urban development and rapid population growth contribute significantly to poor environmental quality. This causes living organisms to be surrounded by heavy metals, pesticides such as deltamethrin and paraquat, or air pollutants such as particulate matter (PM) [104]. The intracellular mechanisms associated with the toxic effects of most of these compounds are related to the generation of RONS [105,106].
Heavy or Transition Metals: Heavy metals are one of the primary contaminants associated with cell damage and refer to elements with a high atomic weight and densities five times higher than water [107]. Some heavy metals such as copper (Cu), Fe, cobalt (Co), selenium (Se), chromium (Cr), nickel (Ni), arsenic (As), and zinc (Zn) are present in living beings and participate in a variety of biological processes [107]. However, the increase in the concentration of these metals, in the form of free ions, generates cell damage. In addition, there are highly toxic metals, many of which do not have a biological function, causing damage even at low concentrations. These metals include lead (Pb), aluminum (Al), mercury (Hg), and cadmium (Cd) [108,109].
Heavy metals, according to their redox function, are divided into two groups: redox-active (Fe, Cu, Cr, Co) and redox inactive (Cd, Zn, Ni, Al, etc.). Redox-active metals are directly involved in cells’ redox reactions and participate in the Haber–Weiss and Fenton reactions together with H2O2, and O2•− to produce •OH [110]. Redox-inactive metals also produce ROS through indirect mechanisms such as ETC disruption, interaction with the antioxidant defense system, or the induction of lipid peroxidation due to increased lipoxygenase (LOX) [111,112]. Furthermore, heavy metals such as Cd inactivate antioxidant enzymes by binding to their Cys residues, leading to misfolding and the inhibition of activity and/or interference with enzymatic redox regulation [113]. Likewise, it is known that there are enzymes that use metal ions as cofactors; therefore, the displacement of one metal ion by another causes the inhibition or loss of enzymatic activity [114]. Heavy metal-mediated free radical formation causes a variety of DNA base modifications, increased lipid peroxidation, and changes in calcium homeostasis and sulfhydryl groups [107].
Pesticides: Pesticides are chemicals used as bactericides, fungicides, herbicides, insecticides, or rodenticides [115]. They have been grouped into different families: organochlorines, organophosphates, organofluorines, carbamates, pyrethroids, bipyridyl herbicides, triazine herbicides, triazoles, and chloroacetanilide herbicides [116]. The World Health Organization reports that three million workers experience severe pesticide poisoning each year, of which approximately 18,000 die [117]. Among its damage mechanisms is the induction of oxidative stress. Pesticides increase ROS and RNS production by activating NOX, NO•, and Ca2+ levels [118]. They also inhibit hydrolases, particularly acetylcholinesterase. The inhibition of acetylcholinesterase is associated with increased ROS in agricultural workers exposed to organophosphate pesticides and bipyridyl herbicides (e.g., paraquat) [119]. Furthermore, cytochrome P450 is a potent source of pesticide-induced ROS. Pesticides are metabolized by the cytochrome P450 enzyme, generating reactive metabolites and increasing ROS production [120,121]. These enzymes also catalyze an organic substrate’s oxygenation, and molecular oxygen reduction can occur. If this process is not strictly controlled, the possibility of mitochondrial uncoupling arises and subsequently leads to the formation of ROS [122,123]. Oxidative stress is also induced by increased lipid peroxidation and a decrease in antioxidant capacity caused by pesticides [124]. The increase in RONS induces the oxidation of lipids, proteins, and DNA. Furthermore, pesticides cause the activation of the Keap1/Nrf2 antioxidant response element (ARE) [125]. For example, permethrin (PER), a compound that kills lice and mites, has been shown to increase Nrf2 gene expression in rat cerebellum and cardiac cells [126]. Nrf2 expression was also increased in rat adrenal pheochromocytoma PC12 cells through 26S proteasome inhibition [127] and in human neural progenitor cells (hNPC) treated with paraquat for 24 h [118]. This effect could be due to the fact that the key cysteine residues in Keap1 are oxidized, and therefore, its ability to act as an E3 ligase adapter is inhibited, leading to the increased nuclear translocation of Nrf2 and the consequent transcription of antioxidant genes [128,129,130]. Pesticides also cause the activation of adapter protein tumor necrosis factor receptor 1 (TNFR1)/tumor necrosis factor (TNF-α), MAPK, NF-κB, and mitochondrial apoptosis pathways [125].
Air Pollutants: Atmospheric pollutants are another of the main environmental causes of ROS production, which is why they represent an environmental risk factor in today’s world [131]. Among these pollutants are PM. PM is made up of solid and liquid particles in the air that are small enough to be inhaled. PM originates from automobile exhausts, combustion, mining, industrial sources, fine dust from the earth, road and tire abrasion, construction work, dust, and agricultural sources [132]. The composition of PM depends on its source and atmospheric chemistry but typically includes transition metal ions, organic compounds (primarily polycyclic aromatic hydrocarbons (PAHs)), and organic matter [133]. Adsorption on the surface of PM causes oxidative stress. PM has been reported to increase the production of ROS, such as H2O2, 1O2, O2•−, and •OH, by lowering the mitochondrial membrane potential and altering calcium homeostasis [134]. In addition, a reduction in the GSH/glutathione disulfide (GSSG) ratio has been observed. The alteration in redox homeostasis induced by PM alters the activation of signaling pathways such as ERK and MAPK, leading to the activation of the transcription factor NF-κβ to produce inflammatory cytokines that can lead to different adverse effects such as cardiovascular, atherosclerotic, and neuronal effects [135,136].
Foods
Currently, it is known that specific dietary patterns lead to an increase in ROS in the body. In particular, Western diets characterized by the excessive consumption of saturated fats, refined sugars, and animal proteins, as well as low consumption of plant-based fiber, increase protein carbonylation and lipid peroxidation products while reducing the state of antioxidant defense, causing oxidative stress. It has also been reported that the permanent availability of oxidizable substrates at rest (such as carbohydrates: glucose and D-galactose) leads to a decrease in oxidative phosphorylation, which generates the possibility that a more significant number of electrons leak from the ETC, causing the generation of ROS [137,138]. Something similar happens with the accumulation of triglycerides, which stimulate β-oxidation and, therefore, ETC. In addition, they produce low-density lipoproteins (LDL) that form oxidized LDL (Ox-LDL) [139,140]. ROS induced by high-fat diets trigger the activation of NF-κB-dependent proinflammatory molecules [141] and increase iNOS activity, causing excessive NO• production [142]. Therefore, high-calorie diets not only cause oxidative stress but are also associated with diseases such as diabetes, obesity, and metabolic syndrome, among others [143].
Alcohol and Smoking
Alcoholism and smoking have also been associated with oxidative stress and, therefore, with various diseases. Alcoholism is a pathology resulting from high alcohol intake [144]. Alcohol consumption, both acute and chronic, causes an increase in ROS, partially related to the acetaldehyde formed as a product during ethanol metabolism. Acetaldehyde alters calcium homeostasis and increases ROS production, which can cause ER stress [145]. In addition, it has been found that the most outstanding production of ROS by alcohol arises from complexes I and III of the mitochondria ETC. Alcohol also raises Fe levels in the body, contributing to the Fenton reaction and •OH production, and also reduces endogenous antioxidant levels, creating a more significant imbalance that promotes oxidative stress [146].
On the other hand, cigarette smoke has been considered a dangerous substance for health that causes premature aging. Cigarette smoke comprises more than 5,000 chemicals, including nicotine, polycyclic aromatic hydrocarbons (PAHs), tar, nitrosamines, carbon monoxide, NO•, and phenolic hydrocarbons. Of these compounds, 150 are toxic, and 60 have been established as carcinogenic [147]. Several studies have reported that cigarette smoke induces the excessive production of ROS. In addition, cigarette smoke contains free radical substances, including H2O2 and •OH, which are inhaled directly into the respiratory tract. Furthermore, like alcohol, smoking reduces the availability of endogenous antioxidants, generating oxidative stress, which, in turn, can cause the activation of NF-κB and the subsequent expression of proinflammatory mediators [148].
Medicines
There is a large amount of clear evidence linking the excessive production of ROS with the consumption of different drugs. These drugs include anticancer therapies, nonsteroidal anti-inflammatory drugs (NSAIDs), antiretroviral agents, antipsychotics, and pain relievers [149]. Medications can generate ROS due to the generation of reactive intermediates during their metabolism that can partially reduce molecular oxygen. For example, chlorpromazine, a neuroleptic drug belonging to the group of phenothiazines widely used in treating some psychiatric disorders, causes the generation of 1O2 and O2•− due to photodechlorination, which transfers electrons from chlorpromazine in an excited state to molecular oxygen [150]. In addition, doxorubicin, an anthracycline antibiotic used in numerous chemotherapy regimens to treat hematologic and solid tumors, has been shown to generate anthracycline semiquinone free radicals during its one-electron reduction by mitochondrial reductases [151]. The reaction of doxorubicin with Fe during its metabolism also generates a Fe II-Dox free radical capable of reducing molecular oxygen [152]. ROS production may also be due to drug-induced skin photosensitivity [153]. Photosensitizing drugs absorb the sun’s radiation, mainly UVA rays, giving rise to chemical reactions such as the excessive production of ROS. Photosensitivity can manifest as a phototoxic or, more rarely, photoallergic reaction. For example, ibuprofen causes dose-dependent phototoxic hemolysis after UVA radiation [154]. In comparison, ketoprofen is the most frequent cause of photoallergy and induces cellular lipid peroxidation and damage to DNA and cell membranes [155,156]. Likewise, amlodipine and nifedipine generate ROS by absorbing UVA radiation [157]. Other drugs associated with photosensitization include nonsteroidal anti-inflammatory drugs (NSAIDs), cardiovascular drugs, psychotropic drugs, antimicrobials, antihyperlipidemic drugs, and antineoplastic drugs [158,159]. For more information on photosensitive drugs, see the work by Kowalska et al. [160].
On the other hand, some drugs increase ROS levels through NOX activation. An example of these drugs is cisplatin, an antineoplastic agent used to treat different cancers [161]. Furthermore, cisplatin causes the depletion of critical antioxidants, mitochondrial membrane potential changes, calcium handling, and apoptosis [162]. Many drugs have also been associated with mitochondrial alterations, mainly in the ETC, which increases ROS [163]. For example, NSAIDs increased ROS production via ETC inhibition in rat cardiomyoblast cells and murine neonates [164]
Oxidative stress induced by drug use leads to different clinical consequences, such as nephrotoxicity, hepatotoxicity, and cardiotoxicity. Hepatotoxicity has been a cause of NSAID use. Diphenylamine, a structural component of most NSAIDs, has been shown to cause mitochondrial dysfunction, causing mitochondrial inflammation and liver damage [165]. In addition, in vitro studies have shown that salicylate (aspirin’s primary metabolite) causes Reye’s syndrome, characterized by liver problems, by increasing sensitivity to open mPTP [166]. Paracetamol also induces hepatotoxicity through mitochondrial dysfunction. The reactive metabolite of acetaminophen binds to mitochondrial proteins and induces oxidative stress, resulting in a mitochondrial permeability transition [167]. On the other hand, it has been reported that 20% of acute renal failure in the community and in hospitals is due to drug use [168]. Mechanisms associated with drug nephrotoxicity include impaired perfusion, the induction of inflammation, the formation of free radicals, and many other mechanisms [169]. For example, NSAIDs and rifampin are common causes of acute inflammation of the interstitial tissue, while acyclovir and antibiotics such as ampicillin have been common causes of insoluble crystal formation in kidney tissue [170]. Finally, some drugs cause heart damage; among the best known is doxorubicin. A study conducted in H9c2 cardiomyoblast cells and rats reported that DOX increases mitochondrial ROS production, which activates ERK-mediated HSF2 nuclear translocation and the upregulation of AT1R, causing heart failure both in vitro and in vivo [171].
4. Oxidative Stress-Induced Oxidative Damage
RONS are frequently generated in regulated amounts; however, dysregulation often leads to toxic oxidative stress, altering biomolecules and redox signaling [19,172]. Both endogenous or exogenous sources of oxidative stress may lead to oxidative stress, causing oxidative damage (oxidation modifications) to nucleic acids, proteins, lipids, and carbohydrates [19]. The damage to membrane lipids, structural proteins, enzymes, and nucleic acids leads to aberrant cell function [172].
4.1. DNA Oxidative Damage
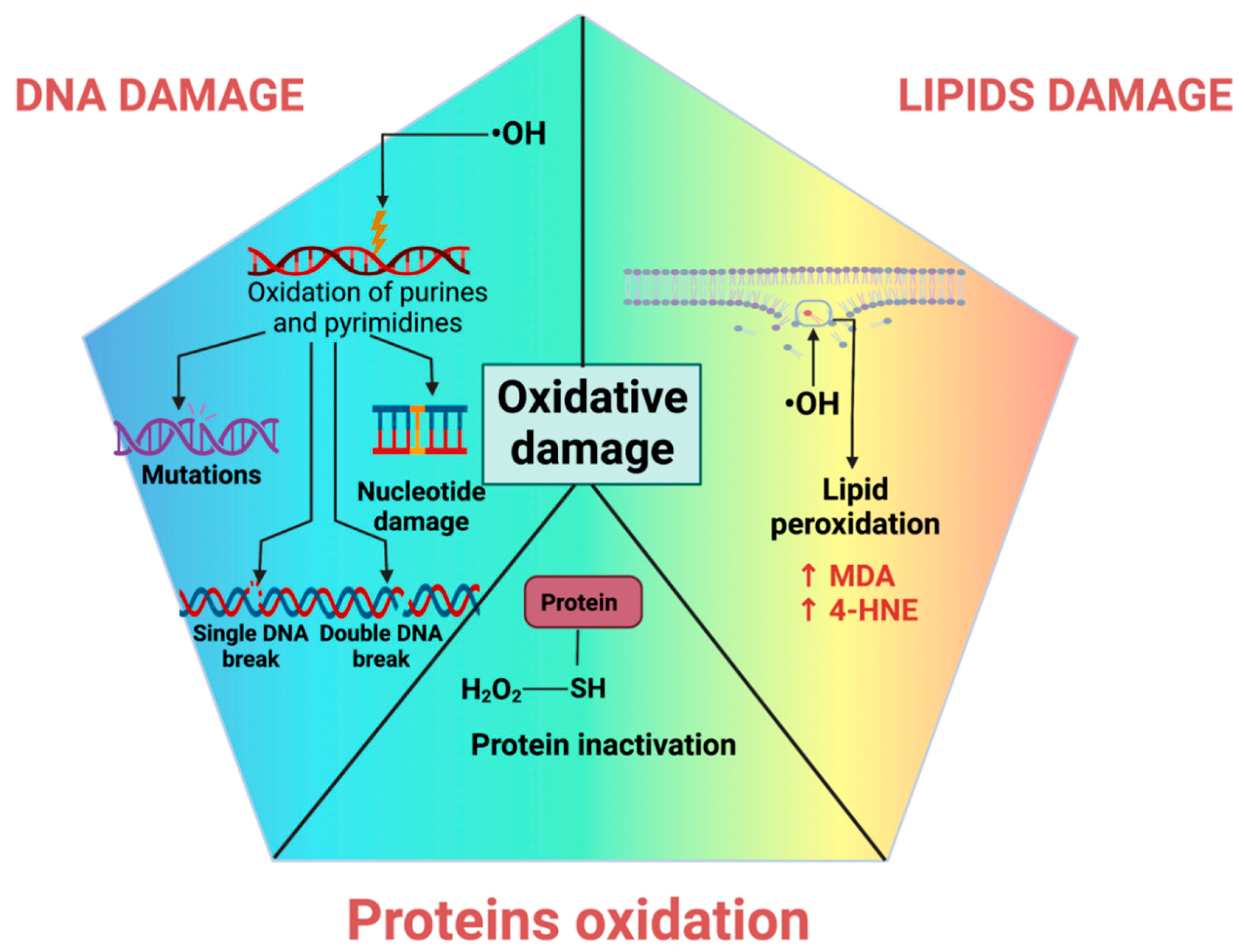
The deoxyribonucleotides that compose DNA are damaged by oxidative stress, resulting in modified products. The ROS that causes direct damage to nucleic acids is •OH, which is highly reactive. •OH reacts with DNA by adding to the double bonds of the purine (adenine, guanine) or pyrimidine (cytosine, thymine) bases of DNA and through the abstraction of a hydrogen atom from the methyl group of thiamine and each of the CH bonds of the sugar 2-deoxyribose [173,174,175]. The generated products of these reactions can lead to chain injuries, cross-linking DNA with proteins, intramolecular cyclization, or clustered sites [175]. It is well known that the •OH preferentially oxidizes the guanine base of DNA, primarily forming 8-hydroxy-2′-deoxyguanosine (8-OHdG; not exclusively, but it is by far the most studied) [173,175]. 8-OHdG affects mechanisms such as replication and transcription (Figure 3) [173,176].
DNA damage has been observed in multiple diseases such as cardiovascular, inflammatory, or neurodegenerative diseases, natural aging, or exposure to drugs and environmental contaminants [173,177,178,179,180]. Therefore, oxidative damage to DNA has frequently been related as an important factor of mutagenic lesions in cancer development [173,181].
4.2. Lipid Peroxidation
Lipid peroxidation results from oxidants (such as RONS) attacking unsaturated lipids, causing the formation of lipid oxidation products. These lipoperoxidation products include 4-hydroxy-2-nonenal (4-HNE), MDA, oxylipins, isoprostanes, and oxysterols, among others [182]. These products are highly reactive and interact with biomolecules such as DNA, proteins, or other lipids, leading to numerous biological effects [182,183,184].
4-HNE is one of the main lipid oxidation products that result from enzymatic and non-enzymatic oxidative pathways from oxidized phospholipids containing polyunsaturated fatty acid (PUFA) n-6 chains [185]. 4-HNE can increase ROS formation and inflammation, alter cell signaling, and cause cell damage and apoptosis [183].
Another product of lipid peroxidation is MDA, which may result as a product of enzymatic arachidonic acid oxygenation or as a final product of n-3 and n-6 fatty acids oxidation [183]. Similarly, it is a highly reactive molecule, so it is typical for cross-linking products to form or to alter cells’ structure, function, and response [182,183]. In addition to MDA and 4-HNE (Figure 3), oxylipins are bioactive lipid metabolites derived from PUFAs through auto-oxidation, cyclooxygenase, lipoxygenase, cytochrome P450, 15-hydroxyprostaglandin dehydrogenase, and soluble epoxide hydrolase enzymes. Oxylipins may exhibit positive and negative effects [186]. These products involve inflammation, pain response, cell adhesion, apoptosis, angiogenesis, blood coagulation, and blood vessel permeability [186,187]. Moreover, linoleic acid (LA) reserves and oxidative stress in the body increase oxidized LA metabolites (OXLAMs) such as 9- and 13-hydroxyoctadecadienoic acid (HODE) and 9- and 13-oxo-octadecadienoic acid (oxo-ODE) [188,189]. HODEs and isoprostanes are associated with coronary artery diseases, dysregulated vascular permeability disease, and nonalcoholic steatohepatitis [182,188,190].
Other crucial oxidized lipid molecules are oxysterols. Oxysterols are cholesterol-oxidized metabolites produced through enzymatic or non-enzymatic mechanisms, primarily during OS conditions [191]. Oxysterols are bioactive lipids involved in cell signaling and metabolic regulations, such as cholesterol, lipid, glucose, brain homeostasis, immune regulation, and cell differentiation [191,192]. Nevertheless, some oxysterols are cytotoxic, such as 7α-hydroxycholesterol [7α-OHC] and 7β-hydroxycholesterol [7β-OHC]). Oxysterol deregulation is associated with age-degenerative and cancer-related diseases that include Alzheimer’s disease, retinal degeneration, breast and colorectal cancer, cardiovascular diseases, and diabetes [193,194].
In summary, many lipoperoxidation products may induce lipid peroxidation, causing oxidative damage to cells and tissues due to promoting initial free radical events and inducing oxidative stress that contributes to the amplification of oxidative cell injury. The latter triggers the formation of protein and DNA adducts, necrosis, apoptosis, and specifically ferroptosis (characterized by the accumulation of lipid peroxides), which leads mainly to aging, neurodegeneration, cancer, and cardiovascular diseases [182,195,196,197].
4.3. Protein Oxidation
The most common oxidant damage to proteins is the oxidation of the sulfhydryl group (-SH) of proteins in amino acids with sulfur, such as Cys and Met, which are the most susceptible (Figure 3). In addition, the formation of protein carbonyls such as aldehydes and ketones is widespread [198]. The protein carbonyls derived from lipoxidation-derived aldehydes such as acrolein, 4-oxo-2-nonenal, 4-HNE, 2,4-decadienal, and MDA are more prevalent than those formed by the direct oxidation of side-chain amino acids [199,200]. The oxidation products of proteins vary since the different radicals have multiple possible sites of attack due to the varied nature of the amino acid side chains of proteins. The amino acid residues most vulnerable to oxidation are lysine, arginine, proline, and threonine [198,201]. The radical reactions with proteins may occur by (1) hydrogen atom abstraction at positions adjacent to electron-delocalizing groups such as -SH, hydroxy groups (in serine and threonine), carboxyl and amide (in aspartic acid, glutamic acid, asparagine, glutamine), and the guanidine group in arginine; (2) electron abstraction from electron-rich sites; and (3) addition to electron-rich centers [201,202].
Protein oxidation generates post-translational modifications that alter amino acid and protein composition, structure, charge, hydrophobicity/hydrophilicity, and folding. These alterations inhibit their functions, such as enzymatic activity, and induce conformational changes, cross-linking, or aggregation [202,203]. The oxidized proteins also increase the risk of developing neurodegenerative, cardiovascular, or pulmonary diseases [182,204,205,206,207].
5. Antioxidants
The biological antioxidant refers to any compound that is capable of delaying or preventing the oxidation of a substrate [208]. Thus, antioxidants decrease oxidants such as RONS, avoiding oxidative stress. In this way, antioxidants attenuate cell biomolecule damage, such as lipids, proteins, and DNA [209]. Since the damaging effects on these biomolecules contribute to cellular toxicity and disease, antioxidants prevent these effects [6]. The antioxidants group includes enzymes such as SOD, CAT, and GPx, which reduce oxidants or free radicals, with the ability to induce the production of other free radicals. In this case, SOD reduces the radical superoxide to H2O2, a ROS more stable that can be reduced to H2O by catalase or GPx. Thus, these enzymes can efficiently degrade ROS. In addition, antioxidants also involve some proteins such as lactoferrin, ferritin, and caeruloplasmin that prevent RONS formation by sequestering prooxidants such as metal ions or the heme group. It is essential to mention that the antioxidants of this group have substrate specificity, which limits their antioxidant activity [210].
Antioxidants, known as “scavengers”, react with radicals by donating or accepting electrons, transforming themselves into less-reactive radicals that can be easily removed.
5.1. Endogenous Antioxidants
Antioxidants can be classified as exogenous and endogenous. Note that there exist other classifications; however, we use this classification according to the source of production. Exogenous antioxidants are obtained from our diet; some examples are vitamins A, C, and E. Endogenous antioxidants are synthesized by the cells and are known to be much more powerful than exogenous antioxidants. For example, it has been found that an endogenous antioxidant, catalase, is an enzyme that is not saturated by its substrate, having high efficiency in the degradation of H2O2. On the other hand, exogenous antioxidants such as Vit C are associated with different antioxidant enzymes that restore their activity in a decrease in these antioxidants, so Vit C loses its activity as an antioxidant. Thus, endogenous antioxidants are the products of cellular metabolism, and these can be enzymatic, such as SOD, CAT, and peroxiredoxin (Prx), or non-enzymatic, such as the GSH. Both enzymatic and non-enzymatic endogenous antioxidants comprise a complex system that overlaps activities to enhance cellular antioxidant defense to reduce ROS, avoiding oxidative stress [6].
