
Molecular Mechanisms of Protein–Lipid Interactions and Protein Folding of Heterogeneous Amylin and Tau Oligomers on Lipid Nanodomains That Link to Alzheimer’s

Abstract
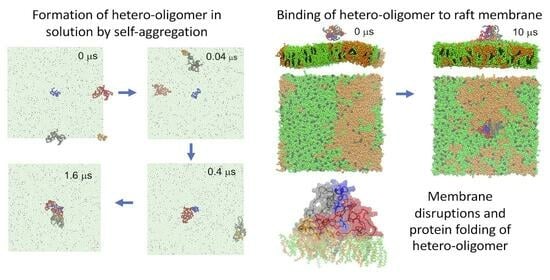
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Modeling Heterogeneous Tau–Amylin Oligomers
2.2. Modeling of the Raft Membrane
2.3. Simulations of Heterogeneous Oligomer Binding to the Raft Membrane
2.4. Classifications of Lipid Domains and Annular Lipids
2.5. Membrane Binding Behaviors of Oligomers
2.6. Characterizations of Disruptions of Lipid Orientational Order by Membrane-Bound Oligomers
2.7. Secondary Structures of Membrane-Bound Oligomers
2.8. Protein–Residue Contact Maps of Membrane-Bound Oligomers
3. Results
3.1. Lipid Binding Kinetics of Hetero-Oligomer Binding to the Raft Membrane
3.2. Lipid Binding Patterns of Hetero-Oligomers
3.3. Disruptions of Lipid Domain Sizes by Hetero- and Homo-Oligomers
3.4. Characterizations of Annular Lipids Surrounding the Hetero- and Homo-Oligomers
3.5. Disruptions of Orientational Orders of Annular Lipids by Hetero- and Homo-Oligomers
3.6. Protein Folding of Hetero- and Homo-Oligomers on Raft Membrane Surfaces
3.7. Residue-Resolved Protein–Protein Contact Map
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AA | all-atom |
| CG | coarse-grained |
| MD | molecular dynamics |
| 1am | monomeric amylin |
| 2am | dimeric amylin oligomer |
| 4am | tetrameric amylin oligomer |
| 1tau | monomeric tau |
| 2tau | dimeric tau oligomer |
| 4tau | tetrameric tau oligomer |
| 1tam | dimeric tau–amylin oligomer |
| 2tam | tetrameric tau–amylin oligomer |
| PC | phosphatidylcholine |
| CHOL | cholesterol |
| DPPC | dipalmitoyl-PC |
| DLPC | dilinoleoyl-PC |
| Lo | liquid-ordered |
| Ld | liquid-disordered |
| Lod | mixed Lo/Ld |
| AL | annular lipid |
| nAL | non-annular lipid |
| mindist | minimum distance |
| DSSP | Define Secondary Structure of Proteins |
References
- Milardi, D.; Gazit, E.; Radford, S.E.; Xu, Y.; Gallardo, R.U.; Caflisch, A.; Westermark, G.T.; Westermark, P.; Rosa, C.; Ramamoorthy, A. Proteostasis of Islet Amyloid Polypeptide: A Molecular Perspective of Risk Factors and Protective Strategies for Type II Diabetes. Chem. Rev. 2021, 121, 1845–1893. [Google Scholar] [CrossRef]
- Gerson, J.E.; Castillo-Carranza, D.L.; Kayed, R. Advances in therapeutics for neurodegenerative tauopathies: Moving toward the specific targeting of the most toxic tau species. ACS Chem. Neurosci. 2014, 5, 752–769. [Google Scholar] [CrossRef]
- Zhang, G.; Meng, L.; Wang, Z.; Peng, Q.; Chen, G.; Xiong, J.; Zhang, Z. Islet amyloid polypeptide cross-seeds tau and drives the neurofibrillary pathology in Alzheimer’s disease. Mol. Neurodegener. 2022, 17, 12. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Tao, Q.; Ang, T.F.A.; Massaro, J.; Gan, Q.; Salim, S.; Zhu, R.Y.; Kolachalama, V.B.; Zhang, X.; Devine, S.; et al. Association of Plasma Amylin Concentration With Alzheimer Disease and Brain Structure in Older Adults. JAMA Netw. Open 2019, 2, e199826. [Google Scholar] [CrossRef] [PubMed]
