CCR6–CCL20-Mediated Immunologic Pathways in Inflammatory Bowel Disease


Abstract
1. Introduction
2. Chemokines
Chemokine Receptor 6 (CCR6) and Chemokine Ligand CCL20
3. Importance of IBD
4. Immune Mechanisms of CCR6–CCL20
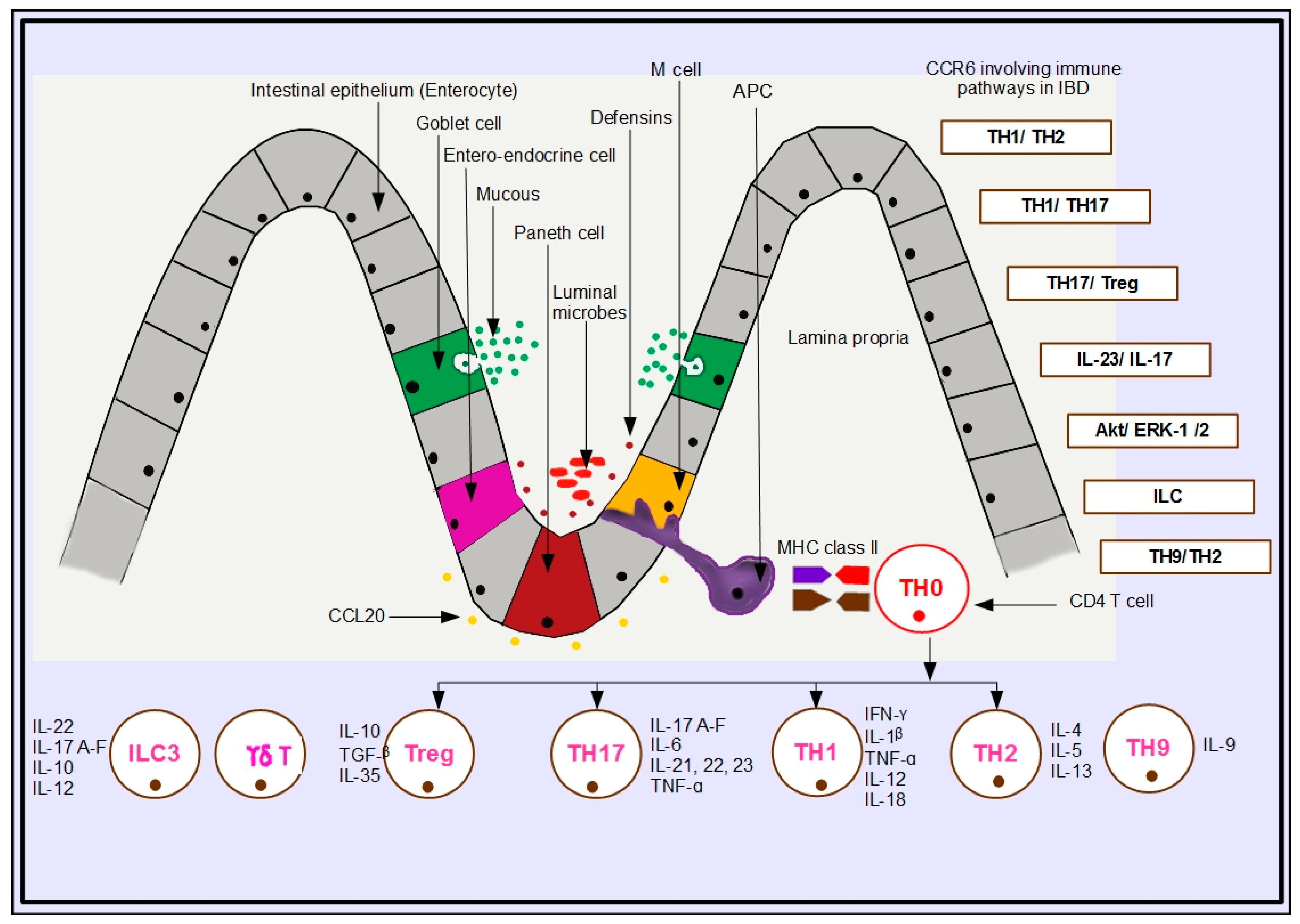
Immune Mechanisms of CCR6–CCL20 Specific to IBD
5. Immunologic Pathways in IBD Involving CCR6
5.1. TH1/TH2 Pathway
5.2. TH1/TH17 Pathway
5.3. TH17/Treg
5.4. IL-23/IL-17 Pathway
5.5. Akt/ERK-1/2 Pathway
5.6. Innate Lymphoid Cells (ILC)
5.7. TH9/TH2 Pathway
5.8. CD4+/CD8α+ Cells (DP8-Alpha Cells)
6. Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| Akt | protein kinase B |
| B | bursa-derived (lymphocyte) |
| β defensin | beta defensin |
| CD | Crohn’s disease |
| CD4+ T | cluster of differentiation 4 positive thymocyte |
| CcR6 | CC chemokine receptor 6 |
| CCL20 | CC chemokine ligand 20 |
| CCr6 | gene for CCR6 |
| CCR6−/− | CCR6-deficient |
| CRC | colorectal carcinoma |
| DC | dendritic cell |
| DNA | deoxyribonucleic acid |
| DSS | dextran sodium sulphate |
| ER | endoplasmic reticulum |
| ERK | extracellular signal-regulated kinases |
| FAE | follicle-associated epithelium |
| FoxP3 | forkhead box P3 |
| GALT | gut-associated lymphoid tissue |
| GATA | transcription factor |
| GI tract | gastrointestinal tract |
| GM-CSF | granulocyte macrophage colony-stimulating factor |
| GWAS | genome-wide association studies |
| γδ T cell | gamma delta T cell |
| HIV | human immunodeficiency virus |
| IBD | inflammatory bowel disease |
| IEC | intestinal epithelial cell |
| IFN-γ | gamma interferon |
| IL | interleukin |
| IL-1β | interleukin one beta |
| ILC3 | innate lymphoid cell 3 |
| IRF | interferon regulatory factor protein |
| JNK | Jun kinase |
| KO | knockout |
| LP | lamina propria |
| LARC | liver and activation-regulated chemokine |
| M cell | microfold cell |
| MAPK | mitogen-activated protein kinase |
| MEK | dual threonine and tyrosine recognition kinase |
| MIP-3α | macrophage inflammatory protein-3 alpha |
| NCR | natural cytotoxicity receptor |
| NF-kB | nuclear factor kappa B |
| NK | natural killer |
| NOD | nucleotide-binding and oligomerization domain |
| p | protein |
| PBMC | peripheral blood mononuclear cells |
| PI3K | phosphoinositide-3-kinase |
| R | receptor |
| Ras/Raf | guanine nucleotide exchange factor |
| RNA | ribonucleic acid |
| RORγt | retinoic-acid-receptor-related orphan nuclear receptor gamma |
| SAPK | stress-activated protein kinase |
| SCID | severe combined immune deficiency |
| SNP | single nucleotide polymorphism |
| STAT3 | signal transducer and activator of transcription 3 |
| T | thymus-derived lymphocyte/thymocyte |
| T-bet | T-box transcription factor |
| Tg | transgenic |