5.1.1. Enzymatic Antioxidants
As mentioned above, endogenous antioxidants can comprise non-enzymatic antioxidants, such as GSH, or enzymatic antioxidants, which include large molecules such as SOD, CAT, Prx, GPx, HO-1, glutathione reductase (GR), GST, thioredoxin (Trx), and thioredoxin reductase (TrxR). Enzymatic antioxidants encompass a complex system that works synergistically to reduce ROS. These antioxidant enzymes will be described in the following paragraphs.
Superoxide Dismutase (SOD)
SODs are present in all organisms that live in the presence of O2 and are a group of metalloenzymes that catalyze the conversion of two molecules of sO2•− into H2O2 and O2. This reaction is accompanied by the oxidation–reduction reactions of the metal ions present in the SODs’ active sites. In mammals, it can be found in three types of SOD: CuZn-SOD (SOD1), present in the cytosol; Mn-SOD (SOD2) in the mitochondrial matrix; and CuZn-SOD (SOD3) in the extracellular fluid. In these antioxidant enzymes, Cu, Zn, and Mn are the metal ions that function as cofactors [211]. The active site of SOD enzymes uses a ping-pong mechanism, where O2•− reduces the metal ion, Cu, Zn, or Mn, and other O2•− oxidizes this reduced metal ion. The latter oxidized metal might be oxidated by another O2•− to start another dismutation of O2•− to produce O2 and H2O2 [212].
SOD enzymes have distinct subcellular localizations, highlighting the need for tight control of ROS homeostasis in the different cell compartments. Changes in SOD activity in a compartment might lead to an H2O2 concentration gradient, increasing H2O2 flux and the activation of redox-sensitive cell-signaling pathway [9]. O2•− is produced by several sources within the cell; for example, in the cytosol, O2•− is produced by NOX, xanthine oxidase, and the cytochrome p450 monooxygenases, which are present mainly in the ER. O2•− can be transformed to H2O2, which diffuses into the cytosol to produce other highly reactive RONS, such as •OH and ONOO−, among others. In this way, the reduction of O2•− by SODs prevents the production of other RONS, avoiding oxidative stress; however, the H2O2 flux could be decreased, inducing the turn-off of redox-sensitive cell-signaling pathways. Thus, SOD regulation is crucial for controlling RONS in this dual role of cellular damage and cell signaling. SOD enzymes are also anti-inflammatory agents that prevent precancerous cell changes and Alzheimer’s disease. Since oxidative stress contributes to cancer development, decreased SOD activity is a key factor in inducing cancer [213]. Moreover, SOD has an essential effect on myocardial ischemia-reperfusion injury, inflammation, and cerebral ischemia-reperfusion injury [214].
SOD consists of three isoforms, Copper/Zinc SOD (CuZnSOD), manganese SOD (MnSOD), and cytosolic CuZnSOD. All SOD isoforms might suffer post-translational modifications such as acetylation, nitration, methylation, and glutathionylation. For instance, MnSOD can suffer glutathionylation, a protective modification implying the transfer of the GSH group to Cys [215]. Other modifications such as nitration in Tyr residues damage the enzyme. This modification has been found in ischemia reperfusion-induced kidney injury, diabetic kidney disease, and aging [216,217,218]. Likewise, Mn-SOD activity reduction is related to decreased mitochondrial S-glutathionylation in the damage induced by folic acid [219].
Catalase (CAT)
H2O2 is degraded by different enzymes, including CAT, GPx, and other peroxidases. CAT is a highly efficient enzyme in H2O2 reduction since this enzyme cannot be saturated by H2O2 levels [220]. CAT breaks down two H2O2 molecules into one molecule of O2 and two molecules of H2O2.
CAT possesses a monofunctional heme-containing prosthetic group of ferric proto- porphyrin IX in its active site, which reacts with H2O2. The heme group is also tightly bound to NADPH in the active conformation of the enzyme, allowing NADPH to obstruct the formation of Fe(IV)O-ligated porphyrin in the inactive form. The first step of the reaction involves the formation of the intermediate “Enzyme[porphyrin Fe(IV)-O]”, which is a covalent oxyferryl species (FeIVO) with a porphyrin π-cation radical through the reduction of one H2O2 molecule [220]. In the second step reaction, “Enzyme[porphyrin Fe(IV)-O]” is reduced through redox reactions by a two-electron transfer from an electron donor (the second molecule of hydrogen peroxide) to produce the free enzyme, O2, and H2O [220]. Thus, these reactions allow CAT to minimize the formation of highly reactive ROS, •OH, by breaking down H2O2 molecules. It is important to mention that •OH is produced by Fenton and Haber–Weiss reactions, which are induced by high levels of H2O2 and O2•−. Thus, when CAT reduces H2O2, it results in a reduction in the production of •OH. In addition, the enzyme can detoxify other proton donor compounds and act as a peroxidase. This enzyme can be found in subcellular tissues such as peroxisomes and mitochondria, diminishing RONS [221]. Note that redox-sensitive cell signaling depends on the levels of H2O2, so CAT must be finely regulated. For example, it has been demonstrated that H2O2 activates kinases but deactivates phosphatases in cell-signaling pathways such as mitogen-activated protein kinase (MAPK) [9]. The regulation of the levels of H2O2 is essential to maintain a redox state and cellular signaling, avoiding diseases associated with these processes [9]. In this regard, CAT deficiency or malfunction is related to the pathogenesis of degenerative diseases such as schizophrenia, Alzheimer’s and Parkinson’s diseases, bipolar disorder, diabetes mellitus, hypertension, anemia, vitiligo, and cancer. Moreover, since oxidative stress is associated with mutagenesis, inflammation, and apoptosis, CAT is an essential enzyme implicated in preventing these processes [222].
CAT promoter is regulated by nuclear factor Y and specificity protein 1 transcription factors, which bind to CCAAT and GGGCGG boxes, promoting gene expression. A regulatory CAT region was identified which requires chromatin remodeling by retinoic acid receptor alpha to induce CAT expression. The importance of this regulation is highlighted by the fact that cancer cells employ this mechanism for H2O2 adaptation [223]. CAT-induced chromatin remodeling has been suggested in MCF7 breast cancer cells [224]. In brain cancer glioblastoma, the overexpression of CAT protects against chemotherapy and radiation due to CAT-induced glioma stem cell enrichment. CAT also decreased the survival in a glioblastoma mouse model [225]. Therefore, CAT is a protective enzyme for cancer cells.
Peroxidase (Prx)
Prx is a group of enzymes responsible for catalyzing the reduction of peroxides, with antioxidant and redox signaling functions. Prx removes H2O2, lipid peroxide, ONOO−, the oxidation of toxic reductants, the biosynthesis and degradation of lignin, auxin catabolism, and responses to environmental stresses such as wounding. Prx has a high capacity to protect proteins from oxidative damage induced by ROS. In addition, Prx 1 plays an essential role as a tumor suppressor since Prx 1 deficiency increases susceptibility to Ras- or ErbB2-induced breast cancer [226]. These enzymes have a conserved Cys residue that serves as the site of oxidation by peroxides. Peroxides oxidize Cys-SH to Cys-SOH, which reacts with another Cys residue to form a disulfide bond. The latter is subsequently reduced by an appropriate electron donor to restore Cys-SH and start another oxidation–reduction cycle at the active site of Prx.
Unlike most ROS-degrading antioxidant enzymes, Prx does not contain any redox cofactors, such as a metal ion, heme, or flavin. Their location depends on the group to which they belong [227]. For example, Prx 2-cysteine (Prx I-IV), Prx 2-atypical cysteine (Prx V), and Prx 1-cysteine (Prx VI) can be found in the cytoplasm, mitochondria, nucleus, peroxisomes, and lysosomes [228].
These Prx locations are associated with H2O2 production and H2O2-dependent intracellular signaling [229]. Because Prx is essential in cell signaling and redox state, the good function of these enzymes protects against cerebral ischemia-reperfusion injury and Parkinson’s, and Alzheimer’s diseases, where oxidative stress and inflammation are involved [230]. Moreover, in inflammation, phagocytes produce high levels of peroxides such as H2O2, lipid hydroperoxide, and ONOO− to kill microorganisms. It has been shown that these peroxides also upregulate cell-inflammatory signaling pathways. However, this burst of ROS must be regulated, and Prxs provide a cytoprotective effect during the inflammation process decrease. Thus, Prx regulates peroxides that serve in cell-signaling pathways and maintains a redox state, avoiding diseases and inflammation [229].
Glutathione Peroxidase (GPx)
Another enzyme that degrades peroxides and is produced endogenously, like CAT and Prx, is GPx. GPx is an enzyme with different subcellular distributions. It has been shown that GPx has a molecular mass estimated to be between 83 and 95 kDa, consisting of a tetramer of identical subunits of 22–23 kDa [231]. GPx is a selenocysteine enzyme that catalyzes the reduction of H2O2 to H2O and other organic peroxides to their corresponding alcohol. GPx uses GSH as a cofactor, an obligate co-substrate in reducing H2O2 to water. Not all GPxs contain selenocysteine, but all use GSH in their active site. These enzymes that do not contain selenocysteine are identified as thioredoxin-dependent peroxidases because they contain a Cys in place of the selenocysteine in the redox-active site.
In humans, there are at least eight mammalian GPx enzymes, GPx1–GPx8. Most of them are selenoproteins (SecGPxs) that are expressed in different cell types; for example, GPx-1, GPx-2, GPx-4, and GPx-6 are mainly expressed in embryos and olfactory epithelium. GPx can be found in the cytosol (GPx1), in the gastrointestinal system (GPx2), in membranes (GPx4), and in the mitochondria and extracellular space (GPx3) [232]. In GPx5, GPx7, and GPx8, the active site Sec residue is replaced by Cys (CysGPxs). GPx-4, GPx-7, and GPx-8 are monomeric, while the others are tetrameric. The alternative use of Cys or Sec would have the biological advantage of having sulfur instead of selenium in these enzymes. However, it has been demonstrated that these GPxs containing Cys are as efficient as their selenocysteine-containing homologs in reducing hydroperoxides. GPx is essential for reducing lipid hydroperoxides and other soluble hydroperoxides after they are released from membrane lipids. Therefore, GPx4 activity is a key enzyme in reducing lipid peroxyl radicals that are associated with the production of aldehydes/ketones such as 4-HNE and MDA, which are highly reactive ROS and can induce DNA damage, forming DNA adducts [6]. The role of GPx4 has been highlighted in the cancer context. For instance, in hepatocellular carcinoma, the expression of GPx4 is related to patient survival, reduced cell proliferation, tissue remodeling, and immune response. The authors also showed that GPx4 overexpression in mice reduced cell proliferation, disturbed angiogenesis, and decreased IL-8 and C-reactive protein, which displayed impaired tumor growth [233].
Since GPx is critical in H2O2 degradation, its deactivation induces oxidative damage and the activation of nuclear factor-κB (NfκB)-related inflammatory pathways [234]. Thus, GPx is implicated in developing and preventing diseases such as cancer, hypertension, vitiligo, neurodegenerative disease, and cardiovascular disease [235]. GPx4 regulates and prevents ferroptosis, a programmed cell death associated with the production of high levels of lipid peroxidation, and as a consequence, GPx4 deficiency will induce ferroptosis and the cell death of brain cells, provoking neurodegenerative diseases [236]. Regarding cancer, GPx2 deficiency is associated with cutaneous squamous cell carcinomas since the production of ROS induces mutations in DNA, carrying out the activation of oncogenes or the loss of the activation of tumor suppressors [237].
Heme Oxygenase-1 (HO-1)
HO-1 is considered a cytoprotective enzyme because it reduces ROS and prevents oxidative stress. HO-1 catalyzes the rate-limiting step in heme degradation, which results in the formation of biliverdin, iron, and carbon monoxide (CO).
HO-1 is the only enzyme that catabolizes heme, as there are housekeeping genes designated to control the heme of the cell. Since HO-1 produces CO, this enzyme is considered the main source of cellular endogenous CO production. It has been demonstrated that the microsomal HO-1 system of mammals consists of three enzymes: HO, NADPH cytochrome c (p450) reductase, and biliverdin reductase (BVR). The latter enzymes use the stoichiometric quantities of NADPH and molecular O2 for biliverdin (BR) production. Three active isoforms of heme oxygenase exist—HO-1, HO-2, and HO-3. HO-1 is the inducible isoform that is upregulated by different stress conditions, such as heavy metals, nitric oxide, UV irradiation, infections, cytokines, and oxidized low-density lipoprotein (LDL). HO-2 and HO-3 are constitutively expressed at basal levels in most human cells; however, the higher concentration levels of these isoforms are expressed in neurons, the spleen, and the liver.
The antioxidant properties of HO-1 are associated with biliverdin since biliverdin and its product, bilirubin, drive a catalytic antioxidant cycle. This cycle involves the reduction of biliverdin to bilirubin by BVR. Bilirubin is a potent antioxidant that reduces lipophilic ROS, such as lipid hydroperoxides and peroxyl radicals, to alcohol or water, respectively. Thus, when ROS oxidizes bilirubin, it is reconverted to biliverdin, which can be used for another catalytic antioxidant cycle.
Interestingly, it has been reported that HO-1 might migrate from the cytosol into the nucleus, which activates transcription factors that respond to an oxidant environment [238]. Note that this action is independent of HO-1 enzymatic activity, and nuclear translocation was only observed under oxidative stress conditions [239]. HO-1 has more non-canonical functions, including mediating protein–protein interactions, acting as a chaperone, immunomodulation, and transcriptional regulation [240]. Since heme degradation products are biologically active, the deregulation of HO-1 is implicated in multiple pathological conditions. For instance, HO-1 deregulation is associated with several diseases such as cancer, inflammation/immune dysfunction, ischemia-reperfusion injury, and transplantation [241].
Glutathione Reductase (GR)
Although different organisms synthesize GSH, it is mainly recycled. When GSH is oxidized to GSSG, the cellular machinery can restore GSH, reducing GSSG. GR is an essential enzyme that recycles GSH, reducing GSSG. Thus, GR is responsible for maintaining the levels of GSH, the most abundant antioxidant in the cells [242]. GR has flavin adenine dinucleotide (FAD) as a cofactor in the reaction of GSSG reduction to GSH. GR is found in the cytoplasm and can regenerate GSH by reducing its oxidized form GSSG [243]. During this process, GSSG reduction to GSH depends on nicotinamide adenine dinucleotide phosphate (NADPH). Interestingly, the reduction of GSSG provides two molecules of GSH.
GSH is highly relevant to maintaining a redox state because GSH is a cofactor for antioxidant enzymes such as GPx, GST, and glyoxalases. Moreover, GSH detoxifies xenobiotics when associated with other antioxidants such as vitamin C or E; sequesters and stores nitric oxide and cysteine, transporting them around the body; and is involved in redox signaling [242]. Therefore, GR maintains the physiological levels of GSH, allowing regulation of the redox state and cellular homeostasis. GR is localized in the cytoplasm, nucleus, ER lumen, lysosomes, and mitochondria. Since RONS are involved in cell signaling for vital processes such as growth, cell cycle, defense against pathogens, and cell death, their regulation is crucial for maintaining homeostasis. GSH is an antioxidant that regulates RONS precisely. Since GSH depends on its products and regeneration, enzymes such as GR are essential for maintaining cellular homeostasis.
In contrast, GSSG cannot be restored in GR dysfunction, and RONS could induce oxidative stress and the dysregulation of cell signaling. The latter is because oxidative stress could keep cell signaling constant, inducing diseases such as cancer. GR deficiency is a rare disorder; however, GR deficiency is associated with red blood cell enzymopathies [244].
Glutathione S-Transferase (GST)
GST is an enzyme related to the metabolism of xenobiotics and endogenous compounds such as antibiotics. Since these enzymes detoxify xenobiotics, they are abundant in the liver, an organ associated with purifying xenobiotics. GSTs are also associated with transporting and detoxifying bilirubin, a toxic heme metabolite. GSTs are related to the biosynthesis of eicosanoids that include prostaglandins and leukotrienes. GST is a cytosolic and microsomal enzyme that participates in the second step of metabolism and is responsible for conjugating toxic electrophilic compounds with GSH for their subsequent elimination. In this reaction, GSTs catalyze the nucleophilic attack of the SH group of GSH on an electrophilic center of a lipophilic second substrate, such as xenobiotics [245].
Thus, GST protects cellular biomolecules from attack by reactive electrophiles, both endogenous and exogenous electrophiles. Since GSH conjugation with toxic electrophiles leads to their elimination, GST protects against toxic compounds, inducing their elimination [246]. It is important to note that GST is able to interact covalently and non-covalently with several toxic compounds, inducing their degradation. GSTs are localized in the cytosol, mitochondria, and in microsomes. Mitochondrial and cytosolic GSTs are soluble enzymes, but microsomal GSTs are associated with membranes and are involved in eicosanoid and GSH metabolism [247]. GSTs also regulate cell signal transduction pathways implicated in cell survival and apoptosis, regulating cell-signaling pathways such as the mitogen-activated protein kinase (MAPK) signaling pathway, which is involved in cell survival and death signaling [242]. Thus, GST decrease adversely affects the function of the nervous system, leading to schizophrenia, diabetic nephropathy, and cancer, among others [248,249,250]. Regarding cancer, it has been shown that GSTs are associated with detoxification, and their overexpression in cancer is a bad prognosis since GSTs induce multidrug resistance involving several mechanisms. For instance, GSTs induce the clearance of anticancer drugs and are related to the overexpression of efflux transporters such as multidrug resistance protein 1 (MRP1) and ATP-binding cassette, increasing the efflux of anticancer drugs. Thus, the overexpression of GST and efflux transporters induces resistance to the cytotoxic action of anticancer drugs [247].
Thioredoxin (Trx)
In mammalian cells, Trx is one of the central antioxidant systems. Trx is a member of a conserved family of redox-active proteins that contain an active site dithiol motif. This enzyme maintains a reducing environment by catalyzing the flow of electrons from nicotinamide adenine dinucleotide phosphate (NADPH) to Trx target proteins using highly conserved thiol groups. Trx has a redox-active dithiol as a cofactor at its active site. The amino acid sequence in the active site containing this dithiol, -Trp-Cys-Gly-Pro-Cys-Lys-, is highly conserved, and the Cys residues are oxidized by the corresponding Trx substrate, as shown in the following reaction:
Trx complements and overlaps the action of the GSH system in protecting against oxidative stress and its toxicity. That is, Trx and GSH reduce peroxides through the activity of multiple Trx peroxidases and GSH peroxidases. Although Trx is present at lower levels than GSH, its role in regulating cellular redox events is more direct, making it an essential enzyme to reduce protein oxidation and toxicity. In this way, Trx reduces oxidized proteins, maintaining the proper functioning of proteins since protein oxidation induces their damage and subsequent degradation [251]. Among these proteins are different peroxidases that Trx reduces; therefore, Trx acts as a cofactor, binding partner, and protein reductant of other peroxidases. This function influences and reduces the cellular toxicity induced by oxidative stress and oxidative damage. For instance, Trx, as a cofactor for ribonucleotide reductase and Met sulfoxide reductase, contributes to DNA repair.
Moreover, Trx reduces thiols of the transcription factors AP-1 and NF-κB, inducing its binding to DNA [252]. Trx can be found in three isoforms: Trx-1, present in the endoplasmic reticulum; trx-2, present in mitochondria; and Trx-3, also in mitochondria. It contains at its active site two thiol groups (Trx-(SH)2), which can be oxidized to their disulfide form (Trx-S2) by reducing compounds such as disulfide proteins (P-S2) or H2O2 [253]. Trx is implicated in human diseases such as cardiovascular diseases, heart failure, stroke, inflammation, metabolic syndrome, neurodegenerative diseases, arthritis, and cancer [254]. Furthermore, Trx also plays a critical role in tumor resistance, as Trx-1 also binds to proteins, modulating folding and resistance to anticancer drugs [252].
Thioredoxin Reductase (TrxR)
TrxR reduces Trx that has been oxidized by reducing its target proteins, thus regulating the intracellular redox environment due to the maintenance of Trx. Mammalian TrxRs are selenium pyridine-disulfide oxidoreductase enzymes containing the amino acid sequence -Cys-Val-Asn-Val-Gly-Cys- at the redox catalytic site. The mechanism of action of TrxR is the following: when TrxSH2 is oxidized to Trx-S2, Trx-S2 can be reduced by NADPH through the catalytic action of the TrxR flavoenzyme. The reaction mechanism of TrxR is shown below:
Therefore, the maintenance of an adequate intracellular redox environment is mainly due to the action of TrxR and the TrxR/Trx system. TrxR complementing the TrxR/Trx system is crucial for cell survival, as they are the central protein systems that control the redox balance of proteins in the cytoplasm [255]. TrxR is an NADPH-dependent homodimer that contains one FAD per subunit and is found in two isoforms: cytosolic TrxR-1 and mitochondrial TrxR-2. It includes a residue of selenocysteine, which allows it to reduce to Trx [253]. Since TrxR maintains Trx in good condition, the role of TrxR as a protector against oxidative injury is essential, albeit indirectly, for cell growth and even cell transformation.
Furthermore, TrxR is crucial for recycling ascorbate from its oxidized form. The latter is critical for humans, who lack the ability to synthesize ascorbic acid, an essential antioxidant scavenger in protecting cells from oxidative stress and oxidative damage, so TrxR activity is vital for maintaining cellular homeostasis and a good redox state [255]. Furthermore, because TrxR can reduce the number of substrates other than Trx, their role in cell physiology likely remains to be discovered. TrxR is vital in human diseases, including cancer, acquired immunodeficiency syndrome (AIDS), and autoimmune disease [255]. Decreasing TrxR levels and activity could be a potential therapy for cancer treatment because TrxR overexpression is associated with resistance to anticancer therapy [256]. Therefore, TrxR inhibitors have a potential antitumor activity that would induce oxidative stress, causing cell-cycle arrest and apoptosis.
5.1.2. Non-Enzymatic Antioxidants
Non-enzymatic antioxidants work by intercepting and terminating free-radical chain reactions [257]. Some of these antioxidants are water-soluble and are found in the cytosol or cytoplasmic matrix, while others are fat-soluble and are present in cell membranes [258]. Examples of non-enzymatic antioxidants are GSH, bilirubin (BR), melatonin, uric acid, coenzyme Q, and others [259].
Glutathione (GSH)
GSH is a tripeptide composed of cysteine, glutamic acid, and glycine residues [260]. In addition, it is the most critical low-molecular-weight soluble antioxidant and is ubiquitously distributed in all cells. It is produced in many different tissues [245]; however, in the liver, GSH synthesis is more intense [243]. The de novo synthesis of GSH begins with the union of cysteine with glutamate to produce γ-glutamylcysteine by the action of the enzyme glutamate cysteine ligase (GCL) called γ-glutamylcysteine synthetase. Subsequently, the enzyme glutathione synthetase adds glycine to the dipeptide, forming GSH (γ-glutamyl cysteinyl glycine). Both enzymes require ATP hydrolysis to perform their function [245]. It should be noted that the rate-limiting step in de novo GSH synthesis is that catalyzed by GCL, a target gene of Nrf2 [261].
GSH can exist in two redox forms, the reduced thiol and the GSSG [262]. Under normal conditions, the reduced form predominantly represents more than 98% of total GSH [263]. GSH can be converted from reduced to oxidized form by the enzymes GPx and GR. GPx oxidizes GSH to GSSG during the detoxification of H2O2 or other organic hydroperoxides. However, GSSG can be reduced back to GSH by the action of GR and in the presence of nicotinamide adenine dinucleotide phosphate in its oxidized form (NADP+) [264].