- Bortoletto, A.S.; Parchem, R.J. A pancreatic player in dementia: Pathological role for islet amyloid polypeptide accumulation in the brain. Neural Regen. Res. 2023, 18, 2141–2146. [Google Scholar] [PubMed]
- Wijesekara, N.; Goncalves, R.A.; Ahrens, R.; Ha, K.; De Felice, F.G.; Fraser, P.E. Combination of human tau and islet amyloid polypeptide exacerbates metabolic dysfunction in transgenic mice. J. Pathol. 2021, 254, 244–253. [Google Scholar] [CrossRef] [PubMed]
- Arya, S.; Claud, S.L.; Cantrell, K.L.; Bowers, M.T. Catalytic Prion-Like Cross-Talk between a Key Alzheimer’s Disease Tau-Fragment R3 and the Type 2 Diabetes Peptide IAPP. ACS Chem. Neurosci. 2019, 10, 4757–4765. [Google Scholar] [CrossRef] [PubMed]
- Akter, R.; Cao, P.; Noor, H.; Ridgway, Z.; Tu, L.H.; Wang, H.; Wong, A.G.; Zhang, X.; Abedini, A.; Schmidt, A.M.; et al. Islet Amyloid Polypeptide: Structure, Function, and Pathophysiology. J. Diabetes Res. 2016, 2016, 2798269. [Google Scholar] [CrossRef] [PubMed]
- Bharadwaj, P.; Solomon, T.; Sahoo, B.R.; Ignasiak, K.; Gaskin, S.; Rowles, J.; Verdile, G.; Howard, M.J.; Bond, C.S.; Ramamoorthy, A.; et al. Amylin and beta amyloid proteins interact to form amorphous heterocomplexes with enhanced toxicity in neuronal cells. Sci. Rep. 2020, 10, 10356. [Google Scholar] [CrossRef]
- Sciacca, M.F.M.; La Rosa, C.; Milardi, D. Amyloid-Mediated Mechanisms of Membrane Disruption. Biophysica 2021, 1, 137–156. [Google Scholar] [CrossRef]
- Shafiei, S.S.; Guerrero-Munoz, M.J.; Castillo-Carranza, D.L. Tau Oligomers: Cytotoxicity, Propagation, and Mitochondrial Damage. Front. Aging Neurosci. 2017, 9, 83. [Google Scholar] [CrossRef]
- Cao, Q.; Boyer, D.R.; Sawaya, M.R.; Ge, P.; Eisenberg, D.S. Cryo-EM structure and inhibitor design of human IAPP (amylin) fibrils. Nat. Struct. Mol. Biol. 2020, 27, 653–659. [Google Scholar] [CrossRef]
- Fitzpatrick, A.W.P.; Falcon, B.; He, S.; Murzin, A.G.; Murshudov, G.; Garringer, H.J.; Crowther, R.A.; Ghetti, B.; Goedert, M.; Scheres, S.H.W. Cryo-EM structures of tau filaments from Alzheimer’s disease. Nature 2017, 547, 185–190. [Google Scholar] [CrossRef]
- Cheng, K.H.; Graf, A.; Lewis, A.; Pham, T.; Acharya, A. Exploring Membrane Binding Targets of Disordered Human Tau Aggregates on Lipid Rafts Using Multiscale Molecular Dynamics Simulations. Membranes 2022, 12, 1098. [Google Scholar] [CrossRef]
- Lewis, A.; Pham, T.; Nguyen, N.; Graf, A.; Cheng, K.H. Lipid domain boundary triggers membrane damage and protein folding of human islet amyloid polypeptide in the early pathogenesis of amyloid diseases. Biophys. Chem. 2023, 296, 106993. [Google Scholar] [CrossRef] [PubMed]
- Wassenaar, T.A.; Pluhackova, K.; Bockmann, R.A.; Marrink, S.J.; Tieleman, D.P. Going Backward: A Flexible Geometric Approach to Reverse Transformation from Coarse Grained to Atomistic Models. J. Chem. Theory Comput. 2014, 10, 676–690. [Google Scholar] [CrossRef] [PubMed]
- Sali, A.; Blundell, T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef] [PubMed]
- Monticelli, L.; Kandasamy, S.K.; Periole, X.; Larson, R.G.; Tieleman, D.P.; Marrink, S.J. The MARTINI Coarse-Grained Force Field: Extension to Proteins. J. Chem. Theory Comput. 2008, 4, 819–834. [Google Scholar] [CrossRef]
