In Search of a Binding Agent: Nano-Scale Evidence of Preferential Carbon Associations with Poorly-Crystalline Mineral Phases in Physically-Stable, Clay-Sized Aggregates

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Soil Samples
2.2. Procedure to Isolate Size Fractions
2.3. Characterization of Each Size Fraction
(sum of SSAoxidized × mass percentage of each fraction) × 100
2.4. Scanning Transmission X-ray Microscopy (STXM) and Near-Edge X-ray Absorption Fine Structure (NEXAFS)
2.5. SEM and TEM Observation and Elemental Analysis
3. Results
3.1. Soil Surface Area before and after OM Removal
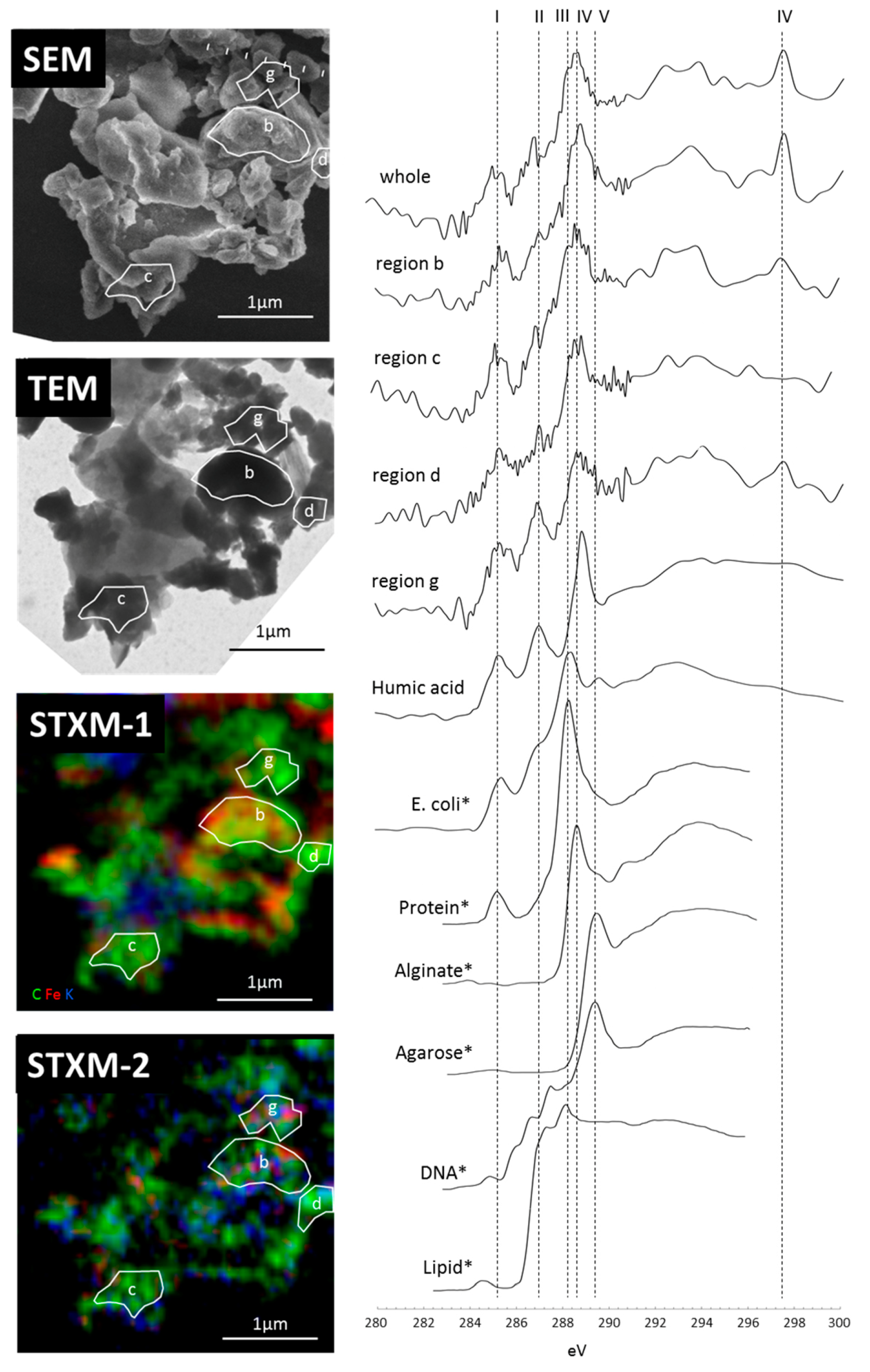