| TH1 | T helper 1 |
| TH2 | T helper 2 |
| TH17 | thymocyte helper 17 |
| TGF-β | transforming growth factor-beta sulphate |
| TNBS | trinitro benzene sulfonic acid |
| TNF-α | tumour necrosis factor-alpha |
| Treg | regulatory thymocyte cell |
| UC | ulcerative colitis |
| WT | wild type |
References
- Ranasinghe, R.; Eri, R. Pleiotropic immune functions of chemokine receptor 6 in health and disease. Medicines 2018, 5, 69. [Google Scholar] [CrossRef] [PubMed]
- Vongsa, R.A.; Zimmerman, N.P.; Dwinell, M.B. CCR6 regulation of the actin cytoskeleton orchestrates human beta defensin-2-and CCL20-mediated restitution of colonic epithelial cells. J. Biol. Chem. 2009, 284, 10034–10045. [Google Scholar] [CrossRef] [PubMed]
- Graham, D.B.; Xavier, R.J. From genetics of inflammatory bowel disease towards mechanistic insights. Trends Immunol. 2013, 34, 371–378. [Google Scholar] [CrossRef] [PubMed]
- Shouval, D.S.; Rufo, P.A. The role of environmental factors in the pathogenesis of inflammatory bowel diseases: A review. JAMA Pediatr. 2017, 171, 999–1005. [Google Scholar] [CrossRef] [PubMed]
- Rosen, M.J.; Dhawan, A.; Saeed, S.A. Inflammatory bowel disease in children and adolescents. JAMA Pediatr. 2015, 169, 1053–1060. [Google Scholar] [CrossRef] [PubMed]
- Kaser, A.; Zeissig, S.; Blumberg, R.S. Inflammatory Bowel Disease. Annu. Rev. Immunol. 2010, 28, 573–621. [Google Scholar] [CrossRef] [PubMed]
- Dahlhamer, J.M.; Zammitti, E.P.; Ward, B.W.; Wheaton, A.G.; Croft, J.B. Prevalence of Inflammatory Bowel Disease among Adults Aged ≥18 Years—United States, 2015. MMWR Morb. Mortal. Wkly. Rep. 2016, 65, 1166–1169. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, M.J. Inflammatory bowel disease goes global. JAMA 2018, 319, 648. [Google Scholar] [CrossRef] [PubMed]
- Niewiadomski, O.; Studd, C.; Hair, C.; Wilson, J.; Ding, N.S.; Heerasing, N.; Ting, A.; McNeill, J.; Knight, R.; Santamaria, J.; et al. Prospective population-based cohort of inflammatory bowel disease in the biologics era: Disease course and predictors of severity. J. Gastroenterol. Hepatol. 2015, 30, 1346–1353. [Google Scholar] [CrossRef] [PubMed]
- Crohn’s and Colitis Foundation Australia Website. 2013. Available online: http://www.crohnsandcolitis.com.au.
- Niewiadomski, O.; Studd, C.; Wilson, J.; Williams, J.; Hair, C.; Knight, R.; Prewett, E.; Dabkowski, P.; Alexander, S.; Allen, B.; et al. Influence of food and lifestyle on the risk of developing inflammatory bowel disease. J. Intern. Med. 2016, 46, 669–676. [Google Scholar] [CrossRef] [PubMed]
- Cader, M.Z.; Kaser, A. Recent advances in inflammatory bowel disease: Mucosal immune cells in intestinal inflammation. Gut 2013, 62, 1653–1664. [Google Scholar] [CrossRef] [PubMed]