GSH fulfills various functions; however, the most important is acting as an antioxidant. The antioxidant function of GSH consists of reducing the reactive oxygen species (ROS) formed during enzymatic and non-enzymatic reactions [265]. In addition, it regenerates other antioxidants, including vitamin C and vitamin E, and participates in the repair of biomolecules with oxidative damage and the maintenance of the sulfhydryl groups of proteins in a reduced state [266,267,268]. However, GSH also has other functions that are not related to ROS. For example, the conjugation of GSH by the enzyme GST with xenobiotic compounds produces non-toxic products, which affects their detoxification [269]. In addition, GSH participates in the metabolism of prostaglandins and leukotrienes and is involved in the transport of amino acids and the absorption of micronutrients from the intestine, mainly Fe and selenium [270,271]. It is also involved in the modulation of cell differentiation and proliferation, apoptosis, enzyme activation, metal transport in cells, neurotransmission, and as a source of cysteine during protein synthesis [272].
Albumin (ALB)
ALB is composed of 585 amino acids, is the main extracellular protein responsible for maintaining the redox state of plasma, and has numerous physiological functions [274]. These functions include regulating osmotic pressure, distributing fluids between the different body compartments, and transporting bile pigments, cholesterol, fatty acids, and drugs [257].
The plasma concentration of ALB in healthy people ranges from 35 g/L to 50 g/L [275]. A lower concentration of ALB is related to the aging process and the development of non-communicable diseases (also known as chronic diseases), which are strongly associated with the action of ROS and the oxidant–antioxidant imbalance [276,277].
ALB exerts its antioxidant effect by binding to many types of molecules and ions to prevent ROS production. For example, free ferrous and cupric ions bind to ALB, preventing them from being catalysts in the Fenton reaction and generating •OH [257,278]. In addition, ALB directly neutralizes free radicals through its sulfur amino acids, Met and Cys [279]. These residues represent between 40% and 80% of the total antioxidant activity of ALB [280]. The free sulfhydryl group of Cys in ALB is capable of trapping several reactive species, such as H2O2, O2.−, HOCl, and ONOO−. When this happens, Cys is oxidized to form sulfenic acid (ALB-SOH), which, in the presence of ROS, can be further oxidized to sulfinic (ALB-SO2H) and sulfonic (ALB-SO3H) acids. These reactions are irreversible and lead to the formation of final products. However, ALB-SOH is also regenerated back to ALB in reduced form (ALB-SH) by reaction with a low-molecular-weight thiol, e.g., free Cys or GSH. Under nitrosative stress, the reduced Cys of albumin is transformed into s-nitroso-albumin (ALB-SNO) [257,281]. This reaction is also reversed by Cys or GSH [282]. ALB also exerts indirect antioxidant effects since it binds to BR and unsaturated fatty acids, preventing the oxidation of these compounds [283]. During pathological conditions such as diabetes, chronic obstructive pulmonary disease (COPD), and kidney disease, glucose- or acrolein-induced structural modifications of ALB can strongly affect its antioxidant properties [284,285]. However, these modifications currently work to monitor the progression of different diseases such as chronic kidney disease [286,287].
Bilirubin (BR)
BR is a highly hydrophobic, water-insoluble tetrapyrrole derivative that is generated from the degradation of hemoglobin and other heme proteins (e.g., myoglobin, cytochromes, CAT) by the mononuclear phagocyte system [288,289]. The formation of BR begins when the heme residue of proteins is enzymatically broken down to biliverdin by the enzyme HO-1. Subsequently, biliverdin is reduced to BR by biliverdin reductase (BVR) in an irreversible process [257].
In plasma, unconjugated BR (uBR) binds to ALB for uptake in the liver by passive transmembrane diffusion combined with active transport mediated by various sinusoidal transporters [288]. Once in the liver, uBR binds to ligandin to be transported to the endoplasmic reticulum, where it is conjugated with glucuronic acid by the action of the enzyme uridine diphosphate glycosyltransferase 1A1 (UGT1A1) and is thus excreted in the bile to the liver to intestine [288,289]. In some diseases where the liver is damaged, the excretion of bile is obstructed, and the level of BR in the blood increases. For this reason, BR is considered a marker of liver function. Various studies have shown that BR can be toxic at high levels, especially in newborns [290]. In addition, hyperbilirubinemia can produce imperceptible effects to severe brain damage and even death [291]. However, moderate concentrations of BR have been associated with antioxidant and anti-inflammatory properties [292,293].
As an antioxidant, BR captures 1O2 with high efficiency, reacts with O2.− and peroxyl radicals, and serves as a reducing substrate for peroxidases in H2O2 or organic hydroperoxides [294]. It should be noted that when BR acts as a reducing agent, it is oxidized to biliverdin; however, BR can be regenerated again by the action of BVR through a cycle that detoxifies excess oxidants up to 10,000 times [288]. There is evidence showing that highly moderate levels of BR are positively associated with a reduced risk of cardiovascular disease, Alzheimer’s disease, ischemia-reperfusion, diabetes, respiratory disease, cancer, metabolic syndrome, and obesity, suggesting that increased BR production is an adaptive response against oxidation [295,296,297,298].
Uric Acid
Uric acid is a low-molecular-weight organic compound generated by the purine degradation pathway. The average concentration of uric acid in humans is 3.5 to 7.5 mg/dL, which is higher than that of other mammals (0.5 to 1.5 mg/dL) since the latter can transform the serum uric acid into allantoin. When uric acid concentrations are much higher than those considered normal, it can lead to obesity, insulin resistance, and hyperuricemia [299,300]. Hyperuricemia can be a risk factor for cardiovascular disease in patients with diabetes mellitus [301] and can cause pulmonary arterial hypertension [302].
On the other hand, uric acid is a hydrophilic antioxidant, providing approximately 60% of the free radical scavenging activity in blood serum [303,304]. Uric acid removes ROS such as ONOO−, 1O2, •OH, H2O2, and lipid peroxides [305]. When uric acid reacts with H2O2, urate radicals are formed, which have a lower oxidizing potential than other free radicals. Urate radicals can react with ascorbic acid and regenerate uric acid [306]. Furthermore, uric acid removes NO2 and carbonate ions [307,308]. It forms stable complexes with Fe ions and copper ions, inhibiting free radical reactions, such as the Fenton reaction and the Haber–Weiss reaction [305]. Uric acid also confers protection and modulates the physiological levels of antioxidant enzymes SOD1 and 3 [309]. Evidence shows that uric acid has beneficial effects in many diseases associated with oxidative stress. Furthermore, uric acid has been proposed to be more effective as an antioxidant than vitamin C [310].
Melatonin (MEL)
MEL (N-acetyl-5-methoxytryptamine) is a low-molecular-weight endogenous hormone derived from tryptophan and released from the pineal gland in the dark. For a long time, MEL was considered a hormone unique to vertebrates. However, recently it has been seen that it is also found in bacteria, algae, higher plants, and invertebrates [311]. In fact, it is now known that due to its bacterial origin, mitochondria also synthesize melatonin [312,313].
MEL is the hormone responsible for regulating different biological functions such as sleep, circadian rhythm, immunity, reproduction, and blood pressure control [314,315]. Furthermore, MEL has been seen to have anti-inflammatory and antioxidant properties. The antioxidant functions of MEL are to (a) directly neutralize free radicals such as hydroxyl, (b) stimulate the activity of antioxidant enzymes, (c) increase the efficiency of mitochondrial oxidative phosphorylation and reduce electron leakage to decrease the generation of RONS, and (d) increase the efficacy of other antioxidants [312,316,317]. Furthermore, due to its small molecular size and specific chemical structure (steroid hormone), MEL can cross biological membranes and react with individual cell components [318]. Therefore, MEL exerts antioxidant protection in both aliphatic and aqueous cellular environments. In fact, MEL also exerts its antioxidant functions through its receptors located in the cell membrane or intracellular organelles [319,320]. These antioxidant actions are indirect and probably involve the stimulation of antioxidant enzyme activities. When MEL acts through its receptors, it can achieve its effect at lower concentrations compared to when it functions as a direct free-radical scavenger because signal transduction pathways associated with the receptors serve to magnify the response. Another essential feature of the MEL is its availability at multiple sites—unlike other antioxidants, such as vitamin E and C, which are only available to humans through their diet [321,322,323].
Coenzyme Q10 (CoQ10)
CoQ10 is a 1,4-benzoquinone derivative that provides antioxidant protection to cell membranes and plasma lipoproteins [327]. It is synthesized in all body tissues, and depending on its oxidation state; it can also be called ubiquinone (CoQ10-oxidized), ubiquinol (CoQ10H2-reduced), and semiquinone (partially reduced, as a radical). CoQ10 is synthesized from tyrosine and a polyprenyl group resulting from acetyl-CoA [328,329]. Moreover, as its name implies, CoQ10 is ubiquitous and present in all human cells. It is mainly found in the mitochondrial respiratory chain, cell membranes, and lipoproteins.
CoQ10 has several biochemical functions, including electron transport in the mitochondrial respiratory chain and electron transport out of mitochondria [330]. The activity of CoQ10 is carried out in the lipid phase and participates in the redox reactions of dehydrogenases, cytochromes, or other non-heme proteins [331]. In addition, CoQ10 works as an antioxidant. Ubiquinol, the fully reduced form of CoQ10, scavenges free radicals from fat-soluble antioxidants by affecting the initiation and propagation of ROS [332,333]. Ubiquinol is also known to reduce oxidized α-tocopherol, and the reduced form of tocopherol shows strong antioxidant properties and regenerates vitamin C [334,335].
The average concentration of CoQ10 in the blood ranges between 0.50 and 1.91 μmol/L [336,337]. Concentrations below these are associated with aging and diseases such as hypertension, coronary artery disease, cardiovascular disease, diabetes mellitus, and cancer [333,338], while CoQ10 supplementation shows decreased oxidative stress and the increased activity of antioxidant enzymes [339], resulting in attenuation of the severity of different diseases and conditions [333,338,340].
5.2. Exogen or Dietary Antioxidants
Exogenous antioxidants or dietary antioxidants can be obtained mainly through the diet by consuming fruits, vegetables, derived products, and food supplements. These antioxidants cannot be synthesized in the human body [341]. Some examples of these dietary antioxidants are vitamins such as A, C, and E, some phenols, flavonoids, carotenoids, and metals such as Zn or Se. These, along with endogenous antioxidants, can counteract the deteriorating effects of oxidative stress and maintain redox homeostasis [342,343]. It has been suggested that an antioxidant-rich diet may benefit health [344,345]. Forman et al. [346] proposed that dietary antioxidants’ primary mechanism of action is through the stimulation of antioxidant enzymes and damage elimination and repair systems (e.g., the oxidative activation of the Nrf2 signaling pathway).
5.2.1. Ascorbic Acid
The L-enantiomer of ascorbic acid (vitamin C) is a hydrosoluble vitamin essential for humans; it acts as a reduction agent and is involved in tissue integrity, collagen biosynthesis, neuroprotection, hematopoiesis, and leukocyte functioning, as an efficient antioxidant eliminating ROS, and as free-radical scavenger [343,347]. The average concentration in plasma of vitamin C is approximately 60 to 90 μmol/L, and intracellular concentrations range from 0.5 to 10 mmol/L [348]. Humans cannot synthesize vitamin C endogenously, so the primary source is through the diet [349,350]. Poor intake of vitamin C through diet can lead to its deficiency. This is known as scurvy, which can induce anemia, myalgia, bone pain, easy bruising, swelling, petechiae, perifollicular hemorrhages, corkscrew hairs, gum disease, poor wound healing, mood changes, and depression [351]. In addition, in a meta-analysis of observational studies by Guo et al. [352], an inverse association was found between the levels of vitamin C in the diet and circulation and metabolic syndrome.
Vitamin C is present in fruits, vegetables, and legumes, such as citrus fruits, broccoli, spinach, potatoes, kiwis, strawberries, tomatoes, and red peppers [348,349]. The micronutrient is absorbed by the intestine and excreted via the kidneys. Moreover, vitamin C is transported across cell membranes by two sodium-dependent vitamin C transporters, sodium-dependent vitamin C transporters (SVCT)1 and SVCT2 [350,353,354]. It has also been mentioned that synthetic sources of vitamin C are chemically identical and have comparable bioavailability with food-derived sources of vitamin C [350].
Vitamin C is a ubiquitous antioxidant located in the aqueous phase of cells and gives stability to radicals by its excellent capability to donate electrons [341,355]. Ascorbic acid undergoes one- and two-electron oxidations to produce the ascorbyl radical and dehydroascorbic acid [350]. Ascorbic acid acts primarily as a donor of single hydrogen atoms. This property allows ascorbic acid to reduce many free radicals; the radical anion monodehydroascorbate reacts mainly with radicals [347]. Ascorbic acid may also have prooxidant effects at higher concentrations. Moreover, this prooxidant activity of ascorbic acid is frequently associated with metal ions (e.g., Fe and Cu) due to catalyzing their reduction, causing ROS formation [347,356,357].
5.2.2. Polyphenols
Phenols are chemical compounds that are widely distributed in foods of plant origin. Plants synthesize them in large quantities as a product of their secondary mechanism; some are essential for plant physiological functions, and others participate in defense functions against stress and environmental stimuli [358]. The main groups of polyphenols are phenolic acids, stilbenes, lignans, phenolic alcohols, and flavonoids [358]. The antioxidant activity of phenolic compounds is mainly due to structural characteristics (benzene ring and number-position of hydroxyl groups), which allow them to participate in metabolic oxidation–reduction reactions [359]. Their structure allows them to scavenge most types of oxidizing species by transferring a hydrogen atom or electrons via proton transfer to free-radical substrates, thus producing non-radical substrates. Although, they can act as antioxidants through the uptake of metal ions through the formation of metal complexes with their multiple hydroxyl and carbonyl groups [360]. In addition, it has been seen that they can interfere with iNOS activity, mitigating the oxidative stress caused by NO•. It also may inhibit the activity of ROS-generating enzymes such as XO and NOX and increase the activation of antioxidant enzymes such as SOD, CAT, and GPx [361,362,363,364]. Phenolic compounds are contained in foods mainly as polyphenols: flavonoids (flavones, catechins, anthocyanins, phytoestrogens) and lignans; these have antioxidant activity, so they are considered natural exogenous antioxidants [359,365,366].
5.2.3. Flavonoids
Flavonoids are the most abundant subclass of polyphenols in plants. They are low-molecular-weight compounds composed of two phenyl rings joined through a heterocyclic pyran ring and are hydroxylated in their aromatic rings [367]. Flavonoids occur predominantly as glycosides (O-glycosides and C-glycosides). More than 4000 varieties of flavonoids have been identified. Several subgroups of flavonoids are classified according to their chemical structural characteristics, such as the presence of functional groups, the oxidation state of the heterocyclic ring, and the number and position of hydroxyl groups, among others [361]. Therefore, it can be mentioned generally that the main subgroups are flavonols, flavones, flavanones, isoflavones, anthocyanidins, and flavanols (catechins and proanthocyanidins) [367,368].
Among the foods with high polyphenols are various berries, cocoa, grapes, and tea; their derived products are also foods rich in antioxidants. Berries include black, blue, straw, and raspberries, particularly rich in anthocyanins [366,369,370]. Moreover, flavanols in cocoa are mainly in the form of epicatechins, catechins, and procyanidins. In the case of wine, the primary polyphenols are catechins and proanthocyanidins [371,372]. Similarly, coffee and tea are relatively high in antioxidants and, due to frequent consumption, are important sources of antioxidants in the diet [369]. In tea, most flavonoids are found as catechins. In black tea, polymerized catechins such as theaflavins and thearubigins are predominant. In contrast, in green tea, the main flavonoids present are epigallocatechin gallate (EGCG), epigallocatechin (EGC), epicatechin gallate (ECG), epigallocatechin gallate (ECG), and epicatechin (EC), all of them monomeric catechins [373,374]. The catechins EGCG, EGC, ECG, and EC have been attributed to possessing antimutagenic, antidiabetic, anti-inflammatory, and cancer preventive properties, all of these probably related to their activity as antioxidants [373].
5.2.4. Resveratrol
3,5,4′-trihydroxy-trans-stilbene, more commonly known as resveratrol, is a phytoalexin-type polyphenol (antimicrobial) belonging to the stilbene family [375]. It has been observed that resveratrol as a phenolic compound can directly remove some RONS such as •OH, O2−, H2O2, and ONOO− [376,377,378]. However, many of the beneficial effects of resveratrol in vivo are mediated through gene regulation. Resveratrol has been identified as an activator of histone sirtuin 1/NAD+-dependent protein deacetylase (SIRT1), which modulates gene expression by targeting transcription factors such as NF-κB, FOXO, and PGC-1α, some proteins such as eNOS, and molecules such as histones [379]. Similarly, it has been observed that resveratrol can improve the expression of enzymes such as SOD, GPx1, and CAT [376]. Furthermore, even though the mechanism through which resveratrol can activate Nrf2 is not clear, there is evidence that resveratrol increases the transcriptional activity of Nrf2 and upregulates Nrf2/ARE-dependent antioxidant enzymes such as NQO1 and HO-1 [376,378,380]. Therefore, resveratrol can help decrease ROS production and accelerate ROS detoxification. Resveratrol has been attributed properties such as antiglycation, antioxidant, anti-inflammatory, neuroprotective, antiaging, cardioprotective, and anticancer [375,381].
6. OS and Diseases
Metabolic disorders and OS are tightly related. Metabolic disorders provided by elevated body weight include metabolic diseases such as diabetes and obesity. According to the World Health Organization (WHO), over 1.9 billion adults suffer from being overweight. One of the principal risk factors for developing metabolic disorders is a high-energy diet, which can overstimulate the mitochondria because of metabolic overload. This stimulation further upregulates ETS, resulting in the excessive production of ROS as byproducts [382]. This suggests a crucial role of mitochondria in metabolic disorders. Supporting the latter, patients with elevated body mass index (BMI) secrete 8-epiprostaglandin F2, an isoprostane produced by the non-enzymatic peroxidation of arachidonic acid in membrane phospholipids, through urine [383].
Furthermore, adipocytes in obese people show an overactivation of NOXs and ER stress, inducing chronic sustained inflammation. Oxidative stress is directly correlated with an increase in LDL proteins and inversely correlated with HDL decrease, proposing that OS regulates lipid metabolism. Accordingly, LDLs are the lipids with the most propensity to oxidize, and they are known as oxidized LDLs (oxLDLs). OxLDLs can be engulfed by macrophages, forming macrophage foaming cells [384]. The antioxidant system is also reduced in obese patients, contributing to metabolic syndrome.
Diabetes is another complication of metabolic alterations. During this disease, advanced glycation end products (AGEs) activate NOXs as described above. This NOX activation generates RONS; thus, high levels of these radicals generate OS (for more details, review [385]). ROS participates in the dysfunction of β-pancreatic cells, promoting type 2 diabetes development. Moreover, diabetes-induced ROS induces the overexpression of p27, an inhibitor of the cell cycle, and represses cyclins D1 and D2, arresting the cell cycle [386]. High glucose levels also produce ROS, activating interleukins such as IL-TNF-α, IL-1β, IL-6, and IL-18, thus contributing to inflammation, characteristic of diabetic patients [387].
In cancer, RONS regulate cancer cell proliferation and differentiation, acting as second messengers in several signal transduction pathways sensitive to cancer [6]. Cancer comprises a multistage cellular process involving initiation, promotion, and progression, and RONS intervene at each step. In initiation, RONS are implicated as mutagenic because of radicals’ ability to interact with DNA bases, triggering mutations in key genes. These mutations might carry out the activation of proteins that induce a proliferative phenotype or the uncontrolled growth of cells, thus producing proliferation. The promotion stage comprises, in fact, proliferation by the activation of signaling pathways by H2O2. The constant production of H2O2 deactivates phosphatases, avoiding the turn-off of signaling pathways such MAPKs [388]. Finally, in the progression stage, genomic instability is present, which involves the production of oxidized DNA pair bases such as 8-OHdG. It has been reported that 8-OHdG traps topoisomerases, causing DNA breaks [389]. The latter, along with deficient DNA system repairing, leads to the formation of chromosomal aberrations, consequently resulting in dysfunctional proteins.
7. Concluding Remarks
RONS are required for several cellular functions; however, high concentrations produce OS, contributing to several pathological conditions. Although mitochondria and NOXs are the principal RONS sources, RONS might provide other internal or external sources. Therefore, antioxidant systems are used by the cells to avoid RONS surpassing the canonical concentrations. Additionally, natural antioxidants obtained from the diet might help to maintain low RONS concentrations.
Author Contributions
Conceptualization, A.K.A.-R., A.C.-G., Y.L.A.-H., E.Y.H.-C. and J.P.-C.; investigation, A.K.A.-R., A.C.-G. and E.Y.H.-C.; writing, A.K.A.-R., A.C.-G. and E.Y.H.-C.; review and editing, A.K.A.-R., A.C.-G., E.Y.H.-C., Y.L.A.-H. and J.P.-C.; figure preparation, A.K.A.-R. and E.Y.H.-C.; supervision, J.P.-C.; project administration, J.P.-C.; funding acquisition, J.P.-C. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Consejo Nacional de Ciencia y Tecnología (CONACyT; grant number A1-S-7495) and by Dirección General de Asuntos del Personal Académico (DGAPA; grant number: IN200922).