- Hess, B.; Kutzner, C.; van der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef] [PubMed]
- Han, B.; Tashjian, A.H. User-friendly and versatile software for analysis of protein hydrophobicity. BioTechniques 1998, 25, 256–263. [Google Scholar] [CrossRef] [PubMed]
- Kyte, J.; Doolittle, R.F. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 1982, 157, 105–132. [Google Scholar] [CrossRef]
- Cebecauer, M.; Amaro, M.; Jurkiewicz, P.; Sarmento, M.J.; Sachl, R.; Cwiklik, L.; Hof, M. Membrane Lipid Nanodomains. Chem. Rev. 2018, 118, 11259–11297. [Google Scholar] [CrossRef]
- De Wit, G.; Danial, J.S.; Kukura, P.; Wallace, M.I. Dynamic label-free imaging of lipid nanodomains. Proc. Natl. Acad. Sci. USA 2015, 112, 12299–12303. [Google Scholar] [CrossRef]
- Sezgin, E.; Levental, I.; Mayor, S.; Eggeling, C. The mystery of membrane organization: Composition, regulation and roles of lipid rafts. Nat. Rev. Mol. Cell Biol. 2017, 18, 361–374. [Google Scholar] [CrossRef]
- Simons, K.; Sampaio, J.L. Membrane organization and lipid rafts. Cold Spring Harb. Perspect. Biol. 2011, 3, a004697. [Google Scholar] [CrossRef]
- Pham, T.; Cheng, K.H. Exploring the binding kinetics and behaviors of self-aggregated beta-amyloid oligomers to phase-separated lipid rafts with or without ganglioside-clusters. Biophys. Chem. 2022, 290, 106874. [Google Scholar] [CrossRef]
- Risselada, H.J.; Marrink, S.J. The molecular face of lipid rafts in model membranes. Proc. Natl. Acad. Sci. USA 2008, 105, 17367–17372. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef]
- Blumer, M.; Harris, S.; Li, M.; Martinez, L.; Untereiner, M.; Saeta, P.N.; Carpenter, T.S.; Ingolfsson, H.I.; Bennett, W.F.D. Simulations of Asymmetric Membranes Illustrate Cooperative Leaflet Coupling and Lipid Adaptability. Front. Cell Dev. Biol. 2020, 8, 575. [Google Scholar] [CrossRef]
- Grote, F.; Lyubartsev, A.P. Optimization of Slipids Force Field Parameters Describing Headgroups of Phospholipids. J. Phys. Chem. B 2020, 124, 8784–8793. [Google Scholar] [CrossRef]
- Kabsch, W.; Sander, C. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983, 22, 2577–2637. [Google Scholar] [CrossRef]
- Mercadante, D.; Grater, F.; Daday, C. CONAN: A Tool to Decode Dynamical Information from Molecular Interaction Maps. Biophys. J. 2018, 114, 1267–1273. [Google Scholar] [CrossRef]
- Brender, J.R.; McHenry, A.J.; Ramamoorthy, A. Does cholesterol play a role in the bacterial selectivity of antimicrobial peptides? Front. Immunol. 2012, 3, 195. [Google Scholar] [CrossRef]
- Hasan, M.; Moghal, M.M.R.; Saha, S.K.; Yamazaki, M. The role of membrane tension in the action of antimicrobial peptides and cell-penetrating peptides in biomembranes. Biophys. Rev. 2019, 11, 431–448. [Google Scholar] [CrossRef]
- Yang, S.T.; Kiessling, V.; Tamm, L.K. Line tension at lipid phase boundaries as driving force for HIV fusion peptide-mediated fusion. Nat. Commun. 2016, 7, 11401. [Google Scholar] [CrossRef]