- Fakhoury, M.; Negruli, R.; Mooranian, A.; Al-salam, H. Inflammatory bowel disease: Clinical aspects and treatments. J. Inflamm. Res. 2014, 7, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Jin, J. Inflammatory bowel disease. JAMA 2014, 311, 2034. [Google Scholar] [CrossRef] [PubMed]
- Blumberg, R.S. Crohn Disease. JAMA 2008, 300, 439–440. [Google Scholar] [PubMed]
- Cima, R.R.; Pemberton, J.H. Medical and surgical management of chronic ulcerative colitis. Arch. Surg. 2005, 140, 300–310. [Google Scholar] [CrossRef] [PubMed]
- Luo, C.; Zhang, H. The role of proinflammatory pathways in the pathogenesis of colitis-associated colorectal cancer. Mediat. Inflamm. 2017, 2017, 5126048. [Google Scholar] [CrossRef] [PubMed]
- Hanauer, S.B. Inflammatory bowel disease: Epidemiology, pathogenesis and therapeutic opportunities. Inflamm. Bowel Dis. 2006, 12, S3–S9. [Google Scholar] [CrossRef] [PubMed]
- Geremia, A.; Biancheri, P.; Allan, P.; Corazza, G.R.; Di Sabatino, A. Innate and adaptive immunity in inflammatory bowel disease. Autoimmun. Rev. 2014, 13, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Kmiec, Z.; Cyman, M.; Slebioda, T.J. Cells of the innate and adaptive immunity and their interactions in inflammatory bowel disease. Adv. Med. Sci. 2017, 62, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Skovdhal, H.K.; Granlund, A.V.; Ostvik, A.E.; Bruland, T.; Bakke, I.; Torp, S.H.; Damas, J.K.; Sandvik, A.K. Expression of CCL20 and its corresponding receptor CCR6 is enhanced in active inflammatory bowel disease and TLR3 mediates CCL20 expression in colonic epithelial cells. PLoS ONE 2015, 10, e0141710. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.Y.S.; Eri, R.; Lyons, A.B.; Grimm, M.C.; Korner, H. CC chemokine ligand CCL20 and its cognate receptor CCR6 in mucosal T cell immunology and inflammatory bowel disease: Odd couple or axis of evil? Front. Immunol. 2013, 4, 194–206. [Google Scholar] [CrossRef] [PubMed]
- Proudfoot, A.E. Chemokine receptors: Multifaceted therapeutic targets. Nat. Rev. Immunol. 2002, 2, 106–115. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, T.; Yang, X.O.; Chung, Y.; Fukunaga, A.; Nurieva, R.; Pappu, B.; Martin-Orozco, N.; Kang, H.S.; Ma, L.; Panopoulos, A.D.; et al. CCR6 regulates the migration of inflammatory and regulatory T cells. J. Immunol. 2008, 181, 8391–8401. [Google Scholar] [CrossRef] [PubMed]
- Moldoveanu, A.C.; Diculescu, M.; Braticevici, C.F. Cytokines in inflammatory bowel disease. Rom. J. Intern. Med. 2015, 53, 118–127. [Google Scholar] [CrossRef] [PubMed]
- Murphy, K.M. Janeway’s Immunobiology, 8th ed.; Garland Publishing Inc.: San Francisco, CA, USA, 2012. [Google Scholar]