Acknowledgments
A.K.A.-R. and E.Y.H.-C. are Ph.D. students from Posgrado en Ciencias Biológicas at the Universidad Nacional Autoónoma de México and they recipient a scholarship from CONACyT, México (CVU 818062 and CVU 853816). We gratefully acknowledge the postdoctoral grant program from the Consejo Nacional de Ciencia y Tecnología (CONACyT) for the postdoctoral fellow position of A.C.-G.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Li, Y.R.; Trush, M. Defining ROS in Biology and Medicine. React. Oxyg. Species 2016, 1, 9–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sies, H.; Jones, D.P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef] [PubMed]
- Lambeth, J.D.; Neish, A.S. Nox Enzymes and New Thinking on Reactive Oxygen: A Double-Edged Sword Revisited. Annu. Rev. Pathol. Mech. Dis. 2014, 9, 119–145. [Google Scholar] [CrossRef]
- Girotti, A.W. Mechanisms of lipid peroxidation. J. Free Radic. Biol. Med. 1985, 1, 87–95. [Google Scholar] [CrossRef]
- Sies, H. Hydrogen peroxide as a central redox signaling molecule in physiological oxidative stress: Oxidative eustress. Redox Biol. 2017, 11, 613–619. [Google Scholar] [CrossRef] [PubMed]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.D.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 2007, 39, 44–84. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.M.; Cheema, A.K.; Zhang, L.; Suzuki, Y.J. Protein Carbonylation as a Novel Mechanism in Redox Signaling. Circ. Res. 2008, 102, 310–318. [Google Scholar] [CrossRef] [Green Version]
- Anand, P.; Stamler, J.S. Enzymatic mechanisms regulating protein S-nitrosylation: Implications in health and disease. J. Mol. Med. 2012, 90, 233–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holmström, K.M.; Finkel, T. Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat. Rev. Mol. Cell Biol. 2014, 15, 411–421. [Google Scholar] [CrossRef] [PubMed]
- Finkel, T. Signal transduction by reactive oxygen species. J. Cell Biol. 2011, 194, 7–15. [Google Scholar] [CrossRef]
- Buetler, T.M.; Krauskopf, A.; Ruegg, U.T. Role of Superoxide as a Signaling Molecule. Physiology 2004, 19, 120–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Wang, X.; Vikash, V.; Ye, Q.; Wu, D.; Liu, Y.; Dong, W. ROS and ROS-Mediated Cellular Signaling. Oxid. Med. Cell. Longev. 2016, 2016, e4350965. [Google Scholar] [CrossRef] [Green Version]
- Giorgio, M.; Trinei, M.; Migliaccio, E.; Pelicci, P.G. Hydrogen peroxide: A metabolic by-product or a common mediator of ageing signals? Nat. Rev. Mol. Cell Biol. 2007, 8, 722–728. [Google Scholar] [CrossRef] [PubMed]
- Gupta, V.; Carroll, K.S. Sulfenic acid chemistry, detection and cellular lifetime. Biochim. Biophys. Acta BBA-Gen. Subj. 2014, 1840, 847–875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madesh, M.; Hajnóczky, G. VDAC-dependent permeabilization of the outer mitochondrial membrane by superoxide induces rapid and massive cytochrome c release. J. Cell Biol. 2001, 155, 1003–1016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griendling, K.K.; Sorescu, D.; Ushio-Fukai, M. NAD(P)H Oxidase: Role in Cardiovascular Biology and Disease. Circ. Res. 2000, 86, 494–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, W.-C.; Chio, C.-C.; Chi, K.-H.; Wu, H.-M.; Lin, W.-W. Superoxide Anion-Dependent Raf/MEK/ERK Activation by Peroxisome Proliferator Activated Receptor γ Agonists 15-Deoxy-Δ12,14-prostaglandin J2, Ciglitazone, and GW1929. Exp. Cell Res. 2002, 277, 192–200. [Google Scholar] [CrossRef]
- Yang, J.-Q.; Buettner, G.R.; Domann, F.E.; Li, Q.; Engelhardt, J.F.; Weydert, C.D.; Oberley, L.W. v-Ha-ras mitogenic signaling through superoxide and derived reactive oxygen species. Mol. Carcinog. 2002, 33, 206–218. [Google Scholar] [CrossRef] [Green Version]
- Sies, H.; Berndt, C.; Jones, D.P. Oxidative Stress. Ann. Rev. Biochem. 2017, 86, 715–748. [Google Scholar] [CrossRef]
- Aranda-Rivera, A.K.; Cruz-Gregorio, A.; Pedraza-Chaverri, J.; Scholze, A. Nrf2 Activation in Chronic Kidney Disease: Promises and Pitfalls. Antioxidants 2022, 11, 1112. [Google Scholar] [CrossRef]
- Horie, Y.; Suzuki, T.; Inoue, J.; Iso, T.; Wells, G.; Moore, T.W.; Mizushima, T.; Dinkova-Kostova, A.T.; Kasai, T.; Kamei, T.; et al. Molecular basis for the disruption of Keap1–Nrf2 interaction via Hinge & Latch mechanism. Commun. Biol. 2021, 4, 576. [Google Scholar] [CrossRef]
- Motohashi, H.; Yamamoto, M. Nrf2–Keap1 defines a physiologically important stress response mechanism. Trends Mol. Med. 2004, 10, 549–557. [Google Scholar] [CrossRef]
- Baird, L.; Yamamoto, M. The Molecular Mechanisms Regulating the KEAP1-NRF2 Pathway. Mol. Cell. Biol. 2020, 40, e00099-20. [Google Scholar] [CrossRef]
- Klotz, L.-O.; Sánchez-Ramos, C.; Prieto-Arroyo, I.; Urbánek, P.; Steinbrenner, H.; Monsalve, M. Redox regulation of FoxO transcription factors. Redox Biol. 2015, 6, 51–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borniquel, S.; García-Quintáns, N.; Valle, I.; Olmos, Y.; Wild, B.; Martínez-Granero, F.; Soria, E.; Lamas, S.; Monsalve, M. Inactivation of Foxo3a and Subsequent Downregulation of PGC-1α Mediate Nitric Oxide-Induced Endothelial Cell Migration. Mol. Cell. Biol. 2010, 30, 4035–4044. [Google Scholar] [CrossRef] [Green Version]
- Wu, M.; Bian, Q.; Liu, Y.; Fernandes, A.; Taylor, A.; Pereira, P.; Shang, F. Sustained oxidative stress inhibits NF-κB activation partially via inactivating the proteasome. Free Radic. Biol. Med. 2009, 46, 62–69. [Google Scholar] [CrossRef] [Green Version]
- Nishi, T.; Shimizu, N.; Hiramoto, M.; Sato, I.; Yamaguchi, Y.; Hasegawa, M.; Aizawa, S.; Tanaka, H.; Kataoka, K.; Watanabe, H.; et al. Spatial Redox Regulation of a Critical Cysteine Residue of NF-κB In Vivo. J. Biol. Chem. 2002, 277, 44548–44556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallace, D.C. Mitochondria and cancer. Nat. Rev. Cancer 2012, 12, 685–698. [Google Scholar] [CrossRef] [Green Version]
- Go, Y.-M.; Chandler, J.D.; Jones, D.P. The cysteine proteome. Free Radic. Biol. Med. 2015, 84, 227–245. [Google Scholar] [CrossRef] [Green Version]
- Zorova, L.D.; Popkov, V.A.; Plotnikov, E.Y.; Silachev, D.N.; Pevzner, I.B.; Jankauskas, S.S.; Babenko, V.A.; Zorov, S.D.; Balakireva, A.V.; Juhaszova, M.; et al. Mitochondrial membrane potential. Anal. Biochem. 2018, 552, 50–59. [Google Scholar] [CrossRef]
- McLain, A.L.; Szweda, P.A.; Szweda, L.I. α-Ketoglutarate dehydrogenase: A mitochondrial redox sensor. Free Radic. Res. 2011, 45, 29–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, S.J.; Jang, H.-S.; Noh, M.R.; Kim, J.; Kong, M.J.; Kim, J.I.; Park, J.-W.; Park, K.M. Mitochondrial NADP +-Dependent Isocitrate Dehydrogenase Deficiency Exacerbates Mitochondrial and Cell Damage after Kidney Ischemia-Reperfusion Injury. J. Am. Soc. Nephrol. 2017, 28, 1200–1215. [Google Scholar] [CrossRef] [Green Version]
- Hurd, T.R.; Collins, Y.; Abakumova, I.; Chouchani, E.T.; Baranowski, B.; Fearnley, I.M.; Prime, T.A.; Murphy, M.P.; James, A.M. Inactivation of Pyruvate Dehydrogenase Kinase 2 by Mitochondrial Reactive Oxygen Species. J. Biol. Chem. 2012, 287, 35153–35160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mapuskar, K.A.; Wen, H.; Holanda, D.G.; Rastogi, P.; Steinbach, E.; Han, R.; Coleman, M.C.; Attanasio, M.; Riley, D.P.; Spitz, D.R.; et al. Persistent increase in mitochondrial superoxide mediates cisplatin-induced chronic kidney disease. Redox Biol. 2019, 20, 98–106. [Google Scholar] [CrossRef]
- Hill, B.G.; Higdon, A.N.; Dranka, B.P.; Darley-Usmar, V.M. Regulation of vascular smooth muscle cell bioenergetic function by protein glutathiolation. Biochim. Biophys. Acta BBA-Bioenerg. 2010, 1797, 285–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishida, H.; Kurahashi, T.; Saito, Y.; Otsuki, N.; Kwon, M.; Ohtake, H.; Yamakawa, M.; Yamada, K.-I.; Miyata, S.; Tomita, Y.; et al. Kidney fibrosis is independent of the amount of ascorbic acid in mice with unilateral ureteral obstruction. Free Radic. Res. 2014, 48, 1115–1124. [Google Scholar] [CrossRef]
- Aranda-Rivera, A.K.; Cruz-Gregorio, A.; Aparicio-Trejo, O.E.; Ortega-Lozano, A.J.; Pedraza-Chaverri, J. Redox signaling pathways in unilateral ureteral obstruction (UUO)-induced renal fibrosis. Free Radic. Biol. Med. 2021, 172, 65–81. [Google Scholar] [CrossRef]
- Shanmugasundaram, K.; Nayak, B.K.; Friedrichs, W.E.; Kaushik, D.; Rodriguez, R.; Block, K. NOX4 functions as a mitochondrial energetic sensor coupling cancer metabolic reprogramming to drug resistance. Nat. Commun. 2017, 8, 997. [Google Scholar] [CrossRef] [Green Version]
- Aranda-Rivera, A.K.; Cruz-Gregorio, A.; Aparicio-Trejo, O.E.; Pedraza-Chaverri, J. Mitochondrial Redox Signaling and Oxidative Stress in Kidney Diseases. Biomolecules 2021, 11, 1144. [Google Scholar] [CrossRef]
- Zhao, R.; Jiang, S.; Zhang, L.; Yu, Z. Mitochondrial electron transport chain, ROS generation and uncoupling (Review). Int. J. Mol. Med. 2019, 44, 3–15. [Google Scholar] [CrossRef]
- Bleier, L.; Wittig, I.; Heide, H.; Steger, M.; Brandt, U.; Dröse, S. Generator-specific targets of mitochondrial reactive oxygen species. Free Radic. Biol. Med. 2015, 78, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Herb, M.; Schramm, M. Functions of ROS in Macrophages and Antimicrobial Immunity. Antioxidants 2021, 10, 313. [Google Scholar] [CrossRef] [PubMed]
- Lambeth, J.D. NOX enzymes and the biology of reactive oxygen. Nat. Rev. Immunol. 2004, 4, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Glass, M.J.; Huang, J.; Oselkin, M.; Tarsitano, M.J.; Wang, G.; Iadecola, C.; Pickel, V.M. Subcellular localization of nicotinamide adenine dinucleotide phosphate oxidase subunits in neurons and astroglia of the rat medial nucleus tractus solitarius: Relationship with tyrosine hydroxylase immunoreactive neurons. Neuroscience 2006, 143, 547–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aparicio-Trejo, O.E.; Tapia, E.; Sánchez-Lozada, L.G.; Pedraza-Chaverri, J. Mitochondrial bioenergetics, redox state, dynamics and turnover alterations in renal mass reduction models of chronic kidney diseases and their possible implications in the progression of this illness. Pharmacol. Res. 2018, 135, 1–11. [Google Scholar] [CrossRef]
- Peroxisomal Hydrogen Peroxide Metabolism and Signaling in Health and Disease. Int. J. Mol. Sci. 2019, 20, 3673. [CrossRef] [Green Version]
- Zhang, J.; Kim, J.; Alexander, A.; Cai, S.; Tripathi, D.N.; Dere, R.; Tee, A.R.; Tait-Mulder, J.; Di Nardo, A.; Han, J.M.; et al. A tuberous sclerosis complex signalling node at the peroxisome regulates mTORC1 and autophagy in response to ROS. Nat. Cell Biol. 2013, 15, 1186–1196. [Google Scholar] [CrossRef]
- Malhotra, J.D.; Kaufman, R.J. ER Stress and Its Functional Link to Mitochondria: Role in Cell Survival and Death. Cold Spring Harb. Perspect. Biol. 2011, 3, a004424. [Google Scholar] [CrossRef]
- Bravo, R.; Vicencio, J.M.; Parra, V.; Troncoso, R.; Munoz, J.P.; Bui, M.; Quiroga, C.; Rodriguez, A.E.; Verdejo, H.E.; Ferreira, J.; et al. Increased ER–mitochondrial coupling promotes mitochondrial respiration and bioenergetics during early phases of ER stress. J. Cell Sci. 2011, 124, 2143–2152. [Google Scholar] [CrossRef] [Green Version]
- Almanza, A.; Carlesso, A.; Chintha, C.; Creedican, S.; Doultsinos, D.; Leuzzi, B.; Luís, A.; McCarthy, N.; Montibeller, L.; More, S.; et al. Endoplasmic reticulum stress signalling—From basic mechanisms to clinical applications. FEBS J. 2019, 286, 241–278. [Google Scholar] [CrossRef]
- Schwarz, D.S.; Blower, M.D. The endoplasmic reticulum: Structure, function and response to cellular signaling. Cell. Mol. Life Sci. 2016, 73, 79–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, C.J.; Kopp, M.C.; Larburu, N.; Nowak, P.R.; Ali, M.M.U. Structure and Molecular Mechanism of ER Stress Signaling by the Unfolded Protein Response Signal Activator IRE1. Front. Mol. Biosci. 2019, 6, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huber, R.; Hof, P.; Duarte, R.O.; Moura, J.J.; Moura, I.; Liu, M.Y.; LeGall, J.; Hille, R.; Archer, M.; Romão, M.J. A structure-based catalytic mechanism for the xanthine oxidase family of molybdenum enzymes. Proc. Natl. Acad. Sci. USA 1996, 93, 8846–8851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ives, A.; Nomura, J.; Martinon, F.; Roger, T.; LeRoy, D.; Miner, J.N.; Simon, G.; Busso, N.; So, A. Xanthine oxidoreductase regulates macrophage IL1β secretion upon NLRP3 inflammasome activation. Nat. Commun. 2015, 6, 6555. [Google Scholar] [CrossRef] [Green Version]
- Bredemeier, M.; Lopes, L.M.; Eisenreich, M.A.; Hickmann, S.; Bongiorno, G.K.; d’Avila, R.; Morsch, A.L.B.; da Silva Stein, F.; Campos, G.G.D. Xanthine oxidase inhibitors for prevention of cardiovascular events: A systematic review and meta-analysis of randomized controlled trials. BMC Cardiovasc. Disord. 2018, 18, 24. [Google Scholar] [CrossRef] [PubMed]
- Osarogiagbon, U.R.; Choong, S.; Belcher, J.D.; Vercellotti, G.M.; Paller, M.S.; Hebbel, R.P. Reperfusion injury pathophysiology in sickle transgenic mice. Blood 2000, 96, 314–320. [Google Scholar] [CrossRef] [PubMed]
- Ndrepepa, G. Myeloperoxidase—A bridge linking inflammation and oxidative stress with cardiovascular disease. Clin. Chim. Acta 2019, 493, 36–51. [Google Scholar] [CrossRef]
- Wang, Z.; Nicholls, S.J.; Rodriguez, E.R.; Kummu, O.; Hörkkö, S.; Barnard, J.; Reynolds, W.F.; Topol, E.J.; DiDonato, J.A.; Hazen, S.L. Protein carbamylation links inflammation, smoking, uremia and atherogenesis. Nat. Med. 2007, 13, 1176–1184. [Google Scholar] [CrossRef]
- Anatoliotakis, N.; Deftereos, S.; Bouras, G.; Giannopoulos, G.; Tsounis, D.; Angelidis, C.; Kaoukis, A.; Stefanadis, C. Myeloperoxidase: Expressing Inflammation and Oxidative Stress in Cardiovascular Disease. Curr. Top. Med. Chem. 2013, 13, 115–138. [Google Scholar] [CrossRef]
- Lehners, A.; Lange, S.; Niemann, G.; Rosendahl, A.; Meyer-Schwesinger, C.; Oh, J.; Stahl, R.; Ehmke, H.; Benndorf, R.; Klinke, A.; et al. Myeloperoxidase deficiency ameliorates progression of chronic kidney disease in mice. Am. J. Physiol.-Ren. Physiol. 2014, 307, F407–F417. [Google Scholar] [CrossRef]
- Haegens, A.; van der Vliet, A.; Butnor, K.J.; Heintz, N.; Taatjes, D.; Hemenway, D.; Vacek, P.; Freeman, B.A.; Hazen, S.L.; Brennan, M.L.; et al. Asbestos-Induced Lung Inflammation and Epithelial Cell Proliferation Are Altered in Myeloperoxidase-Null Mice. Cancer Res. 2005, 65, 9670–9677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lowry, J.L.; Brovkovych, V.; Zhang, Y.; Skidgel, R.A. Endothelial Nitric-oxide Synthase Activation Generates an Inducible Nitric-oxide Synthase-like Output of Nitric Oxide in Inflamed Endothelium. J. Biol. Chem. 2013, 288, 4174–4193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Förstermann, U.; Münzel, T. Endothelial Nitric Oxide Synthase in Vascular Disease: From Marvel to Menace. Circulation 2006, 113, 1708–1714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Jager, T.L.; Cockrell, A.E.; Du Plessis, S.S. Ultraviolet Light Induced Generation of Reactive Oxygen Species. In Ultraviolet Light in Human Health, Diseases and Environment; Ahmad, S.I., Ed.; Advances in Experimental Medicine and Biology; Springer International Publishing: Cham, Germany, 2017; Volume 996, pp. 15–23. ISBN 978-3-319-56016-8. [Google Scholar]
- Holman, D.M.; Ding, H.; Guy, G.P.; Watson, M.; Hartman, A.M.; Perna, F.M. Prevalence of Sun Protection Use and Sunburn and Association of Demographic and Behaviorial Characteristics With Sunburn Among US Adults. JAMA Dermatol. 2018, 154, 561. [Google Scholar] [CrossRef]
- Young, A.R.; Narbutt, J.; Harrison, G.I.; Lawrence, K.P.; Bell, M.; O’Connor, C.; Olsen, P.; Grys, K.; Baczynska, K.A.; Rogowski-Tylman, M.; et al. Optimal sunscreen use, during a sun holiday with a very high ultraviolet index, allows vitamin D synthesis without sunburn. Br. J. Dermatol. 2019, 181, 1052–1062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hussein, A.; Elhassaneen, Y. Natural dye from red onion skins and applied in dyeing cotton fabrics for the production of women’s headwear resistance to ultraviolet radiation (UVR). J. Am. Sci. 2014, 10, 129–139. [Google Scholar]
- Bergman, R.S. Germicidal UV Sources and Systems. Photochem. Photobiol. 2021, 97, 466–470. [Google Scholar] [CrossRef]
- Kimeswenger, S.; Schwarz, A.; Födinger, D.; Müller, S.; Pehamberger, H.; Schwarz, T.; Jantschitsch, C. Infrared A radiation promotes survival of human melanocytes carrying ultraviolet radiation-induced DNA damage. Exp. Dermatol. 2016, 25, 447–452. [Google Scholar] [CrossRef] [Green Version]
- Heck, D.E.; Vetrano, A.M.; Mariano, T.M.; Laskin, J.D. UVB Light Stimulates Production of Reactive Oxygen Species. J. Biol. Chem. 2003, 278, 22432–22436. [Google Scholar] [CrossRef] [Green Version]
- Deliconstantinos, G.; Villiotou, V.; Stavrides, J.C. Alterations of nitric oxide synthase and xanthine oxidase activities of human keratinocytes by ultraviolet B radiation. Biochem. Pharmacol. 1996, 51, 1727–1738. [Google Scholar] [CrossRef]
- Bossi, O.; Gartsbein, M.; Leitges, M.; Kuroki, T.; Grossman, S.; Tennenbaum, T. UV irradiation increases ROS production via PKCδ signaling in primary murine fibroblasts. J. Cell. Biochem. 2008, 105, 194–207. [Google Scholar] [CrossRef]
- Wei, H.; Cai, Q.; Rahn, R.; Zhang, X. Singlet Oxygen Involvement in Ultraviolet (254 nm) Radiation-Induced Formation of 8-Hydroxy-Deoxyguanosine in DNA. Free Radic. Biol. Med. 1997, 23, 148–154. [Google Scholar] [CrossRef]
- Ray, A.J.; Turner, R.; Nikaido, O.; Rees, J.L.; Birch-Machin, M.A. The Spectrum of Mitochondrial DNA Deletions is a Ubiquitous Marker of Ultraviolet Radiation Exposure in Human Skin. J. Invest. Dermatol. 2000, 115, 674–679. [Google Scholar] [CrossRef] [Green Version]
- Bernard, J.J.; Gallo, R.L.; Krutmann, J. Photoimmunology: How ultraviolet radiation affects the immune system. Nat. Rev. Immunol. 2019, 19, 688–701. [Google Scholar] [CrossRef] [PubMed]
- Bouillaguet, S.; Owen, B.; Wataha, J.C.; Campo, M.A.; Lange, N.; Schrenzel, J. Intracellular reactive oxygen species in monocytes generated by photosensitive chromophores activated with blue light. Dent. Mater. 2008, 24, 1070–1076. [Google Scholar] [CrossRef] [PubMed]
- Prasad, A. Ultraweak photon emission induced by visible light and ultraviolet A radiation via photoactivated skin chromophores: In vivo charge coupled device imaging. J. Biomed. Opt. 2012, 17, 085004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boo, Y.C. Emerging Strategies to Protect the Skin from Ultraviolet Rays Using Plant-Derived Materials. Antioxidants 2020, 9, 637. [Google Scholar] [CrossRef]
- Bickers, D.R.; Athar, M. Oxidative Stress in the Pathogenesis of Skin Disease. J. Invest. Dermatol. 2006, 126, 2565–2575. [Google Scholar] [CrossRef] [Green Version]
- Reelfs, O.; Tyrrell, R.M.; Pourzand, C. Ultraviolet A Radiation-Induced Immediate Iron Release Is a Key Modulator of the Activation of NF-κB in Human Skin Fibroblasts. J. Invest. Dermatol. 2004, 122, 1440–1447. [Google Scholar] [CrossRef] [PubMed]
- Bassez, M.-P. Water, Air, Earth and Cosmic Radiation. Orig. Life Evol. Biosph. 2015, 45, 5–13. [Google Scholar] [CrossRef]
- Indriolo, N.; Neufeld, D.A.; Gerin, M.; Schilke, P.; Benz, A.O.; Winkel, B.; Menten, K.M.; Chambers, E.T.; Black, J.H.; Bruderer, S.; et al. Herschel Survey of Galactic OH+, H2O+, and H3O+: Probing the Molecular Hydrogen Fraction and Cosmic-Ray Ionization Rate. Astrophys. J. 2015, 800, 40. [Google Scholar] [CrossRef]
- Zdrojewicz, Z.; Szlagor, A.; Wielogórska, M.; Nowakowska, D.; Nowakowski, J. Influence of ionizing radiation on human body. Fam. Med. Prim. Care Rev. 2016, 2, 174–179. [Google Scholar] [CrossRef] [Green Version]