- Akimov, S.A.; Kuzmin, P.I.; Zimmerberg, J.; Cohen, F.S. Lateral tension increases the line tension between two domains in a lipid bilayer membrane. Phys. Rev. E Stat. Nonlin Soft Matter Phys. 2007, 75 Pt 1, 011919. [Google Scholar] [CrossRef]
- Belicka, M.; Weitzer, A.; Pabst, G. High-resolution structure of coexisting nanoscopic and microscopic lipid domains. Soft Matter 2017, 13, 1823–1833. [Google Scholar] [CrossRef]
- Pinigin, K.V.; Kondrashov, O.V.; Jimenez-Munguia, I.; Alexandrova, V.V.; Batishchev, O.V.; Galimzyanov, T.R.; Akimov, S.A. Elastic deformations mediate interaction of the raft boundary with membrane inclusions leading to their effective lateral sorting. Sci. Rep. 2020, 10, 4087. [Google Scholar] [CrossRef]
- Nanga, R.P.; Brender, J.R.; Vivekanandan, S.; Ramamoorthy, A. Structure and membrane orientation of IAPP in its natively amidated form at physiological pH in a membrane environment. Biochim. Biophys. Acta 2011, 1808, 2337–2342. [Google Scholar] [CrossRef]
- Khemtemourian, L.; Fatafta, H.; Davion, B.; Lecomte, S.; Castano, S.; Strodel, B. Structural Dissection of the First Events Following Membrane Binding of the Islet Amyloid Polypeptide. Front. Mol. Biosci. 2022, 9, 849979. [Google Scholar] [CrossRef]
- Brender, J.R.; Hartman, K.; Reid, K.R.; Kennedy, R.T.; Ramamoorthy, A. A single mutation in the nonamyloidogenic region of islet amyloid polypeptide greatly reduces toxicity. Biochemistry 2008, 47, 12680–12688. [Google Scholar] [CrossRef]
- Engel, M.F.; Khemtemourian, L.; Kleijer, C.C.; Meeldijk, H.J.; Jacobs, J.; Verkleij, A.J.; de Kruijff, B.; Killian, J.A.; Hoppener, J.W. Membrane damage by human islet amyloid polypeptide through fibril growth at the membrane. Proc. Natl. Acad. Sci. USA 2008, 105, 6033–6038. [Google Scholar] [CrossRef]
- Sepehri, A.; Nepal, B.; Lazaridis, T. Distinct Modes of Action of IAPP Oligomers on Membranes. J. Chem. Inf. Model. 2021, 61, 4645–4655. [Google Scholar] [CrossRef]
- Sulatskaya, A.I.; Kosolapova, A.O.; Bobylev, A.G.; Belousov, M.V.; Antonets, K.S.; Sulatsky, M.I.; Kuznetsova, I.M.; Turoverov, K.K.; Stepanenko, O.V.; Nizhnikov, A.A. β-Barrels and Amyloids: Structural Transitions, Biological Functions, and Pathogenesis. Int. J. Mol. Sci. 2021, 22, 11316. [Google Scholar] [CrossRef]










Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santos, N.; Segura, L.; Lewis, A.; Pham, T.; Cheng, K.H. Molecular Mechanisms of Protein–Lipid Interactions and Protein Folding of Heterogeneous Amylin and Tau Oligomers on Lipid Nanodomains That Link to Alzheimer’s. Macromol 2023, 3, 805-827. https://doi.org/10.3390/macromol3040046
Santos N, Segura L, Lewis A, Pham T, Cheng KH. Molecular Mechanisms of Protein–Lipid Interactions and Protein Folding of Heterogeneous Amylin and Tau Oligomers on Lipid Nanodomains That Link to Alzheimer’s. Macromol. 2023; 3(4):805-827. https://doi.org/10.3390/macromol3040046
Chicago/Turabian StyleSantos, Natalia, Luthary Segura, Amber Lewis, Thuong Pham, and Kwan H. Cheng. 2023. "Molecular Mechanisms of Protein–Lipid Interactions and Protein Folding of Heterogeneous Amylin and Tau Oligomers on Lipid Nanodomains That Link to Alzheimer’s" Macromol 3, no. 4: 805-827. https://doi.org/10.3390/macromol3040046