- Ohman, L.; Astrom, R.; Hornquist, E.H. Impaired B cell responses to orally administered antigens in lamina propria but not peyer’s patches of Gαi2 deficient mice. Immunology 2005, 115, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Basheer, W.; Kunde, D.; Eri, R. Role of chemokine ligand CCL20 and its receptor CCR6 in intestinal inflammation. Immunol. Infect. Dis. 2013, 1, 30–37. [Google Scholar]
- Varona, R.; Cadenas, V.; Flores, J.; Martinez-A, C.; Marquez, G. CCR6 has a non-redundant role in the development of inflammatory bowel disease. Eur. J. Immunol. 2003, 33, 2937–2946. [Google Scholar] [CrossRef] [PubMed]
- Varona, R.; Villares, R.; Carramolino, L.; Goya, I.; Zaballos, A.; Gutierrez, J.; Torres, M.; Martinez-A, C.; Marquez, G. CCR6 deficient mice have impaired leukocyte homeostasis and altered contact hypersensitivity and delayed type hypersensitivity responses. J. Clin. Investig. 2001, 107, R37–R45. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.Y.S.; Phan, T.K.; Hulett, M.D.; Korner, H. The relationship between CCR6 and its binding partners: Does the CCR6-CCL20 axis have to be extended? Cytokine 2015, 72, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Parkes, M.; Cortes, A.; van Heel, D.A.; Brown, M.A. Genetic insights into common pathways and complex relationships among immune-mediated diseases. Nat. Rev. Genet. 2013, 14, 661–673. [Google Scholar] [CrossRef] [PubMed]
- Brand, S. Crohn’s disease: Th1, Th17 or both? The change of a paradigm: New immunological and genetic insights implicate Th17 cells in the pathogenesis of Crohn’s disease. Gut 2009, 58, 1152–1167. [Google Scholar] [CrossRef] [PubMed]
- Ueno, A.; Ghosh, A.; Hung, D.; Li, J.; Jijon, H. Th17 plasticity and its changes associated with inflammatory bowel disease. World J. Gastroenterol. 2015, 21, 12283–12295. [Google Scholar] [CrossRef] [PubMed]
- Korn, T.; Bettelli, E.; Oukka, M.; Kuchroo, V.K. IL-17 and Th17 cells. Annu. Rev. Immunol. 2009, 27, 485–517. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Kwon, J.; Chao, M.L. Immunological pathogenesis of inflammatory bowel disease. Intestig. Res. 2018, 16, 26–42. [Google Scholar] [CrossRef] [PubMed]
- Beurel, E.; Lowell, J.A. Th17 cells in depression. Brain Behav. Immun. 2018, 69, 28–34. [Google Scholar] [CrossRef] [PubMed]
- Nadeema, A.; Ahmada, S.F.; Al-Harbia, N.O.; Fardana, A.S.; El-Sherbeenyb, A.M.; Ibrahimc, K.E.; Attiaa, S.M. IL-17A causes depression-like symptoms via NFκB and p38MAPK signalling pathways in mice: Implications for psoriasis associated depression. Cytokine 2017, 97, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Chao, M.L.; Kang, J.W.; Moon, Y.M.; Nam, H.J.; Jhun, J.Y.; Heo, S.B.; Jin, H.T.; Min, S.Y.; Ju, J.H.; Park, K.S.; et al. STAT3 and NF-_B signal pathway is required for IL-23-mediated IL-17 production in spontaneous arthritis animal model IL-1 receptor antagonist-deficient mice. J. Immunol. 2006, 176, 5652–5661, Copyright © 2006 by The American Association of Immunologists, Inc.. [Google Scholar] [CrossRef]