- Alizadeh, E.; Orlando, T.M.; Sanche, L. Biomolecular Damage Induced by Ionizing Radiation: The Direct and Indirect Effects of Low-Energy Electrons on DNA. Annu. Rev. Phys. Chem. 2015, 66, 379–398. [Google Scholar] [CrossRef] [PubMed]
- ICNIRP. Principles for Non-Ionizing Radiation Protection. Health Phys. 2020, 118, 477–482. [Google Scholar] [CrossRef]
- Kirsch, D.G.; Diehn, M.; Kesarwala, A.H.; Maity, A.; Morgan, M.A.; Schwarz, J.K.; Bristow, R.; Demaria, S.; Eke, I.; Griffin, R.J.; et al. The Future of Radiobiology. JNCI J. Natl. Cancer Inst. 2018, 110, 329–340. [Google Scholar] [CrossRef]
- Cui, F.; Ma, N.; Han, X.; Chen, N.; Xi, Y.; Yuan, W.; Xu, Y.; Han, J.; Xu, X.; Tu, Y. Effects of 60Co γ Irradiation on the Reproductive Function of Caenorhabditis elegans. Dose-Response 2019, 17, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santacruz-Gomez, K.; Sarabia-Sainz, A.; Acosta-Elias, M.; Sarabia-Sainz, M.; Janetanakit, W.; Khosla, N.; Melendrez, R.; Montero, M.P.; Lal, R. Antioxidant activity of hydrated carboxylated nanodiamonds and its influence on water γ -radiolysis. Nanotechnology 2018, 29, 125707. [Google Scholar] [CrossRef] [PubMed]
- Rai, Y.; Anita; Kumari, N.; Singh, S.; Kalra, N.; Soni, R.; Bhatt, A.N. Mild mitochondrial uncoupling protects from ionizing radiation induced cell death by attenuating oxidative stress and mitochondrial damage. Biochim. Biophys. Acta BBA-Bioenerg. 2021, 1862, 148325. [Google Scholar] [CrossRef]
- Einor, D.; Bonisoli-Alquati, A.; Costantini, D.; Mousseau, T.A.; Møller, A.P. Ionizing radiation, antioxidant response and oxidative damage: A meta-analysis. Sci. Total Environ. 2016, 548–549, 463–471. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.A.; Yoon, S.H.; Rho, J.K.; Jang, B.; Choi, D.S.; Lee, D.-E.; Byun, E.-B.; Jeon, J.; Park, S.H. Radioprotective effect of hesperetin against γ-irradiation-induced DNA damage and immune dysfunction in murine splenocytes. Food Sci. Biotechnol. 2016, 25, 163–168. [Google Scholar] [CrossRef]
- Rezaeyan, A.; Haddadi, G.; Hosseinzadeh, M.; Moradi, M.; Najafi, M. Radioprotective effects of hesperidin on oxidative damages and histopathological changes induced by X-irradiation in rats heart tissue. J. Med. Phys. 2016, 41, 182. [Google Scholar] [CrossRef]
- Shaban, N.Z.; Ahmed Zahran, A.M.; El-Rashidy, F.H.; Abdo Kodous, A.S. Protective role of hesperidin against γ-radiation-induced oxidative stress and apoptosis in rat testis. J. Biol. Res.-Thessalon. 2017, 24, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sage, E.; Shikazono, N. Radiation-induced clustered DNA lesions: Repair and mutagenesis. Free Radic. Biol. Med. 2017, 107, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Sylvester, C.B.; Abe, J.; Patel, Z.S.; Grande-Allen, K.J. Radiation-Induced Cardiovascular Disease: Mechanisms and Importance of Linear Energy Transfer. Front. Cardiovasc. Med. 2018, 5, 5. [Google Scholar] [CrossRef] [Green Version]
- Helm, J.S.; Rudel, R.A. Adverse outcome pathways for ionizing radiation and breast cancer involve direct and indirect DNA damage, oxidative stress, inflammation, genomic instability, and interaction with hormonal regulation of the breast. Arch. Toxicol. 2020, 94, 1511–1549. [Google Scholar] [CrossRef] [PubMed]
- Doukali, H.; Ben Salah, G.; Hamdaoui, L.; Hajjaji, M.; Tabebi, M.; Ammar-Keskes, L.; Masmoudi, M.-E.; Kamoun, H. Oxidative stress and glutathione S-transferase genetic polymorphisms in medical staff professionally exposed to ionizing radiation. Int. J. Radiat. Biol. 2017, 93, 697–704. [Google Scholar] [CrossRef] [PubMed]
- Schonfeld, S.J.; Bhatti, P.; Brown, E.E.; Linet, M.S.; Simon, S.L.; Weinstock, R.M.; Hutchinson, A.A.; Stovall, M.; Preston, D.L.; Alexander, B.H.; et al. Polymorphisms in oxidative stress and inflammation pathway genes, low-dose ionizing radiation, and the risk of breast cancer among US radiologic technologists. Cancer Causes Control 2010, 21, 1857–1866. [Google Scholar] [CrossRef] [Green Version]
- Møller, A.P.; Mousseau, T.A. Strong effects of ionizing radiation from Chernobyl on mutation rates. Sci. Rep. 2015, 5, 8363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belli, M.; Tabocchini, M.A. Ionizing Radiation-Induced Epigenetic Modifications and Their Relevance to Radiation Protection. Int. J. Mol. Sci. 2020, 21, 5993. [Google Scholar] [CrossRef]
- Su, S.; Jin, Y.; Zhang, W.; Yang, L.; Shen, Y.; Cao, Y.; Tong, J. Aberrant Promoter Methylation of p16 INK4a and O 6 -Methylguanine-DNA Methyltransferase Genes in Workers at a Chinese Uranium Mine. J. Occup. Health 2006, 48, 261–266. [Google Scholar] [CrossRef] [Green Version]
- Romanenko, A.; Morell-Quadreny, L.; Lopez-Guerrero, J.A.; Pellin, A.; Nepomnyaschy, V.; Vozianov, A.; Llombart-Bosch, A. The INK4a/ARF locus: Role in cell cycle control for renal cell epithelial tumor growth after the Chernobyl accident. Virchows Arch. 2004, 445, 298–304. [Google Scholar] [CrossRef]
- Szumiel, I. Ionizing radiation-induced oxidative stress, epigenetic changes and genomic instability: The pivotal role of mitochondria. Int. J. Radiat. Biol. 2015, 91, 1–12. [Google Scholar] [CrossRef]
- Zheng, F.; Gonçalves, F.M.; Abiko, Y.; Li, H.; Kumagai, Y.; Aschner, M. Redox toxicology of environmental chemicals causing oxidative stress. Redox Biol. 2020, 34, 101475. [Google Scholar] [CrossRef]
- Sun, H.; He, F.; Choi, W. Production of Reactive Oxygen Species by the Reaction of Periodate and Hydroxylamine for Rapid Removal of Organic Pollutants and Waterborne Bacteria. Environ. Sci. Technol. 2020, 54, 6427–6437. [Google Scholar] [CrossRef]
- Lelieveld, S.; Wilson, J.; Dovrou, E.; Mishra, A.; Lakey, P.S.J.; Shiraiwa, M.; Pöschl, U.; Berkemeier, T. Hydroxyl Radical Production by Air Pollutants in Epithelial Lining Fluid Governed by Interconversion and Scavenging of Reactive Oxygen Species. Environ. Sci. Technol. 2021, 55, 14069–14079. [Google Scholar] [CrossRef]
- Paithankar, J.G.; Saini, S.; Dwivedi, S.; Sharma, A.; Chowdhuri, D.K. Heavy metal associated health hazards: An interplay of oxidative stress and signal transduction. Chemosphere 2021, 262, 128350. [Google Scholar] [CrossRef]
- Balali-Mood, M.; Naseri, K.; Tahergorabi, Z.; Khazdair, M.R.; Sadeghi, M. Toxic Mechanisms of Five Heavy Metals: Mercury, Lead, Chromium, Cadmium, and Arsenic. Front. Pharmacol. 2021, 12, 643972. [Google Scholar] [CrossRef]
- Exley, C. Aluminum in Biological Systems. In Encyclopedia of Metalloproteins; Kretsinger, R.H., Uversky, V.N., Permyakov, E.A., Eds.; Springer New York: New York, NY, USA, 2013; pp. 33–34. ISBN 978-1-4614-1532-9. [Google Scholar]
- Hossain, M.A.; Piyatida, P.; da Silva, J.A.T.; Fujita, M. Molecular Mechanism of Heavy Metal Toxicity and Tolerance in Plants: Central Role of Glutathione in Detoxification of Reactive Oxygen Species and Methylglyoxal and in Heavy Metal Chelation. J. Bot. 2012, 2012, 1–37. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Wang, Y.; Luo, N.; Chen, Z.; Wu, K.; Yin, G. Redox inactive metal ion triggered N-dealkylation by an iron catalyst with dioxygen activation: A lesson from lipoxygenases. Dalton Trans. 2015, 44, 9847–9859. [Google Scholar] [CrossRef]
- Niu, P.Y.; Niu, Q.; Zhang, Q.L.; Wang, L.P.; He, S.C.; Wu, T.C.; Conti, P.; Di Gioacchino, M.; Boscolo, P. Aluminum Impairs Rat Neural Cell Mitochondria In Vitro. Int. J. Immunopathol. Pharmacol. 2005, 18, 683–689. [Google Scholar] [CrossRef] [Green Version]
- Wei, W.; Smith, N.; Wu, X.; Kim, H.; Seravalli, J.; Khalimonchuk, O.; Lee, J. YCF1-Mediated Cadmium Resistance in Yeast Is Dependent on Copper Metabolism and Antioxidant Enzymes. Antioxid. Redox Signal. 2014, 21, 1475–1489. [Google Scholar] [CrossRef]
- Saito, K.; Nakagawa, M.; Mandal, M.; Ishikita, H. Role of redox-inactive metals in controlling the redox potential of heterometallic manganese–oxido clusters. Photosynth. Res. 2021, 148, 153–159. [Google Scholar] [CrossRef]
- Jayaraj, R.; Megha, P.; Sreedev, P. Review Article. Organochlorine pesticides, their toxic effects on living organisms and their fate in the environment. Interdiscip. Toxicol. 2016, 9, 90–100. [Google Scholar] [CrossRef] [Green Version]
- Georgiadis, N.; Tsarouhas, K.; Tsitsimpikou, C.; Vardavas, A.; Rezaee, R.; Germanakis, I.; Tsatsakis, A.; Stagos, D.; Kouretas, D. Pesticides and cardiotoxicity. Where do we stand? Toxicol. Appl. Pharmacol. 2018, 353, 1–14. [Google Scholar] [CrossRef]
- Litchfield, M.H. Estimates of Acute Pesticide Poisoning in Agricultural Workers in Less Developed Countries: Toxicol. Rev. 2005, 24, 271–278. [Google Scholar] [CrossRef]
- Dou, T.; Yan, M.; Wang, X.; Lu, W.; Zhao, L.; Lou, D.; Wu, C.; Chang, X.; Zhou, Z. Nrf2/ARE Pathway Involved in Oxidative Stress Induced by Paraquat in Human Neural Progenitor Cells. Oxid. Med. Cell Longev. 2016, 2016, 8923860. [Google Scholar] [CrossRef] [Green Version]
- Ranjbar, A.; Pasalar, P.; Sedighi, A.; Abdollahi, M. Induction of oxidative stress in paraquat formulating workers. Toxicol. Lett. 2002, 131, 191–194. [Google Scholar] [CrossRef]
- Abass, K.; Lämsä, V.; Reponen, P.; Küblbeck, J.; Honkakoski, P.; Mattila, S.; Pelkonen, O.; Hakkola, J. Characterization of human cytochrome P450 induction by pesticides. Toxicology 2012, 294, 17–26. [Google Scholar] [CrossRef]
- Delescluse, C.; Ledirac, N.; Li, R.; Piechocki, M.P.; Hines, R.N.; Gidrol, X.; Rahmani, R. Induction of cytochrome P450 1A1 gene expression, oxidative stress, and genotoxicity by carbaryl and thiabendazole in transfected human HepG2 and lymphoblastoid cells. Biochem. Pharmacol. 2001, 61, 399–407. [Google Scholar] [CrossRef]
- Puntarulo, S.; Cederbaum, A.I. Production of Reactive Oxygen Species by Microsomes Enriched in Specific Human Cytochrome P450 Enzymes. Free Radic. Biol. Med. 1998, 24, 1324–1330. [Google Scholar] [CrossRef]
- Veith, A.; Moorthy, B. Role of cytochrome P450s in the generation and metabolism of reactive oxygen species. Curr. Opin. Toxicol. 2018, 7, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Prakasam, A.; Sethupathy, S.; Lalitha, S. Plasma and RBCs antioxidant status in occupational male pesticide sprayers. Clin. Chim. Acta 2001, 310, 107–112. [Google Scholar] [CrossRef]
- Sule, R.O.; Condon, L.; Gomes, A.V. A Common Feature of Pesticides: Oxidative Stress—The Role of Oxidative Stress in Pesticide-Induced Toxicity. Oxid. Med. Cell. Longev. 2022, 2022, 5563759. [Google Scholar] [CrossRef] [PubMed]
- Carloni, M.; Nasuti, C.; Fedeli, D.; Montani, M.; Vadhana, M.S.D.; Amici, A.; Gabbianelli, R. Early life permethrin exposure induces long-term brain changes in Nurr1, NF-kB and Nrf-2. Brain Res. 2013, 1515, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Izumi, Y.; Yamamoto, N.; Matsushima, S.; Yamamoto, T.; Takada-Takatori, Y.; Akaike, A.; Kume, T. Compensatory role of the Nrf2–ARE pathway against paraquat toxicity: Relevance of 26S proteasome activity. J. Pharmacol. Sci. 2015, 129, 150–159. [Google Scholar] [CrossRef]
- Saito, R.; Suzuki, T.; Hiramoto, K.; Asami, S.; Naganuma, E.; Suda, H.; Iso, T.; Yamamoto, H.; Morita, M.; Baird, L.; et al. Characterizations of Three Major Cysteine Sensors of Keap1 in Stress Response. Mol. Cell Biol. 2015, 36, 271–284. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, T.; Muramatsu, A.; Saito, R.; Iso, T.; Shibata, T.; Kuwata, K.; Kawaguchi, S.; Iwawaki, T.; Adachi, S.; Suda, H.; et al. Molecular Mechanism of Cellular Oxidative Stress Sensing by Keap1. Cell Rep. 2019, 28, 746–758.e4. [Google Scholar] [CrossRef] [Green Version]
- Kansanen, E.; Kuosmanen, S.M.; Leinonen, H.; Levonen, A.-L. The Keap1-Nrf2 pathway: Mechanisms of activation and dysregulation in cancer. Redox Biol. 2013, 1, 45–49. [Google Scholar] [CrossRef] [Green Version]
- Gangwar, R.S.; Bevan, G.H.; Palanivel, R.; Das, L.; Rajagopalan, S. Oxidative stress pathways of air pollution mediated toxicity: Recent insights. Redox Biol. 2020, 34, 101545. [Google Scholar] [CrossRef]
- Mukherjee, A.; Agrawal, M. World air particulate matter: Sources, distribution and health effects. Environ. Chem. Lett. 2017, 15, 283–309. [Google Scholar] [CrossRef]
- Li, N.; Sioutas, C.; Cho, A.; Schmitz, D.; Misra, C.; Sempf, J.; Wang, M.; Oberley, T.; Froines, J.; Nel, A. Ultrafine particulate pollutants induce oxidative stress and mitochondrial damage. Environ. Health Perspect. 2003, 111, 455–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazuryk, O.; Stochel, G.; Brindell, M. Variations in Reactive Oxygen Species Generation by Urban Airborne Particulate Matter in Lung Epithelial Cells—Impact of Inorganic Fraction. Front. Chem. 2020, 8, 581752. [Google Scholar] [CrossRef] [PubMed]
- Fagundes, L.S.; Fleck, A.d.S.; Zanchi, A.C.; Saldiva, P.H.N.; Rhoden, C.R. Direct contact with particulate matter increases oxidative stress in different brain structures. Inhal. Toxicol. 2015, 27, 462–467. [Google Scholar] [CrossRef] [PubMed]
- Lawal, A.O. Air particulate matter induced oxidative stress and inflammation in cardiovascular disease and atherosclerosis: The role of Nrf2 and AhR-mediated pathways. Toxicol. Lett. 2017, 270, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Görlach, A.; Dimova, E.Y.; Petry, A.; Martínez-Ruiz, A.; Hernansanz-Agustín, P.; Rolo, A.P.; Palmeira, C.M.; Kietzmann, T. Reactive oxygen species, nutrition, hypoxia and diseases: Problems solved? Redox Biol. 2015, 6, 372–385. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, T.; Li, M.; Shi, D.; Tan, X.; Qiu, F. Lycopene attenuates d -galactose-induced insulin signaling impairment by enhancing mitochondrial function and suppressing the oxidative stress/inflammatory response in mouse kidneys and livers. Food Funct. 2022, 13, 7720–7729. [Google Scholar] [CrossRef]
- Ference, B.A.; Ginsberg, H.N.; Graham, I.; Ray, K.K.; Packard, C.J.; Bruckert, E.; Hegele, R.A.; Krauss, R.M.; Raal, F.J.; Schunkert, H.; et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease. 1. Evidence from genetic, epidemiologic, and clinical studies. A consensus statement from the European Atherosclerosis Society Consensus Panel. Eur. Heart J. 2017, 38, 2459–2472. [Google Scholar] [CrossRef] [Green Version]
- Motamed, M.; Nargesi, A.; Heidari, B.; Mirmiranpour, H.; Esteghamati, A.; Nakhjavani, M. Oxidized Low-Density Lipoprotein (ox-LDL) to LDL Ratio (ox-LDL/LDL) and ox-LDL to High-Density Lipoprotein Ratio (ox-LDL/HDL). Clin. Lab. 2016, 62, 1609–1617. [Google Scholar] [CrossRef]
- Dalvi, P.S.; Chalmers, J.A.; Luo, V.; Han, D.-Y.; Wellhauser, L.; Liu, Y.; Tran, D.Q.; Castel, J.; Luquet, S.; Wheeler, M.B.; et al. High fat induces acute and chronic inflammation in the hypothalamus: Effect of high-fat diet, palmitate and TNF-α on appetite-regulating NPY neurons. Int. J. Obes. 2017, 41, 149–158. [Google Scholar] [CrossRef]
- Tapias, V.; Hu, X.; Luk, K.C.; Sanders, L.H.; Lee, V.M.; Greenamyre, J.T. Synthetic alpha-synuclein fibrils cause mitochondrial impairment and selective dopamine neurodegeneration in part via iNOS-mediated nitric oxide production. Cell Mol. Life Sci. 2017, 74, 2851–2874. [Google Scholar] [CrossRef]
- Jiang, S.; Liu, H.; Li, C. Dietary Regulation of Oxidative Stress in Chronic Metabolic Diseases. Foods 2021, 10, 1854. [Google Scholar] [CrossRef] [PubMed]
- Dguzeh, U.; Haddad, N.; Smith, K.; Johnson, J.; Doye, A.; Gwathmey, J.; Haddad, G. Alcoholism: A Multi-Systemic Cellular Insult to Organs. Int. J. Environ. Res. Public. Health 2018, 15, 1083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, J.; Liu, Y.; Chang, X.; Gou, W.; Zhou, X.; Liu, Z.; Li, Z.; Wu, Y.; Zuo, D. Acetaldehyde Induces Neurotoxicity In Vitro via Oxidative Stress- and Ca 2+ Imbalance-Mediated Endoplasmic Reticulum Stress. Oxid. Med. Cell Longev. 2019, 2019, 2593742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, D.; Cederbaum, A.I. Alcohol, Oxidative Stress, and Free Radical Damage. Alcohol Res. Health 2003, 27, 277–284. [Google Scholar] [PubMed]
- Baker, R.R.; Massey, E.D.; Smith, G. An overview of the effects of tobacco ingredients on smoke chemistry and toxicity. Food Chem. Toxicol. 2004, 42, 53–83. [Google Scholar] [CrossRef] [PubMed]
- Lakshmi, S.P.; Reddy, A.T.; Kodidhela, L.D.; Varadacharyulu, N.C. Epigallocatechin gallate diminishes cigarette smoke-induced oxidative stress, lipid peroxidation, and inflammation in human bronchial epithelial cells. Life Sci. 2020, 259, 118260. [Google Scholar] [CrossRef] [PubMed]
- Deavall, D.G.; Martin, E.A.; Horner, J.M.; Roberts, R. Drug-Induced Oxidative Stress and Toxicity. J. Toxicol. 2012, 2012, 645460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chignell, C.F.; Motten, A.G.; Buettner, G.R. Photoinduced free radicals from chlorpromazine and related phenothiazines: Relationship to phenothiazine-induced photosensitization. Environ. Health Perspect. 1985, 64, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Davies, K.J.; Doroshow, J.H. Redox cycling of anthracyclines by cardiac mitochondria. I. Anthracycline radical formation by NADH dehydrogenase. J. Biol. Chem. 1986, 261, 3060–3067. [Google Scholar] [CrossRef]
- Micallef, I.; Baron, B. Doxorubicin: An Overview of the Anti-Cancer and Chemoresistance Mechanisms. Ann. Clin. Toxicol. 2020, 3, 12. [Google Scholar]
- Miyake, M.; Nakai, Y.; Hori, S.; Morizawa, Y.; Hirao, Y.; Fujimoto, K. Transient liver toxicity as a result of the oral administration of 5-aminolevulinic acid for photodynamic diagnosis in patients with bladder cancer. Int. J. Urol. 2019, 26, 315–317. [Google Scholar] [CrossRef] [Green Version]
- Sánchez-Borges, M.; Capriles-Hulett, A.; Caballero-Fonseca, F. Risk of skin reactions when using ibuprofen-based medicines. Expert Opin. Drug Saf. 2005, 4, 837–848. [Google Scholar] [CrossRef]
- Bagheri, H.; Lhiaubet, V.; Montastruc, J.L.; Chouini-Lalanne, N. Photosensitivity to Ketoprofen: Mechanisms and Pharmacoepidemiological Data. Drug Saf. 2000, 22, 339–349. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, I.; Persson, E.; Ekebergh, A.; Mårtensson, J.; Börje, A. Ketoprofen-Induced Formation of Amino Acid Photoadducts: Possible Explanation for Photocontact Allergy to Ketoprofen. Chem. Res. Toxicol. 2014, 27, 1294–1303. [Google Scholar] [CrossRef]
- Onoue, S.; Igarashi, N.; Yamada, S.; Tsuda, Y. High-throughput reactive oxygen species (ROS) assay: An enabling technology for screening the phototoxic potential of pharmaceutical substances. J. Pharm. Biomed. Anal. 2008, 46, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Akat, P. Severe photosensitivity reaction induced by topical diclofenac. Indian J. Pharmacol. 2013, 45, 408. [Google Scholar] [CrossRef]
- Lhiaubet, V.; Paillous, N.; Chouini-Lalanne, N. Comparison of DNA Damage Photoinduced by Ketoprofen, Fenofibric Acid and Benzophenone via Electron and Energy Transfer. Photochem. Photobiol. 2007, 74, 670–678. [Google Scholar] [CrossRef]
- Kowalska, J.; Rok, J.; Rzepka, Z.; Wrześniok, D. Drug-Induced Photosensitivity—From Light and Chemistry to Biological Reactions and Clinical Symptoms. Pharmaceuticals 2021, 14, 723. [Google Scholar] [CrossRef] [PubMed]
- Köroğlu, K.M.; Çevik, Ö.; Şener, G.; Ercan, F. Apocynin alleviates cisplatin-induced testicular cytotoxicity by regulating oxidative stress and apoptosis in rats. Andrologia 2019, 51, e13227. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Yan, M.-H.; Liu, Y.; Liu, Z.; Wang, Z.; Chen, C.; Zhang, J.; Sun, Y.-S. Ginsenoside Rg5 Ameliorates Cisplatin-Induced Nephrotoxicity in Mice through Inhibition of Inflammation, Oxidative Stress, and Apoptosis. Nutrients 2016, 8, 566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorini, S.; De Angelis, A.; Berrino, L.; Malara, N.; Rosano, G.; Ferraro, E. Chemotherapeutic Drugs and Mitochondrial Dysfunction: Focus on Doxorubicin, Trastuzumab, and Sunitinib. Oxid. Med. Cell Longev. 2018, 2018, 7582730. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, R.; Hwang, S.M.; Cui, Z.; Gilda, J.E.; Gomes, A.V. Different effects of the nonsteroidal anti-inflammatory drugs meclofenamate sodium and naproxen sodium on proteasome activity in cardiac cells. J. Mol. Cell Cardiol. 2016, 94, 131–144. [Google Scholar] [CrossRef]
- Щекіна, К.Г.; Belik, G.; Semeniv, D.; Ulanova, V. A comparative study of the hepatotrophic properties of non-steroidal anti-inflammatory drugs. Ukr. Biopharm. J. 2021, 66, 36–41. [Google Scholar] [CrossRef]
- Martens, M.E.; Lee, C.-P. Reye’s syndrome: Salicylates and mitochondrial functions. Biochem. Pharmacol. 1984, 33, 2869–2876. [Google Scholar] [CrossRef]
- Jiang, J.; Briedé, J.J.; Jennen, D.G.J.; Van Summeren, A.; Saritas-Brauers, K.; Schaart, G.; Kleinjans, J.C.S.; de Kok, T.M.C.M. Increased mitochondrial ROS formation by acetaminophen in human hepatic cells is associated with gene expression changes suggesting disruption of the mitochondrial electron transport chain. Toxicol. Lett. 2015, 234, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Bellomo, R. The epidemiology of acute renal failure: 1975 versus 2005: Curr. Opin. Crit. Care 2006, 12, 557–560. [Google Scholar] [CrossRef] [PubMed]
- Ghane Shahrbaf, F. Drug-induced renal disorders. Drug-Induc. Ren. Disord. 2015, 4, 57–60. [Google Scholar] [CrossRef]
- Perneger, T.V.; Whelton, P.K.; Klag, M.J. Risk of Kidney Failure Associated with the Use of Acetaminophen, Aspirin, and Nonsteroidal Antiinflammatory Drugs. N. Engl. J. Med. 1994, 331, 1675–1679. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.; Chen, J.; Kuo, C.; Pai, P.; Ho, T.; Chen, T.; Tsai, F.; Padma, V.V.; Kuo, W.; Huang, C. Mitochondrial ROS-induced ERK1/2 activation and HSF2-mediated AT 1 R upregulation are required for doxorubicin-induced cardiotoxicity. J. Cell Physiol. 2018, 233, 463–475. [Google Scholar] [CrossRef]
- Forman, H.J.; Zhang, H. Targeting oxidative stress in disease: Promise and limitations of antioxidant therapy. Nat. Rev. Drug Discov. 2021, 20, 689–709. [Google Scholar] [CrossRef] [PubMed]
- Cooke, M.S.; Evans, M.D.; Dizdaroglu, M.; Lunec, J. Oxidative DNA damage: Mechanisms, mutation, and disease. FASEB J. 2003, 17, 1195–1214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chao, M.-R.; Rossner, P.; Haghdoost, S.; Jeng, H.A.; Hu, C.-W. Nucleic Acid Oxidation in Human Health and Disease. Oxid. Med. Cell Longev. 2013, 2013, 368651. [Google Scholar] [CrossRef] [PubMed]
- Dizdaroglu, M.; Jaruga, P. Mechanisms of free radical-induced damage to DNA. Free Radic. Res. 2012, 46, 382–419. [Google Scholar] [CrossRef]
- Gaillard, H.; García-Muse, T.; Aguilera, A. Replication stress and cancer. Nat. Rev. Cancer 2015, 15, 276–289. [Google Scholar] [CrossRef] [PubMed]
- Van Remmen, H.; Hamilton, M.L.; Richardson, A. Oxidative Damage to DNA and Aging. Exerc. Sport Sci. Rev. 2003, 31, 149–153. [Google Scholar] [CrossRef]
- Bruic, M.; Grujic-Milanovic, J.; Miloradovic, Z.; Jovovic, D.; Zivkovic, L.; Mihailovic-Stanojevic, N.; Karanovic, D.; Spremo-Potparevic, B. DNA, protein and lipid oxidative damage in tissues of spontaneously hypertensive versus normotensive rats. Int. J. Biochem. Cell Biol. 2021, 141, 106088. [Google Scholar] [CrossRef]
- Besaratinia, A.; Caliri, A.W.; Tommasi, S. Hydroxychloroquine induces oxidative DNA damage and mutation in mammalian cells. DNA Repair 2021, 106, 103180. [Google Scholar] [CrossRef]
- Fu, Z.; Xi, S. The effects of heavy metals on human metabolism. Toxicol. Mech. Methods 2020, 30, 167–176. [Google Scholar] [CrossRef]
- Kudryavtseva, A.V.; Krasnov, G.S.; Dmitriev, A.A.; Alekseev, B.Y.; Kardymon, O.L.; Sadritdinova, A.F.; Fedorova, M.S.; Pokrovsky, A.V.; Melnikova, N.V.; Kaprin, A.D.; et al. Mitochondrial dysfunction and oxidative stress in aging and cancer. Oncotarget 2016, 7, 44879–44905. [Google Scholar] [CrossRef] [Green Version]
- Gianazza, E.; Brioschi, M.; Martinez Fernandez, A.; Casalnuovo, F.; Altomare, A.; Aldini, G.; Banfi, C. Lipid Peroxidation in Atherosclerotic Cardiovascular Diseases. Antioxid. Redox Signal. 2021, 34, 49–98. [Google Scholar] [CrossRef]
- Ayala, A.; Muñoz, M.F.; Argüelles, S. Lipid Peroxidation: Production, Metabolism, and Signaling Mechanisms of Malondialdehyde and 4-Hydroxy-2-Nonenal. Oxid. Med. Cell Longev. 2014, 2014, 360438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartsch, H.; Nair, J. Chronic inflammation and oxidative stress in the genesis and perpetuation of cancer: Role of lipid peroxidation, DNA damage, and repair. Langenbecks Arch. Surg. 2006, 391, 499–510. [Google Scholar] [CrossRef] [PubMed]
- Spickett, C.M. The lipid peroxidation product 4-hydroxy-2-nonenal: Advances in chemistry and analysis. Redox Biol. 2013, 1, 145–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dyall, S.C.; Balas, L.; Bazan, N.G.; Brenna, J.T.; Chiang, N.; da Costa Souza, F.; Dalli, J.; Durand, T.; Galano, J.-M.; Lein, P.J.; et al. Polyunsaturated fatty acids and fatty acid-derived lipid mediators: Recent advances in the understanding of their biosynthesis, structures, and functions. Prog. Lipid Res. 2022, 86, 101165. [Google Scholar] [CrossRef] [PubMed]
- Vangaveti, V.; Baune, B.T.; Kennedy, R.L. Hydroxyoctadecadienoic acids: Novel regulators of macrophage differentiation and atherogenesis. Ther. Adv. Endocrinol. Metab. 2010, 1, 51–60. [Google Scholar] [CrossRef] [Green Version]
- Schuster, S.; Johnson, C.D.; Hennebelle, M.; Holtmann, T.; Taha, A.Y.; Kirpich, I.A.; Eguchi, A.; Ramsden, C.E.; Papouchado, B.G.; McClain, C.J.; et al. Oxidized linoleic acid metabolites induce liver mitochondrial dysfunction, apoptosis, and NLRP3 activation in mice. J. Lipid Res. 2018, 59, 1597–1609. [Google Scholar] [CrossRef] [Green Version]
- Ramsden, C.E.; Ringel, A.; Feldstein, A.E.; Taha, A.Y.; MacIntosh, B.A.; Hibbeln, J.R.; Majchrzak-Hong, S.F.; Faurot, K.R.; Rapoport, S.I.; Cheon, Y.; et al. Lowering dietary linoleic acid reduces bioactive oxidized linoleic acid metabolites in humans. Prostaglandins Leukot. Essent. Fat. Acids 2012, 87, 135–141. [Google Scholar] [CrossRef] [Green Version]
- Shishehbor, M.H.; Zhang, R.; Medina, H.; Brennan, M.-L.; Brennan, D.M.; Ellis, S.G.; Topol, E.J.; Hazen, S.L. Systemic elevations of free radical oxidation products of arachidonic acid are associated with angiographic evidence of coronary artery disease. Free Radic. Biol. Med. 2006, 41, 1678–1683. [Google Scholar] [CrossRef] [Green Version]
- Mutemberezi, V.; Guillemot-Legris, O.; Muccioli, G.G. Oxysterols: From cholesterol metabolites to key mediators. Prog. Lipid Res. 2016, 64, 152–169. [Google Scholar] [CrossRef]
- Reichert, C.O.; de Freitas, F.A.; Levy, D.; Bydlowski, S.P. Oxysterols and mesenchymal stem cell biology. In Vitamins and Hormones; Elsevier: Amsterdam, The Netherlands, 2021; Volume 116, pp. 409–436. ISBN 978-0-323-85550-1. [Google Scholar]
- Zhang, X.; Alhasani, R.H.; Zhou, X.; Reilly, J.; Zeng, Z.; Strang, N.; Shu, X. Oxysterols and retinal degeneration. Br. J. Pharmacol. 2021, 178, 3205–3219. [Google Scholar] [CrossRef]
- Samadi, A.; Sabuncuoglu, S.; Samadi, M.; Isikhan, S.Y.; Chirumbolo, S.; Peana, M.; Lay, I.; Yalcinkaya, A.; Bjørklund, G. A Comprehensive Review on Oxysterols and Related Diseases. Curr. Med. Chem. 2020, 28, 110–136. [Google Scholar] [CrossRef] [PubMed]
- Angelova, P.R.; Esteras, N.; Abramov, A.Y. Mitochondria and lipid peroxidation in the mechanism of neurodegeneration: Finding ways for prevention. Med. Res. Rev. 2021, 41, 770–784. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Jiang, W.; Wang, W.; Xiong, R.; Wu, X.; Geng, Q. Ferroptosis and its emerging roles in cardiovascular diseases. Pharmacol. Res. 2021, 166, 105466. [Google Scholar] [CrossRef] [PubMed]
- Tudek, B.; Zdżalik-Bielecka, D.; Tudek, A.; Kosicki, K.; Fabisiewicz, A.; Speina, E. Lipid peroxidation in face of DNA damage, DNA repair and other cellular processes. Free Radic. Biol. Med. 2017, 107, 77–89. [Google Scholar] [CrossRef]
- Bayr, H. Reactive oxygen species. Crit. Care Med. 2005, 33, S498. [Google Scholar] [CrossRef]
- Yuan, Q.; Zhu, X.; Sayre, L.M. Chemical nature of stochastic generation of protein-based carbonyls: Metal-catalyzed oxidation versus modification by products of lipid oxidation. Chem. Res. Toxicol. 2007, 20, 129–139. [Google Scholar] [CrossRef]
- Grimsrud, P.A.; Xie, H.; Griffin, T.J.; Bernlohr, D.A. Oxidative Stress and Covalent Modification of Protein with Bioactive Aldehydes. J. Biol. Chem. 2008, 283, 21837–21841. [Google Scholar] [CrossRef] [Green Version]
- Hawkins, C.L.; Davies, M.J. Generation and propagation of radical reactions on proteins. Biochim. Biophys. Acta 2001, 1504, 196–219. [Google Scholar] [CrossRef] [Green Version]
- Hawkins, C.L.; Davies, M.J. Detection, identification, and quantification of oxidative protein modifications. J. Biol. Chem. 2019, 294, 19683–19708. [Google Scholar] [CrossRef] [Green Version]
- Fuentes-Lemus, E.; Hägglund, P.; López-Alarcón, C.; Davies, M.J. Oxidative Crosslinking of Peptides and Proteins: Mechanisms of Formation, Detection, Characterization and Quantification. Molecules 2021, 27, 15. [Google Scholar] [CrossRef]
- Angelova, P.R. Sources and triggers of oxidative damage in neurodegeneration. Free Radic. Biol. Med. 2021, 173, 52–63. [Google Scholar] [CrossRef] [PubMed]
- Starosta, V.; Griese, M. Oxidative damage to surfactant protein D in pulmonary diseases. Free Radic. Res. 2006, 40, 419–425. [Google Scholar] [CrossRef] [PubMed]
- Martínez, A.; Portero-Otin, M.; Pamplona, R.; Ferrer, I. Protein Targets of Oxidative Damage in Human Neurodegenerative Diseases with Abnormal Protein Aggregates. Brain Pathol. 2009, 20, 281–297. [Google Scholar] [CrossRef]
- Krisko, A.; Radman, M. Protein damage, ageing and age-related diseases. Open Biol. 2019, 9, 180249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Godic, A.; Poljšak, B.; Adamic, M.; Dahmane, R. The role of antioxidants in skin cancer prevention and treatment. Oxid. Med. Cell Longev. 2014, 2014, 860479. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Gregorio, A.; Manzo-Merino, J.; Lizano, M. Cellular redox, cancer and human papillomavirus. Virus Res. 2018, 246, 35–45. [Google Scholar] [CrossRef]
- Ighodaro, O.M.; Akinloye, O.A. First line defence antioxidants-superoxide dismutase (SOD), catalase (CAT) and glutathione peroxidase (GPX): Their fundamental role in the entire antioxidant defence grid. Alex. J. Med. 2018, 54, 287–293. [Google Scholar] [CrossRef] [Green Version]
- He, L.; He, T.; Farrar, S.; Ji, L.; Liu, T.; Ma, X. Antioxidants Maintain Cellular Redox Homeostasis by Elimination of Reactive Oxygen Species. Cell Physiol. Biochem. 2017, 44, 532–553. [Google Scholar] [CrossRef]
- Sheng, Y.; Abreu, I.A.; Cabelli, D.E.; Maroney, M.J.; Miller, A.-F.; Teixeira, M.; Valentine, J.S. Superoxide dismutases and superoxide reductases. Chem. Rev. 2014, 114, 3854–3918. [Google Scholar] [CrossRef]
- Robbins, D.; Zhao, Y. Manganese superoxide dismutase in cancer prevention. Antioxid. Redox Signal. 2014, 20, 1628–1645. [Google Scholar] [CrossRef] [Green Version]
- Younus, H. Therapeutic potentials of superoxide dismutase. Int. J. Health Sci. 2018, 12, 88–93. [Google Scholar]
- Castellano, I.; Cecere, F.; De Vendittis, A.; Cotugno, R.; Chambery, A.; Di Maro, A.; Michniewicz, A.; Parlato, G.; Masullo, M.; Avvedimento, E.V.; et al. Rat mitochondrial manganese superoxide dismutase: Amino acid positions involved in covalent modifications, activity, and heat stability. Biopolymers 2009, 91, 1215–1226. [Google Scholar] [CrossRef] [PubMed]
- MacMillan-Crow, L.A.; Crow, J.P.; Kerby, J.D.; Beckman, J.S.; Thompson, J.A. Nitration and inactivation of manganese superoxide dismutase in chronic rejection of human renal allografts. Proc. Natl. Acad. Sci. USA 1996, 93, 11853–11858. [Google Scholar] [CrossRef] [PubMed]
- MacMillan-Crow, L.A.; Cruthirds, D.L.; Ahki, K.M.; Sanders, P.W.; Thompson, J.A. Mitochondrial tyrosine nitration precedes chronic allograft nephropathy. Free Radic. Biol. Med. 2001, 31, 1603–1608. [Google Scholar] [CrossRef]
- MacMillan-Crow, L.A.; Crow, J.P.; Thompson, J.A. Peroxynitrite-Mediated Inactivation of Manganese Superoxide Dismutase Involves Nitration and Oxidation of Critical Tyrosine Residues. Biochemistry 1998, 37, 1613–1622. [Google Scholar] [CrossRef] [PubMed]
- Aparicio-Trejo, O.E.; Reyes-Fermín, L.M.; Briones-Herrera, A.; Tapia, E.; León-Contreras, J.C.; Hernández-Pando, R.; Sánchez-Lozada, L.G.; Pedraza-Chaverri, J. Protective effects of N-acetyl-cysteine in mitochondria bioenergetics, oxidative stress, dynamics and S-glutathionylation alterations in acute kidney damage induced by folic acid. Free Radic. Biol. Med. 2019, 130, 379–396. [Google Scholar] [CrossRef] [PubMed]
- Aebi, H. Catalase In Vitro. Methods Enzymol. 1984, 105, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Glorieux, C.; Zamocky, M.; Sandoval, J.M.; Verrax, J.; Calderon, P.B. Regulation of catalase expression in healthy and cancerous cells. Free Radic. Biol. Med. 2015, 87, 84–97. [Google Scholar] [CrossRef]
- Nandi, A.; Yan, L.-J.; Jana, C.K.; Das, N. Role of Catalase in Oxidative Stress- and Age-Associated Degenerative Diseases. Oxid. Med. Cell Longev. 2019, 2019, 9613090. [Google Scholar] [CrossRef] [Green Version]
- Glorieux, C.; Sandoval, J.M.; Fattaccioli, A.; Dejeans, N.; Garbe, J.C.; Dieu, M.; Verrax, J.; Renard, P.; Huang, P.; Calderon, P.B. Chromatin remodeling regulates catalase expression during cancer cells adaptation to chronic oxidative stress. Free Radic. Biol. Med. 2016, 99, 436–450. [Google Scholar] [CrossRef]
- Glorieux, C.; Sandoval, J.M.; Dejeans, N.; Nonckreman, S.; Bahloula, K.; Poirel, H.A.; Calderon, P.B. Evaluation of Potential Mechanisms Controlling the Catalase Expression in Breast Cancer Cells. Oxid. Med. Cell Longev. 2018, 2018, 5351967. [Google Scholar] [CrossRef] [Green Version]
- Flor, S.; Oliva, C.R.; Ali, M.Y.; Coleman, K.L.; Greenlee, J.D.; Jones, K.A.; Monga, V.; Griguer, C.E. Catalase Overexpression Drives an Aggressive Phenotype in Glioblastoma. Antioxid. Basel Switz. 2021, 10, 1988. [Google Scholar] [CrossRef]
- Cao, J.; Schulte, J.; Knight, A.; Leslie, N.R.; Zagozdzon, A.; Bronson, R.; Manevich, Y.; Beeson, C.; Neumann, C.A. Prdx1 inhibits tumorigenesis via regulating PTEN/AKT activity. EMBO J. 2009, 28, 1505–1517. [Google Scholar] [CrossRef] [PubMed]
- Cárdenas-Rodríguez, N.; Pedraza-Chaverri, J. Especies reactivas de oxígeno y sistemas antioxidantes: Aspectos básicos. Profr. Al Día Biomed. 2005, 17, 164–173. [Google Scholar]
- Rhee, S.G.; Kil, I.S. Multiple Functions and Regulation of Mammalian Peroxiredoxins. Annu. Rev. Biochem. 2017, 86, 749–775. [Google Scholar] [CrossRef]
- Rhee, S.G. Overview on Peroxiredoxin. Mol. Cells 2016, 39, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Zhu, H.; Santo, A.; Li, Y. The antioxidant enzyme peroxiredoxin and its protective role in neurological disorders. Exp. Biol. Med. Maywood NJ 2012, 237, 143–149. [Google Scholar] [CrossRef]
- Lubos, E.; Loscalzo, J.; Handy, D.E. Glutathione peroxidase-1 in health and disease: From molecular mechanisms to therapeutic opportunities. Antioxid. Redox Signal. 2011, 15, 1957–1997. [Google Scholar] [CrossRef] [Green Version]
- Flohé, L.; Brigelius-Flohé, R. Selenoproteins of the Glutathione Peroxidase Family. In Selenium; Springer: New York, NY, USA, 2011; pp. 167–180. [Google Scholar]
- Rohr-Udilova, N.; Bauer, E.; Timelthaler, G.; Eferl, R.; Stolze, K.; Pinter, M.; Seif, M.; Hayden, H.; Reiberger, T.; Schulte-Hermann, R.; et al. Impact of glutathione peroxidase 4 on cell proliferation, angiogenesis and cytokine production in hepatocellular carcinoma. Oncotarget 2018, 9, 10054–10068. [Google Scholar] [CrossRef]
- Espinoza, S.E.; Guo, H.; Fedarko, N.; DeZern, A.; Fried, L.P.; Xue, Q.-L.; Leng, S.; Beamer, B.; Walston, J.D. Glutathione peroxidase enzyme activity in aging. J. Gerontol. A. Biol. Sci. Med. Sci. 2008, 63, 505–509. [Google Scholar] [CrossRef] [Green Version]
- Sarıkaya, E.; Doğan, S. Glutathione Peroxidase in Health and Diseases; IntechOpen: London, UK, 2020; ISBN 978-1-83880-126-7. [Google Scholar]
- Zhou, Y.; Lin, W.; Rao, T.; Zheng, J.; Zhang, T.; Zhang, M.; Lin, Z. Ferroptosis and Its Potential Role in the Nervous System Diseases. J. Inflamm. Res. 2022, 15, 1555–1574. [Google Scholar] [CrossRef]
- Saunders, N.; Walshe, J.; Serewko-Auret, M.; Chu, F.-F.; Esworthy, S.; Burcham, P.; Reeve, V. Glutathione peroxidase deficiency is a predisposing factor for UV-induced SCC in humans. Cancer Res. 2007, 67, 2632. [Google Scholar]
- Consoli, V.; Sorrenti, V.; Grosso, S.; Vanella, L. Heme Oxygenase-1 Signaling and Redox Homeostasis in Physiopathological Conditions. Biomolecules 2021, 11, 589. [Google Scholar] [CrossRef]
- Lin, Q.; Weis, S.; Yang, G.; Weng, Y.-H.; Helston, R.; Rish, K.; Smith, A.; Bordner, J.; Polte, T.; Gaunitz, F.; et al. Heme oxygenase-1 protein localizes to the nucleus and activates transcription factors important in oxidative stress. J. Biol. Chem. 2007, 282, 20621–20633. [Google Scholar] [CrossRef] [Green Version]
- Vanella, L.; Barbagallo, I.; Tibullo, D.; Forte, S.; Zappalà, A.; Li Volti, G. The non-canonical functions of the heme oxygenases. Oncotarget 2016, 7, 69075–69086. [Google Scholar] [CrossRef]
- Deshane, J.; Wright, M.; Agarwal, A. Heme oxygenase-1 expression in disease states. Acta Biochim. Pol. 2005, 52, 273–284. [Google Scholar] [CrossRef] [Green Version]
- Couto, N.; Wood, J.; Barber, J. The role of glutathione reductase and related enzymes on cellular redox homoeostasis network. Free Radic. Biol. Med. 2016, 95, 27–42. [Google Scholar] [CrossRef]
- Lu, S.C. Glutathione synthesis. Biochim. Biophys. Acta BBA-Gen. Subj. 2013, 1830, 3143–3153. [Google Scholar] [CrossRef] [Green Version]
- Gregg, X.T.; Prchal, J.T. Chapter 44—Red Blood Cell Enzymopathies. In Hematology, 7th ed.; Hoffman, R., Benz, E.J., Silberstein, L.E., Heslop, H.E., Weitz, J.I., Anastasi, J., Salama, M.E., Abutalib, S.A., Eds.; Elsevier: Amsterdam, The Netherlands, 2018; pp. 616–625. ISBN 978-0-323-35762-3. [Google Scholar]
- Forman, H.J.; Zhang, H.; Rinna, A. Glutathione: Overview of its protective roles, measurement, and biosynthesis. Mol. Asp. Med. 2009, 30, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Townsend, D.M.; Tew, K.D. The role of glutathione-S-transferase in anti-cancer drug resistance. Oncogene 2003, 22, 7369–7375. [Google Scholar] [CrossRef] [Green Version]
- Allocati, N.; Masulli, M.; Di Ilio, C.; Federici, L. Glutathione transferases: Substrates, inihibitors and pro-drugs in cancer and neurodegenerative diseases. Oncogenesis 2018, 7, 8. [Google Scholar] [CrossRef] [Green Version]
- Yan, C.; Duan, L.; Fu, C.; Tian, C.; Zhang, B.; Shao, X.; Zhu, G. Association Between Glutathione S-Transferase (GST) Polymorphisms and Schizophrenia in a Chinese Han Population. Neuropsychiatr. Dis. Treat. 2020, 16, 479–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tesauro, M.; Nisticò, S.; Noce, A.; Tarantino, A.; Marrone, G.; Costa, A.; Rovella, V.; Di Cola, G.; Campia, U.; Lauro, D.; et al. The possible role of glutathione-S-transferase activity in diabetic nephropathy. Int. J. Immunopathol. Pharmacol. 2015, 28, 129–133. [Google Scholar] [CrossRef]
- Singh, R.R.; Reindl, K.M. Glutathione S-Transferases in Cancer. Antioxidants 2021, 10, 701. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Kim, S.M.; Lee, R.T. Thioredoxin and Thioredoxin Target Proteins: From Molecular Mechanisms to Functional Significance. Antioxid. Redox Signal. 2013, 18, 1165–1207. [Google Scholar] [CrossRef] [PubMed]
- Watson, W.H.; Yang, X.; Choi, Y.E.; Jones, D.P.; Kehrer, J.P. Thioredoxin and its role in toxicology. Toxicol. Sci. Off. J. Soc. Toxicol. 2004, 78, 3–14. [Google Scholar] [CrossRef] [Green Version]