- Griffin, M.D.; Ritter, T.; Mahon, B.P. Immunological Aspects of Allogeneic Mesenchymal Stem Cell Therapies. Hum. Gene Ther. 2010, 21, 1641–1655. [Google Scholar] [CrossRef] [PubMed]
- Boden, E.K.; Snapper, S.B. Regulatory T cells in inflammatory bowel disease. Cur. Opin. Gastroenterol. 2008, 24, 733–741. [Google Scholar] [CrossRef]
- Mebratu, Y.; Tesfaigzi, Y. How ERK1/2 Activation Controls Cell Proliferation and Cell Death Is Subcellular Localization the Answer? Cell Cycle 2009, 8, 1168–1175. [Google Scholar] [CrossRef] [PubMed]
- Brand, S.; Olszak, T.; Beigel, F.; Diebold, J.; Otte, J.M.; Eichhorst, S.T.; Goke, B.; Dambacher, J. Cell differentiation dependent expressed CCR6 mediates ERK-1/2, SAPK/JNK, and Akt signalling resulting in proliferation and migration of colorectal cancer cells. J. Cell. Biochem. 2006, 97, 709–723. [Google Scholar] [CrossRef] [PubMed]
- Geremia, A.; Arancibia-Carcamo, C.V. Innate lymphoid cells in intestinal inflammation. Front. Immunol. 2017, 8, 1296. [Google Scholar] [CrossRef] [PubMed]
- Geremia, A.; Arancibia-Carcamo, C.V.; Fleming, M.P.; Rust, N.; Singh, B.; Mortensen, N.J.; Travis, S.P.; Powrie, F. IL-23-responsive innate lymphoid cells are increased in inflammatory bowel disease. J. Exp. Med. 2011, 208, 1127–1133. [Google Scholar] [CrossRef] [PubMed]
- Buonocore, S.; Ahern, P.P.; Uhlig, H.H.; Ivanov, I.I.; Littman, D.R.; Maloy, K.J.; Powrie, F. Innate immune lymphoid cells drive IL-23 dependent innate intestinal pathology. Nature 2010, 464, 1371–1375. [Google Scholar] [CrossRef] [PubMed]
- Kara, E.E.; Comerford, I.; Bastow, C.R.; Fenix, K.A.; Litchfield, W.; Handel, T.M.; McColl, S.R. Distinct chemokine receptor axes regulate T helper 9 cell trafficking to allergic and autoimmune inflammatory sites. J. Immunol. 2013, 191, 1110–1117. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Qian, J.; Yi, Q. Th9 cells promote antitumor immune responses in vivo. J. Clin. Investig. 2012, 122, 4160–4171. [Google Scholar] [CrossRef] [PubMed]
- Godefroy, E.; Alameddine, J.; Montassier, E.; Mathé, J.; Desfrançois-Noël, J.; Marec, N.; Bossard, C.; Jarry, A.; Bridonneau, C.; Le Roy, A.; et al. Expression of CCR6 and CXCR6 by gut-derived CD4+/CD8alpha+ T-regulatory cells which are decreased in blood samples from patients with inflammatory bowel diseases. Gastroenterology 2018. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Pathway | Link to CCR6 | Mechanism | Outcome | Reference |
|---|---|---|---|---|