- Nordberg, J.; Arnér, E.S.J. Reactive oxygen species, antioxidants, and the mammalian thioredoxin system1 1This review is based on the licentiate thesis “Thioredoxin reductase—Interactions with the redox active compounds 1-chloro-2,4-dinitrobenzene and lipoic acid” by Jonas Nordberg. Free Radic. Biol. Med. 2001, 31, 1287–1312. [Google Scholar] [CrossRef]
- Mahmood, D.F.D.; Abderrazak, A.; El Hadri, K.; Simmet, T.; Rouis, M. The thioredoxin system as a therapeutic target in human health and disease. Antioxid. Redox Signal. 2013, 19, 1266–1303. [Google Scholar] [CrossRef]
- Mustacich, D.; Powis, G. Thioredoxin reductase. Biochem. J. 2000, 346 Pt 1, 1–8. [Google Scholar] [CrossRef]
- Patwardhan, R.S.; Sharma, D.; Sandur, S.K. Thioredoxin reductase: An emerging pharmacologic target for radiosensitization of cancer. Transl. Oncol. 2022, 17, 101341. [Google Scholar] [CrossRef]
- Mirończuk-Chodakowska, I.; Witkowska, A.M.; Zujko, M.E. Endogenous non-enzymatic antioxidants in the human body. Adv. Med. Sci. 2018, 63, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Moussa, Z.; Judeh, Z.M.A.; Ahmed, S.A. Nonenzymatic Exogenous and Endogenous Antioxidants. In Free Radical Medicine and Biology; Das, K., Das, S., Shivanagouda Biradar, M., Bobbarala, V., Subba Tata, S., Eds.; IntechOpen: London, UK, 2020; ISBN 978-1-78985-143-4. [Google Scholar]
- Janciauskiene, S. The Beneficial Effects of Antioxidants in Health and Diseases. Chronic Obstr. Pulm. Dis. J. COPD Found. 2020, 7, 182–202. [Google Scholar] [CrossRef] [PubMed]
- Noctor, G.; Queval, G.; Mhamdi, A.; Chaouch, S.; Foyer, C.H. Glutathione. Arab. Book 2011, 9, 1–32. [Google Scholar] [CrossRef] [PubMed]
- Moinova, H.R.; Mulcahy, R.T. Up-Regulation of the Human γ-Glutamylcysteine Synthetase Regulatory Subunit Gene Involves Binding of Nrf-2 to an Electrophile Responsive Element. Biochem. Biophys. Res. Commun. 1999, 261, 661–668. [Google Scholar] [CrossRef]
- Giustarini, D.; Colombo, G.; Garavaglia, M.L.; Astori, E.; Portinaro, N.M.; Reggiani, F.; Badalamenti, S.; Aloisi, A.M.; Santucci, A.; Rossi, R.; et al. Assessment of glutathione/glutathione disulphide ratio and S-glutathionylated proteins in human blood, solid tissues, and cultured cells. Free Radic. Biol. Med. 2017, 112, 360–375. [Google Scholar] [CrossRef]
- Ballatori, N.; Krance, S.M.; Notenboom, S.; Shi, S.; Tieu, K.; Hammond, C.L. Glutathione dysregulation and the etiology and progression of human diseases. bchm 2009, 390, 191–214. [Google Scholar] [CrossRef] [Green Version]
- Iskusnykh, I.Y.; Zakharova, A.A.; Pathak, D. Glutathione in Brain Disorders and Aging. Molecules 2022, 27, 324. [Google Scholar] [CrossRef]
- Korge, P.; Calmettes, G.; Weiss, J.N. Increased reactive oxygen species production during reductive stress: The roles of mitochondrial glutathione and thioredoxin reductases. Biochim. Biophys. Acta BBA-Bioenerg. 2015, 1847, 514–525. [Google Scholar] [CrossRef] [Green Version]
- Alli, J.A.; Kehinde, A.O.; Kosoko, A.M.; Ademowo, O.G. Oxidative Stress and Reduced Vitamins C and E Levels Are Associated with Multi-Drug Resistant Tuberculosis. J. Tuberc. Res. 2014, 02, 52–58. [Google Scholar] [CrossRef]
- Chatterjee, A. Reduced Glutathione: A Radioprotector or a Modulator of DNA-Repair Activity? Nutrients 2013, 5, 525–542. [Google Scholar] [CrossRef] [Green Version]
- Tram, N.K.; McLean, R.M.; Swindle-Reilly, K.E. Glutathione Improves the Antioxidant Activity of Vitamin C in Human Lens and Retinal Epithelial Cells: Implications for Vitreous Substitutes. Curr. Eye Res. 2021, 46, 470–481. [Google Scholar] [CrossRef] [PubMed]
- Lushchak, V.I. Glutathione Homeostasis and Functions: Potential Targets for Medical Interventions. J. Amino Acids 2012, 2012, 736837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christ-Hazelhof, E.; Nugteren, D.H. Purification and characterisation of prostaglandin endoperoxide D-isomerase, a cytoplasmic, glutathione-requiring enzymE. Biochim. Biophys. Acta BBA-Lipids Lipid Metab. 1979, 572, 43–51. [Google Scholar] [CrossRef]
- Layrisse, M.; Martínez-Torres, C.; Leets, I.; Taylor, P.; Ramírez, J. Effect of Histidine, Cysteine, Glutathione or Beef on Iron Absorption in Humans. J. Nutr. 1984, 114, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Gu, F.; Chauhan, V.; Chauhan, A. Glutathione redox imbalance in brain disorders: Curr. Opin. Clin. Nutr. Metab. Care 2015, 18, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Ghezzi, P. Role of glutathione in immunity and inflammation in the lung. Int. J. Gen. Med. 2011, 4, 105–113. [Google Scholar] [CrossRef] [Green Version]
- Quinlan, G.J.; Martin, G.S.; Evans, T.W. Albumin: Biochemical properties and therapeutic potential. Hepatology 2005, 41, 1211–1219. [Google Scholar] [CrossRef]
- Ronit, A.; Kirkegaard-Klitbo, D.M.; Dohlmann, T.L.; Lundgren, J.; Sabin, C.A.; Phillips, A.N.; Nordestgaard, B.G.; Afzal, S. Plasma Albumin and Incident Cardiovascular Disease: Results From the CGPS and an Updated Meta-Analysis. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 473–482. [Google Scholar] [CrossRef] [Green Version]
- Health, Aging and Body Composition Study; Visser, M.; Kritchevsky, S.B.; Newman, A.B.; Goodpaster, B.H.; Tylavsky, F.A.; Nevitt, M.C.; Harris, T.B. Lower serum albumin concentration and change in muscle mass: The Health, Aging and Body Composition Study. Am. J. Clin. Nutr. 2005, 82, 531–537. [Google Scholar] [CrossRef]
- Gomi, I.; Fukushima, H.; Shiraki, M.; Miwa, Y.; Ando, T.; Takai, K.; Moriwaki, H. Relationship between Serum Albumin Level and Aging in Community-Dwelling Self-Supported Elderly Population. J. Nutr. Sci. Vitaminol. 2007, 53, 37–42. [Google Scholar] [CrossRef] [Green Version]
- Lloyd, D.R.; Phillips, D.H. Oxidative DNA damage mediated by copper(II), iron(II) and nickel(II) Fenton reactions: Evidence for site-specific mechanisms in the formation of double-strand breaks, 8-hydroxydeoxyguanosine and putative intrastrand cross-links. Mutat. Res. Mol. Mech. Mutagen. 1999, 424, 23–36. [Google Scholar] [CrossRef]
- Di Simplicio, P.; Cheeseman, K.H.; Slater, T.F. The Reactivity of the Sh Group of Bovine Serum Albumin with Free Radicals. Free Radic. Res. Commun. 1991, 14, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Otagiri, M.; Chuang, V.T.G. Pharmaceutically Important Pre- and Posttranslational Modifications on Human Serum Albumin. Biol. Pharm. Bull. 2009, 32, 527–534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Simone, G.; di Masi, A.; Ascenzi, P. Serum Albumin: A Multifaced Enzyme. Int. J. Mol. Sci. 2021, 22, 10086. [Google Scholar] [CrossRef]
- Taverna, M.; Marie, A.-L.; Mira, J.-P.; Guidet, B. Specific antioxidant properties of human serum albumin. Ann. Intensive Care 2013, 3, 4. [Google Scholar] [CrossRef]
- Roche, M.; Rondeau, P.; Singh, N.R.; Tarnus, E.; Bourdon, E. The antioxidant properties of serum albumin. FEBS Lett. 2008, 582, 1783–1787. [Google Scholar] [CrossRef]
- Rondeau, P.; Bourdon, E. The glycation of albumin: Structural and functional impacts. Biochimie 2011, 93, 645–658. [Google Scholar] [CrossRef]
- McGuinness, A.; Sapey, E. Oxidative Stress in COPD: Sources, Markers, and Potential Mechanisms. J. Clin. Med. 2017, 6, 21. [Google Scholar] [CrossRef] [Green Version]
- Bomholt, T.; Adrian, T.; Nørgaard, K.; Ranjan, A.G.; Almdal, T.; Larsson, A.; Vadstrup, M.; Rix, M.; Feldt-Rasmussen, B.; Hornum, M. The Use of HbA1c, Glycated Albumin and Continuous Glucose Monitoring to Assess Glucose Control in the Chronic Kidney Disease Population Including Dialysis. Nephron 2021, 145, 14–19. [Google Scholar] [CrossRef]
- Peacock, T.P.; Shihabi, Z.K.; Bleyer, A.J.; Dolbare, E.L.; Byers, J.R.; Knovich, M.A.; Calles-Escandon, J.; Russell, G.B.; Freedman, B.I. Comparison of glycated albumin and hemoglobin A1c levels in diabetic subjects on hemodialysis. Kidney Int. 2008, 73, 1062–1068. [Google Scholar] [CrossRef] [Green Version]
- Nocentini, A.; Bonardi, A.; Pratesi, S.; Gratteri, P.; Dani, C.; Supuran, C.T. Pharmaceutical strategies for preventing toxicity and promoting antioxidant and anti-inflammatory actions of bilirubin. J. Enzym. Inhib. Med. Chem. 2022, 37, 487–501. [Google Scholar] [CrossRef] [PubMed]
- Sticova, E. New insights in bilirubin metabolism and their clinical implications. World J. Gastroenterol. 2013, 19, 6398. [Google Scholar] [CrossRef]
- Bhutani, V.K.; Wong, R.J.; Stevenson, D.K. Hyperbilirubinemia in Preterm Neonates. Clin. Perinatol. 2016, 43, 215–232. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Wei, X.; Bales, K.R.; Paul, A.B.C.; Ma, Z.; Yan, G.; Paul, S.M.; Du, Y. Minocycline blocks bilirubin neurotoxicity and prevents hyperbilirubinemia-induced cerebellar hypoplasia in the Gunn rat. Eur. J. Neurosci. 2005, 22, 21–27. [Google Scholar] [CrossRef]
- Zhou, Z.-X.; Chen, J.-K.; Hong, Y.-Y.; Zhou, R.; Zhou, D.-M.; Sun, L.-Y.; Qin, W.-L.; Wang, T.-C. Relationship Between the Serum Total Bilirubin and Inflammation in Patients With Psoriasis Vulgaris: Relationship Between the Serum Total Bilirubin and Inflammation. J. Clin. Lab. Anal. 2016, 30, 768–775. [Google Scholar] [CrossRef] [PubMed]
- Ziberna, L.; Martelanc, M.; Franko, M.; Passamonti, S. Bilirubin is an Endogenous Antioxidant in Human Vascular Endothelial Cells. Sci. Rep. 2016, 6, 29240. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.-T. Relationship between bilirubin free radical and formation of pigment gallstone. World J. Gastroenterol. 2002, 8, 413. [Google Scholar] [CrossRef] [PubMed]
- DiNicolantonio, J.J.; McCarty, M.F.; O’Keefe, J.H. Antioxidant bilirubin works in multiple ways to reduce risk for obesity and its health complications. Open Heart 2018, 5, e000914. [Google Scholar] [CrossRef] [Green Version]
- Hinds, T.D.; Stec, D.E. Bilirubin Safeguards Cardiorenal and Metabolic Diseases: A Protective Role in Health. Curr. Hypertens. Rep. 2019, 21, 87. [Google Scholar] [CrossRef]
- Lacko, M.; Roelofs, H.M.J.; te Morsche, R.H.M.; Voogd, A.C.; Ophuis, M.B.O.; Peters, W.H.M.; Manni, J.J. Genetic polymorphism in the conjugating enzyme UGT1A1 and the risk of head and neck cancer. Int. J. Cancer 2010, 127, 2815–2821. [Google Scholar] [CrossRef] [PubMed]
- Zhong, X.; Liao, Y.; Chen, X.; Mai, N.; Ouyang, C.; Chen, B.; Zhang, M.; Peng, Q.; Liang, W.; Zhang, W.; et al. Abnormal Serum Bilirubin/Albumin Concentrations in Dementia Patients With Aβ Deposition and the Benefit of Intravenous Albumin Infusion for Alzheimer’s Disease Treatment. Front. Neurosci. 2020, 14, 859. [Google Scholar] [CrossRef] [PubMed]
- Fabbrini, E.; Serafini, M.; Colic Baric, I.; Hazen, S.L.; Klein, S. Effect of Plasma Uric Acid on Antioxidant Capacity, Oxidative Stress, and Insulin Sensitivity in Obese Subjects. Diabetes 2014, 63, 976–981. [Google Scholar] [CrossRef] [Green Version]
- Klein, B.E.K.; Klein, R.; Lee, K.E. Components of the Metabolic Syndrome and Risk of Cardiovascular Disease and Diabetes in Beaver Dam. Diabetes Care 2002, 25, 1790–1794. [Google Scholar] [CrossRef] [Green Version]
- Zurlo, A.; Veronese, N.; Giantin, V.; Maselli, M.; Zambon, S.; Maggi, S.; Musacchio, E.; Toffanello, E.D.; Sartori, L.; Perissinotto, E.; et al. High serum uric acid levels increase the risk of metabolic syndrome in elderly women: The PRO.V.A study. Nutr. Metab. Cardiovasc. Dis. 2016, 26, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Dhaun, N.; Vachiery, J.-L.; Benza, R.L.; Naeije, R.; Hwang, L.-J.; Liu, X.; Teal, S.; Webb, D.J. Endothelin antagonism and uric acid levels in pulmonary arterial hypertension: Clinical associations. J. Heart Lung Transplant. 2014, 33, 521–527. [Google Scholar] [CrossRef]
- Ames, B.N.; Cathcart, R.; Schwiers, E.; Hochstein, P. Uric acid provides an antioxidant defense in humans against oxidant- and radical-caused aging and cancer: A hypothesis. Proc. Natl. Acad. Sci. USA 1981, 78, 6858–6862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lippi, G.; Montagnana, M.; Franchini, M.; Favaloro, E.J.; Targher, G. The paradoxical relationship between serum uric acid and cardiovascular disease. Clin. Chim. Acta 2008, 392, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Stinefelt, B.; Leonard, S.S.; Blemings, K.P.; Shi, X.; Klandorf, H. Free Radical Scavenging, DNA Protection, and Inhibition of Lipid Peroxidation Mediated by Uric Acid. Ann. Clin. Lab. Sci. 2005, 35, 37–45. [Google Scholar]
- Bowman, G.L.; Shannon, J.; Frei, B.; Kaye, J.A.; Quinn, J.F. Uric Acid as a CNS Antioxidant. J. Alzheimers Dis. 2010, 19, 1331–1336. [Google Scholar] [CrossRef] [Green Version]
- Reyes, A.J. The increase in serum uric acid concentration caused by diuretics might be beneficial in heart failure. Eur. J. Heart Fail. 2005, 7, 461–467. [Google Scholar] [CrossRef]
- Sautin, Y.Y.; Nakagawa, T.; Zharikov, S.; Johnson, R.J. Adverse effects of the classic antioxidant uric acid in adipocytes: NADPH oxidase-mediated oxidative/nitrosative stress. Am. J. Physiol.-Cell Physiol. 2007, 293, C584–C596. [Google Scholar] [CrossRef]
- Hink, H.U.; Santanam, N.; Dikalov, S.; McCann, L.; Nguyen, A.D.; Parthasarathy, S.; Harrison, D.G.; Fukai, T. Peroxidase Properties of Extracellular Superoxide Dismutase: Role of Uric Acid in Modulating In Vivo Activity. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 1402–1408. [Google Scholar] [CrossRef] [Green Version]
- Mahajan, M.; Kaur, S.; Mahajan, S.; Kant, R. Uric acid a better scavenger of free radicals than vitamin C in rheumatoid arthritis. Indian J. Clin. Biochem. 2009, 24, 205–207. [Google Scholar] [CrossRef] [Green Version]
- Laudon, M.; Frydman-Marom, A. Therapeutic Effects of Melatonin Receptor Agonists on Sleep and Comorbid Disorders. Int. J. Mol. Sci. 2014, 15, 15924–15950. [Google Scholar] [CrossRef] [Green Version]
- Reiter, R.; Tan, D.; Rosales-Corral, S.; Galano, A.; Zhou, X.; Xu, B. Mitochondria: Central Organelles for Melatonin′s Antioxidant and Anti-Aging Actions. Molecules 2018, 23, 509. [Google Scholar] [CrossRef] [Green Version]
- Ding, M.; Ning, J.; Feng, N.; Li, Z.; Liu, Z.; Wang, Y.; Wang, Y.; Li, X.; Huo, C.; Jia, X.; et al. Dynamin-related protein 1-mediated mitochondrial fission contributes to post-traumatic cardiac dysfunction in rats and the protective effect of melatonin. J. Pineal Res. 2018, 64, e12447. [Google Scholar] [CrossRef] [PubMed]
- Quera Salva, M.A.; Hartley, S.; Barbot, F.; Alvarez, J.C.; Lofaso, F.; Guilleminault, C. Circadian Rhythms, Melatonin and Depression. Curr. Pharm. Des. 2011, 17, 1459–1470. [Google Scholar] [CrossRef] [PubMed]
- Pandiperumal, S.; Trakht, I.; Srinivasan, V.; Spence, D.; Maestroni, G.; Zisapel, N.; Cardinali, D. Physiological effects of melatonin: Role of melatonin receptors and signal transduction pathways. Prog. Neurobiol. 2008, 85, 335–353. [Google Scholar] [CrossRef]
- Reiter, R.J.; Tan, D.-X.; Mayo, J.C.; Sainz, R.M.; Leon, J.; Czarnocki, Z. Melatonin as an antioxidant: Biochemical mechanisms and pathophysiological implications in humans. Acta Biochim. Pol. 2003, 50, 1129–1146. [Google Scholar] [CrossRef] [Green Version]
- Miller, E.; Mrowicka, M.; Malinowska, K.; Mrowicki, J.; Saluk-Juszczak, J.; Kędziora, J. The effects of whole-body cryotherapy on oxidative stress in multiple sclerosis patients. J. Therm. Biol. 2010, 35, 406–410. [Google Scholar] [CrossRef]
- Othman, A.I.; El-Missiry, M.A.; Amer, M.A.; Arafa, M. Melatonin controls oxidative stress and modulates iron, ferritin, and transferrin levels in adriamycin treated rats. Life Sci. 2008, 83, 563–568. [Google Scholar] [CrossRef] [PubMed]
- Dubocovich, M.L.; Delagrange, P.; Krause, D.N.; Sugden, D.; Cardinali, D.P.; Olcese, J. International Union of Basic and Clinical Pharmacology. LXXV. Nomenclature, Classification, and Pharmacology of G Protein-Coupled Melatonin Receptors. Pharmacol. Rev. 2010, 62, 343–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Sirianni, A.; Pei, Z.; Cormier, K.; Smith, K.; Jiang, J.; Zhou, S.; Wang, H.; Zhao, R.; Yano, H.; et al. The Melatonin MT1 Receptor Axis Modulates Mutant Huntingtin-Mediated Toxicity. J. Neurosci. 2011, 31, 14496–14507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Back, K.; Tan, D.-X.; Reiter, R.J. Melatonin biosynthesis in plants: Multiple pathways catalyze tryptophan to melatonin in the cytoplasm or chloroplasts. J. Pineal Res. 2016, 61, 426–437. [Google Scholar] [CrossRef]
- Tan, D.-X.; Manchester, L.; Qin, L.; Reiter, R. Melatonin: A Mitochondrial Targeting Molecule Involving Mitochondrial Protection and Dynamics. Int. J. Mol. Sci. 2016, 17, 2124. [Google Scholar] [CrossRef] [Green Version]
- Zheng, X.; Tan, D.X.; Allan, A.C.; Zuo, B.; Zhao, Y.; Reiter, R.J.; Wang, L.; Wang, Z.; Guo, Y.; Zhou, J.; et al. Chloroplastic biosynthesis of melatonin and its involvement in protection of plants from salt stress. Sci. Rep. 2017, 7, 41236. [Google Scholar] [CrossRef]
- Bonnefont-Rousselot, D.; Collin, F. Melatonin: Action as antioxidant and potential applications in human disease and aging. Toxicology 2010, 278, 55–67. [Google Scholar] [CrossRef]
- Devore, E.E.; Harrison, S.L.; Stone, K.L.; Holton, K.F.; Barrett-Connor, E.; Ancoli-Israel, S.; Yaffe, K.; Ensrud, K.; Cawthon, P.M.; Redline, S.; et al. Association of urinary melatonin levels and aging-related outcomes in older men. Sleep Med. 2016, 23, 73–80. [Google Scholar] [CrossRef] [Green Version]
- Navarro-Alarcon, M.; Ruiz-Ojeda, F.J.; Blanca-Herrera, R.M.; Agil, A. Antioxidant activity of melatonin in diabetes in relation to the regulation and levels of plasma Cu, Zn, Fe, Mn, and Se in Zucker diabetic fatty rats. Nutrition 2013, 29, 785–789. [Google Scholar] [CrossRef]
- López-Lluch, G.; Rodríguez-Aguilera, J.C.; Santos-Ocaña, C.; Navas, P. Is coenzyme Q a key factor in aging? Mech. Ageing Dev. 2010, 131, 225–235. [Google Scholar] [CrossRef]
- Jankowski, J.; Korzeniowska, K.; Cieślewicz, A.; Jabłecka, A. Coenzyme Q10—A new player in the treatment of heart failure? Pharmacol. Rep. 2016, 68, 1015–1019. [Google Scholar] [CrossRef] [PubMed]
- Petrova, E.V.; Korotkova, E.I.; Kratochvil, B.; Voronova, O.A.; Dorozhko, E.V.; Bulycheva, E.V. Investigation of Coenzyme Q10 by Voltammetry. Procedia Chem. 2014, 10, 173–178. [Google Scholar] [CrossRef] [Green Version]
- Bentinger, M.; Tekle, M.; Dallner, G. Coenzyme Q—Biosynthesis and functions. Biochem. Biophys. Res. Commun. 2010, 396, 74–79. [Google Scholar] [CrossRef]
- Acosta, M.J.; Vazquez Fonseca, L.; Desbats, M.A.; Cerqua, C.; Zordan, R.; Trevisson, E.; Salviati, L. Coenzyme Q biosynthesis in health and disease. Biochim. Biophys. Acta BBA-Bioenerg. 2016, 1857, 1079–1085. [Google Scholar] [CrossRef] [PubMed]
- Navas, P.; Villalba, J.M.; de Cabo, R. The importance of plasma membrane coenzyme Q in aging and stress responses. Mitochondrion 2007, 7, S34–S40. [Google Scholar] [CrossRef]
- Rodick, T.C.; Seibels, D.R.; Babu, J.R.; Huggins, K.W.; Ren, G.; Mathews, S.T. Potential role of coenzyme Q10 in health and disease conditions. Nutr. Diet. Suppl. 2018, 10, 1325. [Google Scholar] [CrossRef]
- Potgieter, M.; Pretorius, E.; Pepper, M.S. Primary and secondary coenzyme Q10 deficiency: The role of therapeutic supplementation. Nutr. Rev. 2013, 71, 180–188. [Google Scholar] [CrossRef] [PubMed]
- Turunen, M.; Olsson, J.; Dallner, G. Metabolism and function of coenzyme Q. Biochim. Biophys. Acta BBA-Biomembr. 2004, 1660, 171–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donnino, M.W.; Cocchi, M.N.; Salciccioli, J.D.; Kim, D.; Naini, A.B.; Buettner, C.; Akuthota, P. Coenzyme Q10 levels are low and may be associated with the inflammatory cascade in septic shock. Crit. Care 2011, 15, R189. [Google Scholar] [CrossRef] [Green Version]