| TH1/TH2 | CCR6+ TH1 cells | Migration of CCR6+ TH1 cells to the intestine, attracted by CCL20 produced by the IEC, given their release of proinflammatory cytokines induces inflammation in the intestine. | Inflammation | [33] |
| TH1/TH17 | CCR6+ TH17 cells | Induced by IL-23, RORγt, and TGF-β, TH17 cells, upon differentiation, release IL-17A–F, which drives CCR6+ TH17 cell recruitment towards the intestinal epithelium, attracted by CCL20 produced by the IEC. | Inflammation | [32,33,34,35,36,37,38,39,40] |
| TH17/Treg | CCR6+ Treg cells | Induced by TGF-β and FoxP3, regulatory Treg cells, after differentiation, release IL-10, which drives CCR6+ Treg cells towards the intestinal epithelium, attracted by CCL20 produced by the IEC. | Resolution | [40,41,42] |
| IL-23/IL-17 | CCR6+ TH17 | IL-12, STAT3, and IL-23-induced CCR6+ TH17 cells migrate towards the intestinal epithelium, stimulated by IL-17A–F and attracted by CCL20 produced by the IEC. | Inflammation | [27,41,42] |
| Akt/ERK-1/2 | CCR6+ TH17/Treg | CCL20 activates Akt/ERK-1/2 and SAPK/JNK MAP kinases to increase cell proliferation and cell mobilization in the intestinal epithelium. | Homeostasis | [42,43] |
| ILC | CCR6+ NCR+ ILC3 | Induced by IL-22, CCR6+ NCR+ ILC3 release IL-12 and activate IFN-γ-producing ILC1 cells. | Inflammation | [44,45,46] |
| IL-23-producing ILC1 activate NCR+ ILC3 to express MHC class II epitopes to initiate transformation of naïve T helper cells into effectors. | Inflammation/Resolution | |||
| Induced by IL-22, CCR6+ ILC3 migrate to Peyer’s patches, attracted by CCL20 produced by the IEC. Decrease in IL-22 results in loss of immune tolerance. | ||||
| TH9/TH2 | CCR6+ TH9 cells | Induced by retinoic acid, TGF-β and IL-10 released by ILC3 effect differentiation of Treg cells in the intestine. | Inflammation | [47,48] |
| Induction by IL-9 elicits antiparasitic or allergic immunity, evoking a TH2-type immune response. |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ranasinghe, R.; Eri, R. CCR6–CCL20-Mediated Immunologic Pathways in Inflammatory Bowel Disease. Gastrointest. Disord. 2019, 1, 15-29. https://doi.org/10.3390/gidisord1010003
Ranasinghe R, Eri R. CCR6–CCL20-Mediated Immunologic Pathways in Inflammatory Bowel Disease. Gastrointestinal Disorders. 2019; 1(1):15-29. https://doi.org/10.3390/gidisord1010003
Chicago/Turabian StyleRanasinghe, Ranmali, and Rajaraman Eri. 2019. "CCR6–CCL20-Mediated Immunologic Pathways in Inflammatory Bowel Disease" Gastrointestinal Disorders 1, no. 1: 15-29. https://doi.org/10.3390/gidisord1010003
APA StyleRanasinghe, R., & Eri, R. (2019). CCR6–CCL20-Mediated Immunologic Pathways in Inflammatory Bowel Disease. Gastrointestinal Disorders, 1(1), 15-29. https://doi.org/10.3390/gidisord1010003