- Noh, Y.H.; Kim, K.-Y.; Shim, M.S.; Choi, S.-H.; Choi, S.; Ellisman, M.H.; Weinreb, R.N.; Perkins, G.A.; Ju, W.-K. Inhibition of oxidative stress by coenzyme Q10 increases mitochondrial mass and improves bioenergetic function in optic nerve head astrocytes. Cell Death Dis. 2013, 4, e820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooney, R.V.; Dai, Q.; Gao, Y.-T.; Chow, W.-H.; Shu, X.-O.; Li, H.; Ji, B.; Cai, Q.; Franke, A.A.; Zheng, W. Abstract 874: Low plasma coenzyme Q10 levels and breast cancer risk in Chinese women: Influence of menopausal status. Cancer Res. 2010, 70, 874. [Google Scholar] [CrossRef]
- Blatt, T.; Littarru, G.P. Biochemical rationale and experimental data on the antiaging properties of CoQ10 at skin level. BioFactors 2011, 37, 381–385. [Google Scholar] [CrossRef]
- Lee, B.-J.; Huang, Y.-C.; Chen, S.-J.; Lin, P.-T. Coenzyme Q10 supplementation reduces oxidative stress and increases antioxidant enzyme activity in patients with coronary artery disease. Nutrition 2012, 28, 250–255. [Google Scholar] [CrossRef]
- Pisoschi, A.M.; Pop, A.; Iordache, F.; Stanca, L.; Predoi, G.; Serban, A.I. Oxidative stress mitigation by antioxidants—An overview on their chemistry and influences on health status. Eur. J. Med. Chem. 2021, 209, 112891. [Google Scholar] [CrossRef]
- Neha, K.; Haider, M.R.; Pathak, A.; Yar, M.S. Medicinal prospects of antioxidants: A review. Eur. J. Med. Chem. 2019, 178, 687–704. [Google Scholar] [CrossRef]
- Pisoschi, A.M.; Pop, A. The role of antioxidants in the chemistry of oxidative stress: A review. Eur. J. Med. Chem. 2015, 97, 55–74. [Google Scholar] [CrossRef] [PubMed]
- Jahns, L.; Conrad, Z.; Johnson, L.K.; Whigham, L.D.; Wu, D.; Claycombe-Larson, K.J. A diet high in carotenoid-rich vegetables and fruits favorably impacts inflammation status by increasing plasma concentrations of IFN-α2 and decreasing MIP-1β and TNF-α in healthy individuals during a controlled feeding trial. Nutr. Res. 2018, 52, 98–104. [Google Scholar] [CrossRef]
- Silva, S.; Costa, E.M.; Veiga, M.; Morais, R.M.; Calhau, C.; Pintado, M. Health promoting properties of blueberries: A review. Crit. Rev. Food Sci. Nutr. 2020, 60, 181–200. [Google Scholar] [CrossRef]
- Forman, H.J.; Davies, K.J.A.; Ursini, F. How Do Nutritional Antioxidants Really Work: Nucleophilic Tone and Para-Hormesis Versus Free Radical Scavenging In Vivo. Free Radic. Biol. Med. 2014, 66, 24–35. [Google Scholar] [CrossRef] [Green Version]
- Njus, D.; Kelley, P.M.; Tu, Y.-J.; Schlegel, H.B. Ascorbic acid: The chemistry underlying its antioxidant properties. Free Radic. Biol. Med. 2020, 159, 37–43. [Google Scholar] [CrossRef]
- Lykkesfeldt, J.; Michels, A.J.; Frei, B. Vitamin C. Adv. Nutr. 2014, 5, 16–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valdés, F. Vitamina C. Actas Dermo-Sifiliográficas 2006, 97, 557–568. [Google Scholar] [CrossRef]
- Carr, A.C.; Vissers, M.C.M. Synthetic or Food-Derived Vitamin C—Are They Equally Bioavailable? Nutrients 2013, 5, 4284–4304. [Google Scholar] [CrossRef] [Green Version]
- Ishizuka, K.; Ohira, Y. Scurvy. QJM Int. J. Med. 2022, 115, 475. [Google Scholar] [CrossRef]
- Guo, H.; Ding, J.; Liu, Q.; Li, Y.; Liang, J.; Zhang, Y. Vitamin C and Metabolic Syndrome: A Meta-Analysis of Observational Studies. Front. Nutr. 2021, 8, 728880. [Google Scholar] [CrossRef]
- Michels, A.J.; Hagen, T.M.; Frei, B. Human Genetic Variation Influences Vitamin C Homeostasis by Altering Vitamin C Transport and Antioxidant Enzyme Function. Annu. Rev. Nutr. 2013, 33, 45–70. [Google Scholar] [CrossRef] [PubMed]
- Lykkesfeldt, J.; Tveden-Nyborg, P. The Pharmacokinetics of Vitamin C. Nutrients 2019, 11, 2412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aziz, M.A.; Diab, A.S.; Mohammed, A.A. Antioxidant Categories and Mode of Action; IntechOpen: London, UK, 2019; ISBN 978-1-78923-920-1. [Google Scholar]
- Kaźmierczak-Barańska, J.; Boguszewska, K.; Adamus-Grabicka, A.; Karwowski, B.T. Two Faces of Vitamin C—Antioxidative and Pro-Oxidative Agent. Nutrients 2020, 12, 1501. [Google Scholar] [CrossRef]
- Chakraborthy, A.; Ramani, P.; Sherlin, H.J.; Premkumar, P.; Natesan, A. Antioxidant and pro-oxidant activity of Vitamin C in oral environment. Indian J. Dent. Res. 2014, 25, 499. [Google Scholar] [CrossRef] [PubMed]
- Bravo, L. Polyphenols: Chemistry, dietary sources, metabolism, and nutritional significance. Nutr. Rev. 1998, 56, 317–333. [Google Scholar] [CrossRef]
- Zeb, A. Concept, mechanism, and applications of phenolic antioxidants in foods. J. Food Biochem. 2020, 44, e13394. [Google Scholar] [CrossRef]
- Santos-Buelga, C.; González-Paramás, A.M.; Oludemi, T.; Ayuda-Durán, B.; González-Manzano, S. Chapter Four—Plant phenolics as functional food ingredients. In Advances in Food and Nutrition Research; Ferreira, I.C.F.R., Barros, L., Eds.; Functional Food Ingredients from Plants; Academic Press: Cambridge, MA, USA, 2019; Volume 90, pp. 183–257. [Google Scholar]
- Nijveldt, R.J.; van Nood, E.; van Hoorn, D.E.; Boelens, P.G.; van Norren, K.; van Leeuwen, P.A. Flavonoids: A review of probable mechanisms of action and potential applications. Am. J. Clin. Nutr. 2001, 74, 418–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orallo, F.; Álvarez, E.; Camiña, M.; Leiro, J.M.; Gómez, E.; Fernández, P. The Possible Implication of trans-Resveratrol in the Cardioprotective Effects of Long-Term Moderate Wine Consumption. Mol. Pharmacol. 2002, 61, 294–302. [Google Scholar] [CrossRef] [Green Version]
- Shoskes, D.A. Effect of Bioflavonoids Quercetin and Curcumin on Ischemic Renal Injury: A New Class of Renoprotective Agents: 1. Transplantation 1998, 66, 147–152. [Google Scholar] [CrossRef] [PubMed]
- Shen, N.; Wang, T.; Gan, Q.; Liu, S.; Wang, L.; Jin, B. Plant flavonoids: Classification, distribution, biosynthesis, and antioxidant activity. Food Chem. 2022, 383, 132531. [Google Scholar] [CrossRef] [PubMed]
- Porras-Loaiza, A.P.; López-Malo, A. Importancia de los grupos fenólicos en los alimentos. Temas Sel. Ing. Aliment. 2009, 31, 121–134. [Google Scholar]
- Singh, R.B.; Fedacko, J.; Fatima, G.; Magomedova, A.; Watanabe, S.; Elkilany, G. Why and How the Indo-Mediterranean Diet May Be Superior to Other Diets: The Role of Antioxidants in the Diet. Nutrients 2022, 14, 898. [Google Scholar] [CrossRef]
- Manach, C.; Scalbert, A.; Morand, C.; Rémésy, C.; Jiménez, L. Polyphenols: Food sources and bioavailability. Am. J. Clin. Nutr. 2004, 79, 727–747. [Google Scholar] [CrossRef] [Green Version]
- Di Lorenzo, C.; Colombo, F.; Biella, S.; Stockley, C.; Restani, P. Polyphenols and Human Health: The Role of Bioavailability. Nutrients 2021, 13, 273. [Google Scholar] [CrossRef]
- Carlsen, M.H.; Halvorsen, B.L.; Holte, K.; Bøhn, S.K.; Dragland, S.; Sampson, L.; Willey, C.; Senoo, H.; Umezono, Y.; Sanada, C.; et al. The total antioxidant content of more than 3100 foods, beverages, spices, herbs and supplements used worldwide. Nutr. J. 2010, 9, 3. [Google Scholar] [CrossRef]
- Liu, J.; Hefni, M.E.; Witthöft, C.M. Characterization of Flavonoid Compounds in Common Swedish Berry Species. Foods 2020, 9, 358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waterhouse, A.L. Wine Phenolics. Ann. N. Y. Acad. Sci. 2002, 957, 21–36. [Google Scholar] [CrossRef]
- Miller, K.B.; Hurst, W.J.; Flannigan, N.; Ou, B.; Lee, C.Y.; Smith, N.; Stuart, D.A. Survey of Commercially Available Chocolate- and Cocoa-Containing Products in the United States. 2. Comparison of Flavan-3-ol Content with Nonfat Cocoa Solids, Total Polyphenols, and Percent Cacao. J. Agric. Food Chem. 2009, 57, 9169–9180. [Google Scholar] [CrossRef]
- Cabrera, C.; Artacho, R.; Giménez, R. Beneficial Effects of Green Tea—A Review. J. Am. Coll. Nutr. 2006, 25, 79–99. [Google Scholar] [CrossRef] [PubMed]
- McKay, D.L.; Blumberg, J.B. The Role of Tea in Human Health: An Update. J. Am. Coll. Nutr. 2002, 21, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Galiniak, S.; Aebisher, D.; Bartusik-Aebisher, D. Health benefits of resveratrol administration. Acta Biochim. Pol. 2019, 66, 13–21. [Google Scholar] [CrossRef] [Green Version]
- Ungvari, Z.; Orosz, Z.; Rivera, A.; Labinskyy, N.; Xiangmin, Z.; Olson, S.; Podlutsky, A.; Csiszar, A. Resveratrol increases vascular oxidative stress resistance. Am. J. Physiol.-Heart Circ. Physiol. 2007, 292, H2417–H2424. [Google Scholar] [CrossRef]
- Leonard, S.S.; Xia, C.; Jiang, B.-H.; Stinefelt, B.; Klandorf, H.; Harris, G.K.; Shi, X. Resveratrol scavenges reactive oxygen species and effects radical-induced cellular responses. Biochem. Biophys. Res. Commun. 2003, 309, 1017–1026. [Google Scholar] [CrossRef]
- Xia, N.; Daiber, A.; Förstermann, U.; Li, H. Antioxidant effects of resveratrol in the cardiovascular system. Br. J. Pharmacol. 2017, 174, 1633–1646. [Google Scholar] [CrossRef] [Green Version]
- Ungvari, Z.; Labinskyy, N.; Mukhopadhyay, P.; Pinto, J.T.; Bagi, Z.; Ballabh, P.; Zhang, C.; Pacher, P.; Csiszar, A. Resveratrol attenuates mitochondrial oxidative stress in coronary arterial endothelial cells. Am. J. Physiol.-Heart Circ. Physiol. 2009, 297, H1876–H1881. [Google Scholar] [CrossRef] [Green Version]
- Ungvari, Z.; Bagi, Z.; Feher, A.; Recchia, F.A.; Sonntag, W.E.; Pearson, K.; de Cabo, R.; Csiszar, A. Resveratrol confers endothelial protection via activation of the antioxidant transcription factor Nrf2. Am. J. Physiol.-Heart Circ. Physiol. 2010, 299, H18–H24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, A.P.; Singh, R.; Verma, S.S.; Rai, V.; Kaschula, C.H.; Maiti, P.; Gupta, S.C. Health benefits of resveratrol: Evidence from clinical studies. Med. Res. Rev. 2019, 39, 1851–1891. [Google Scholar] [CrossRef] [PubMed]
- Balaban, R.S.; Nemoto, S.; Finkel, T. Mitochondria, Oxidants, and Aging. Cell 2005, 120, 483–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hutcheson, R.; Rocic, P. The metabolic syndrome, oxidative stress, environment, and cardiovascular disease: The great exploration. Exp. Diabetes Res. 2012, 2012, 271028. [Google Scholar] [CrossRef] [Green Version]
- Mauthe, M.; Orhon, I.; Rocchi, C.; Zhou, X.; Luhr, M.; Hijlkema, K.-J.; Coppes, R.P.; Engedal, N.; Mari, M.; Reggiori, F. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy 2018, 14, 1435–1455. [Google Scholar] [CrossRef] [Green Version]
- Hallan, S.; Sharma, K. The Role of Mitochondria in Diabetic Kidney Disease. Curr. Diab. Rep. 2016, 16, 61. [Google Scholar] [CrossRef]
- Medema, R.H.; Kops, G.J.P.L.; Bos, J.L.; Burgering, B.M.T. AFX-like Forkhead transcription factors mediate cell-cycle regulation by Ras and PKB through p27(kip1). Nature 2000, 404, 782–787. [Google Scholar] [CrossRef]
- Hotamisligil, G.S. Inflammation and metabolic disorders. Nature 2006, 444, 860–867. [Google Scholar] [CrossRef]
- Bae, Y.S.; Kang, S.W.; Seo, M.S.; Baines, I.C.; Tekle, E.; Chock, P.B.; Rhee, S.G. Epidermal Growth Factor (EGF)-induced Generation of Hydrogen Peroxide: Role in EGF Receptor-Mediated Tyrosine Phosphorylation. J. Biol. Chem. 1997, 272, 217–221. [Google Scholar] [CrossRef] [Green Version]
- Daroui, P.; Desai, S.D.; Li, T.-K.; Liu, A.A.; Liu, L.F. Hydrogen Peroxide Induces Topoisomerase I-mediated DNA Damage and Cell Death. J. Biol. Chem. 2004, 279, 14587–14594. [Google Scholar] [CrossRef]
Figure 1.
Endogenous sources of RONS. The binding of growth factor (GF) to growth factor receptor (GFR) leads to the activation and assembly of nicotinamide adenine dinucleotide phosphate hydrogen (NADPH) oxidases (NOXs) through Rac1. The activation of NOXs principally produces superoxide anion radical (O2•−) that is rapidly dismuted to hydrogen peroxide (H2O2) by cytosolic superoxide dismutase 3 (SOD3). H2O2 can easily cross the cytosolic membrane through aquaporin present in the cytosolic membrane and can modify protein-containing sulfhydryl groups (-SH). The oxidation of -SH can lead to protein inactivation, which produces oxidative stress. Mitochondria produce ROS by the electron transfer system via complex I (CI) and CIII. The main products are O2•− and H2O2, transferred from CI to the mitochondrial matrix and CIII towards the cristae lumen and intermembrane space. These ROS can alter redox-sensitive proteins into mitochondria, such as aconitase 2 (ACO2), α-ketoglutarate dehydrogenase (α-KDH), and isocitrate dehydrogenase (IDH), members of the tricarboxylic acid (TCA) cycle. Also, O2•− is reduced to H2O2 by SOD2. NOX4 is localized in the mitochondrial membrane and produces H2O2, which causes the opening of mitochondrial adenosine triphosphate (ATP)-sensitive potassium K channel (mtKATP) to decrease the mitochondrial membrane potential (↓ΔΨm), causing mitochondrial dysfunction and later oxidative stress. Peroxisome also generates H2O2 through β-oxidation, eliminated by CAT and peroxiredoxin 5 (Prx5), but the high production of H2O2 causes oxidative stress. Protein folding in ER is an essential source of H2O2 and uncontrolled conditions activate ER stress, promoting the activation of the unfolding protein response (UPR). UPR activation induces Ca2+ release, which can damage mitochondria. Oxidative stress causes the activation of signaling pathways to control it, such as nuclear factor erythroid 2-related factor 2 (Nrf2) and Forkhead box O3 (FOXO3), activating an antioxidant response. Asterisks (*) represent TCA cycle enzymes producing ROS. Created with biorender.com, accessed on 30 August 2022 (published with permission from biorender.com).
Figure 1.
Endogenous sources of RONS. The binding of growth factor (GF) to growth factor receptor (GFR) leads to the activation and assembly of nicotinamide adenine dinucleotide phosphate hydrogen (NADPH) oxidases (NOXs) through Rac1. The activation of NOXs principally produces superoxide anion radical (O2•−) that is rapidly dismuted to hydrogen peroxide (H2O2) by cytosolic superoxide dismutase 3 (SOD3). H2O2 can easily cross the cytosolic membrane through aquaporin present in the cytosolic membrane and can modify protein-containing sulfhydryl groups (-SH). The oxidation of -SH can lead to protein inactivation, which produces oxidative stress. Mitochondria produce ROS by the electron transfer system via complex I (CI) and CIII. The main products are O2•− and H2O2, transferred from CI to the mitochondrial matrix and CIII towards the cristae lumen and intermembrane space. These ROS can alter redox-sensitive proteins into mitochondria, such as aconitase 2 (ACO2), α-ketoglutarate dehydrogenase (α-KDH), and isocitrate dehydrogenase (IDH), members of the tricarboxylic acid (TCA) cycle. Also, O2•− is reduced to H2O2 by SOD2. NOX4 is localized in the mitochondrial membrane and produces H2O2, which causes the opening of mitochondrial adenosine triphosphate (ATP)-sensitive potassium K channel (mtKATP) to decrease the mitochondrial membrane potential (↓ΔΨm), causing mitochondrial dysfunction and later oxidative stress. Peroxisome also generates H2O2 through β-oxidation, eliminated by CAT and peroxiredoxin 5 (Prx5), but the high production of H2O2 causes oxidative stress. Protein folding in ER is an essential source of H2O2 and uncontrolled conditions activate ER stress, promoting the activation of the unfolding protein response (UPR). UPR activation induces Ca2+ release, which can damage mitochondria. Oxidative stress causes the activation of signaling pathways to control it, such as nuclear factor erythroid 2-related factor 2 (Nrf2) and Forkhead box O3 (FOXO3), activating an antioxidant response. Asterisks (*) represent TCA cycle enzymes producing ROS. Created with biorender.com, accessed on 30 August 2022 (published with permission from biorender.com).

Figure 2.
Exogenous sources of reactive oxygen species. (ROS). ROS can be generated through exposure to external factors such as ionizing (IR) and ultraviolet (UV) radiation, pollutants, food, medications, and drugs such as alcohol and tobacco. The mechanisms of ROS production by external factors include mitochondrial damage, the decreased activity of antioxidant enzymes and the glutathione (GSH)/glutathione disulfide (GSSG) ratio, the activation of nuclear factor kappa-light-chain-enhancer of activated B (NF-kB), the increased activity of enzymes such as nicotinamide adenine dinucleotide phosphate hydrogen (NADPH) oxidase (NOX) and nitric oxide synthase (NOS), an increased concentration of redox-active metals such as iron (Fe2+), the incomplete reduction of molecular oxygen (O2), and the radiolysis of water (H2O). The production of ROS causes damage to deoxyribonucleic acid (DNA) and the oxidation of lipids such as malondialdehyde (MDA) and 4-hydroxynonenal (4-HNE). UV: ultraviolet light; UVA: 320–400 nm; UVB: 290–320 nm; and UVC: 220–290 nm. Created with biorender.com, accessed on 3 September 2022 (published with permission from biorender.com).
Figure 2.
Exogenous sources of reactive oxygen species. (ROS). ROS can be generated through exposure to external factors such as ionizing (IR) and ultraviolet (UV) radiation, pollutants, food, medications, and drugs such as alcohol and tobacco. The mechanisms of ROS production by external factors include mitochondrial damage, the decreased activity of antioxidant enzymes and the glutathione (GSH)/glutathione disulfide (GSSG) ratio, the activation of nuclear factor kappa-light-chain-enhancer of activated B (NF-kB), the increased activity of enzymes such as nicotinamide adenine dinucleotide phosphate hydrogen (NADPH) oxidase (NOX) and nitric oxide synthase (NOS), an increased concentration of redox-active metals such as iron (Fe2+), the incomplete reduction of molecular oxygen (O2), and the radiolysis of water (H2O). The production of ROS causes damage to deoxyribonucleic acid (DNA) and the oxidation of lipids such as malondialdehyde (MDA) and 4-hydroxynonenal (4-HNE). UV: ultraviolet light; UVA: 320–400 nm; UVB: 290–320 nm; and UVC: 220–290 nm. Created with biorender.com, accessed on 3 September 2022 (published with permission from biorender.com).

Figure 3.
Oxidative stress-induced oxidative damage. Oxidation in purines and pyrimidines of DNA by hydroxyl radical (•OH) leads to mutations, nucleotide damage, and single- and double-strand breaks. •OH also oxidizes lipids to produce 4-hydroxy-2-nonenal (4-HNE) and malondialdehyde (MDA), aldehydes and ketones that are highly active lipid peroxidation products. The persistent oxidation of proteins by hydrogen peroxide (H2O2) in its sulfhydryl group (-SH) leads to protein inactivation. The thunder symbol indicates damage; arrows indicate augment. Created with biorender.com, accessed on 2 September 2022 (published with permission from biorender.com).
Figure 3.
Oxidative stress-induced oxidative damage. Oxidation in purines and pyrimidines of DNA by hydroxyl radical (•OH) leads to mutations, nucleotide damage, and single- and double-strand breaks. •OH also oxidizes lipids to produce 4-hydroxy-2-nonenal (4-HNE) and malondialdehyde (MDA), aldehydes and ketones that are highly active lipid peroxidation products. The persistent oxidation of proteins by hydrogen peroxide (H2O2) in its sulfhydryl group (-SH) leads to protein inactivation. The thunder symbol indicates damage; arrows indicate augment. Created with biorender.com, accessed on 2 September 2022 (published with permission from biorender.com).

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Aranda-Rivera, A.K.; Cruz-Gregorio, A.; Arancibia-Hernández, Y.L.; Hernández-Cruz, E.Y.; Pedraza-Chaverri, J. RONS and Oxidative Stress: An Overview of Basic Concepts. Oxygen 2022, 2, 437-478. https://doi.org/10.3390/oxygen2040030
AMA Style
Aranda-Rivera AK, Cruz-Gregorio A, Arancibia-Hernández YL, Hernández-Cruz EY, Pedraza-Chaverri J. RONS and Oxidative Stress: An Overview of Basic Concepts. Oxygen. 2022; 2(4):437-478. https://doi.org/10.3390/oxygen2040030
Chicago/Turabian StyleAranda-Rivera, Ana Karina, Alfredo Cruz-Gregorio, Yalith Lyzet Arancibia-Hernández, Estefani Yaquelin Hernández-Cruz, and José Pedraza-Chaverri. 2022. "RONS and Oxidative Stress: An Overview of Basic Concepts" Oxygen 2, no. 4: 437-478. https://doi.org/10.3390/oxygen2040030