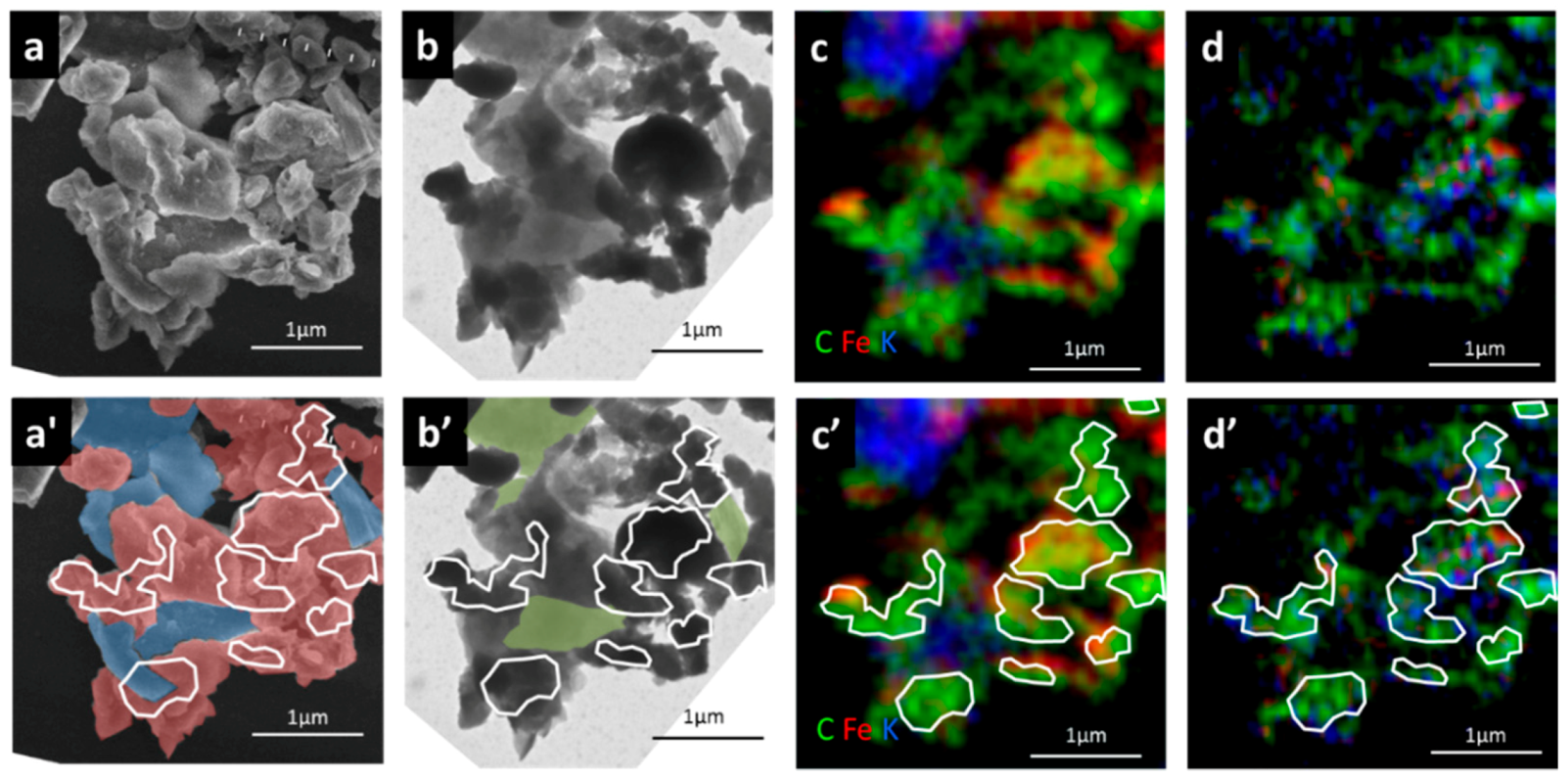
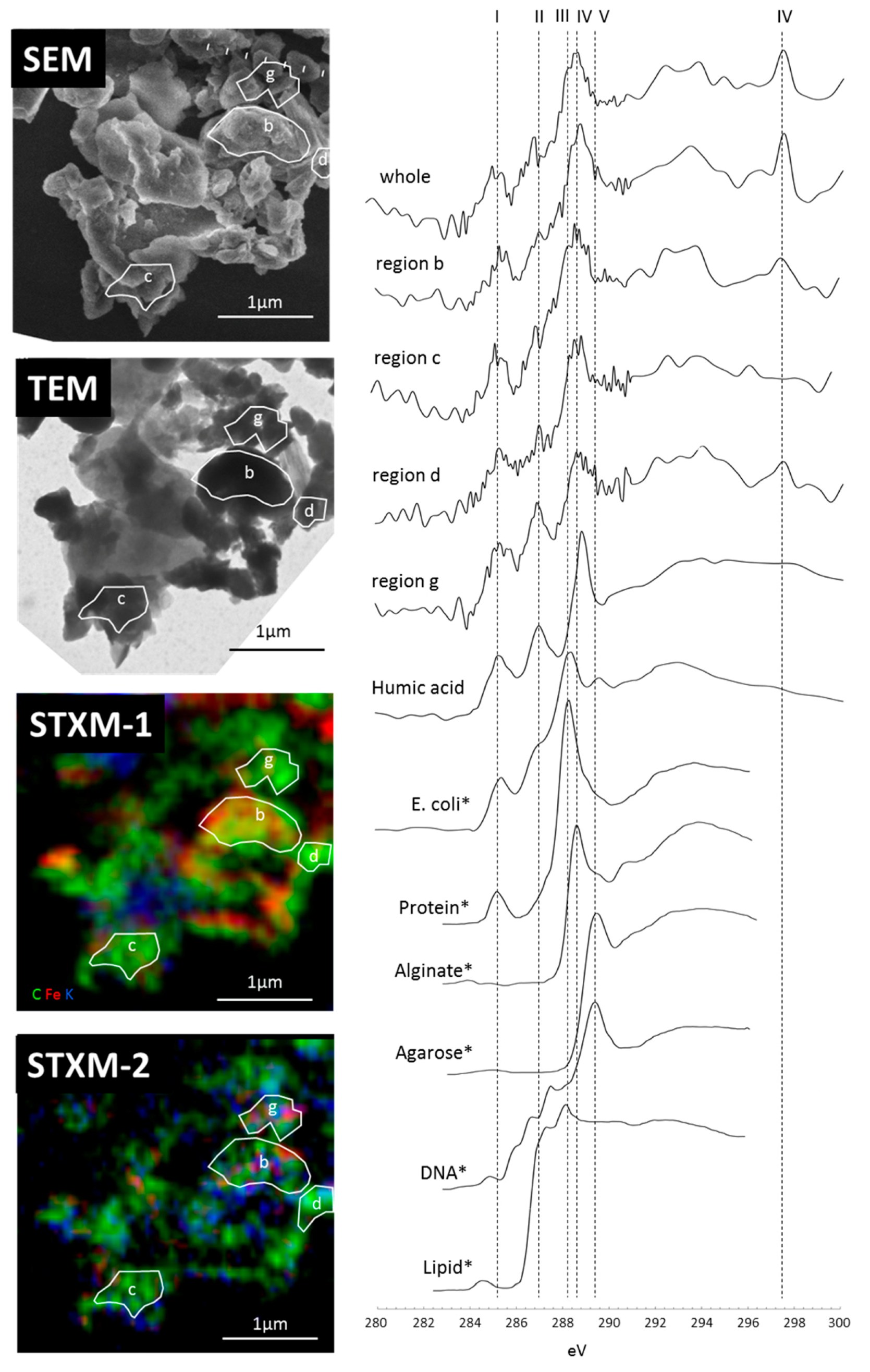
3.2. Carbon Distribution and Chemical Composition within the Selected Organo-Mineralaggregate
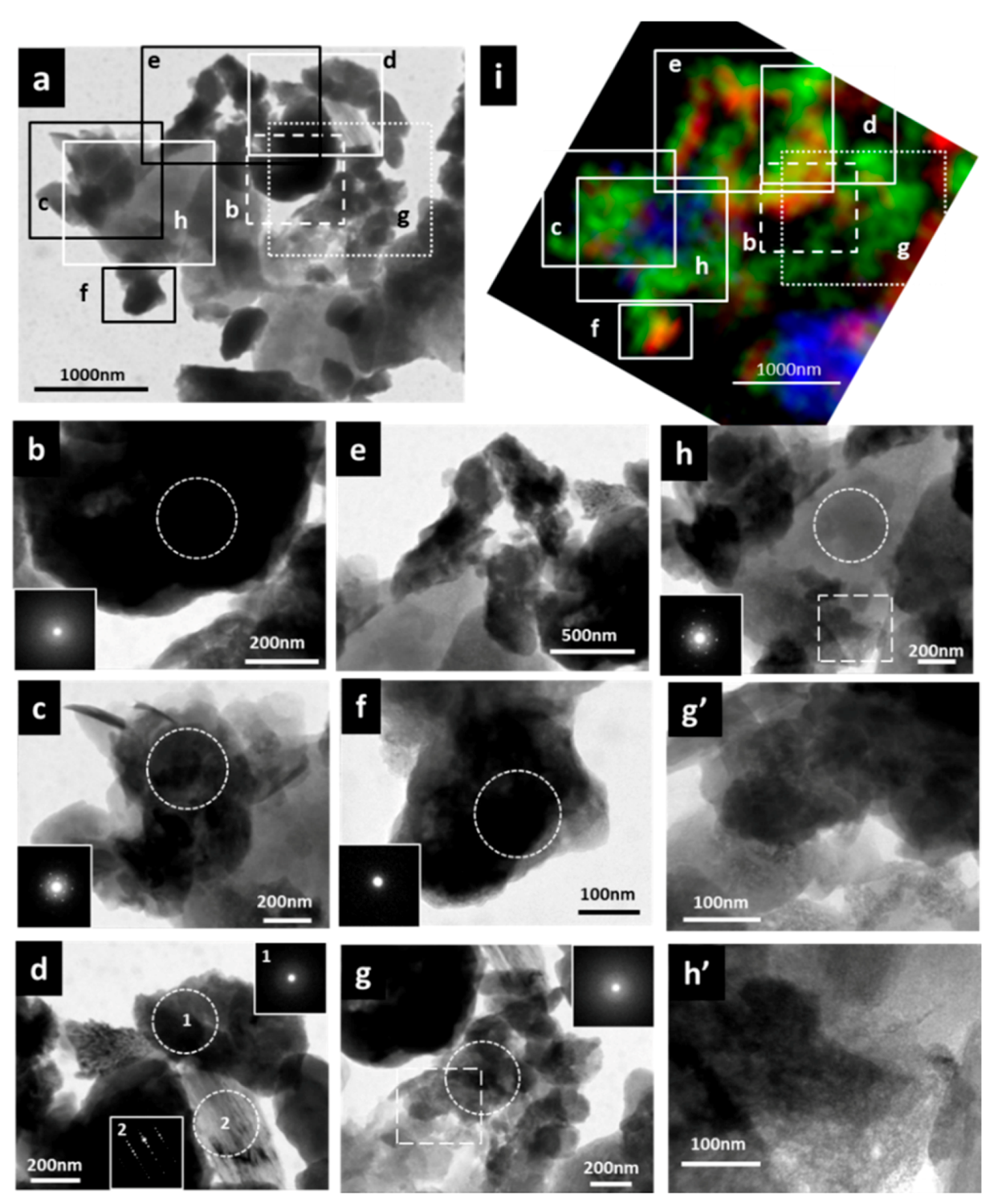
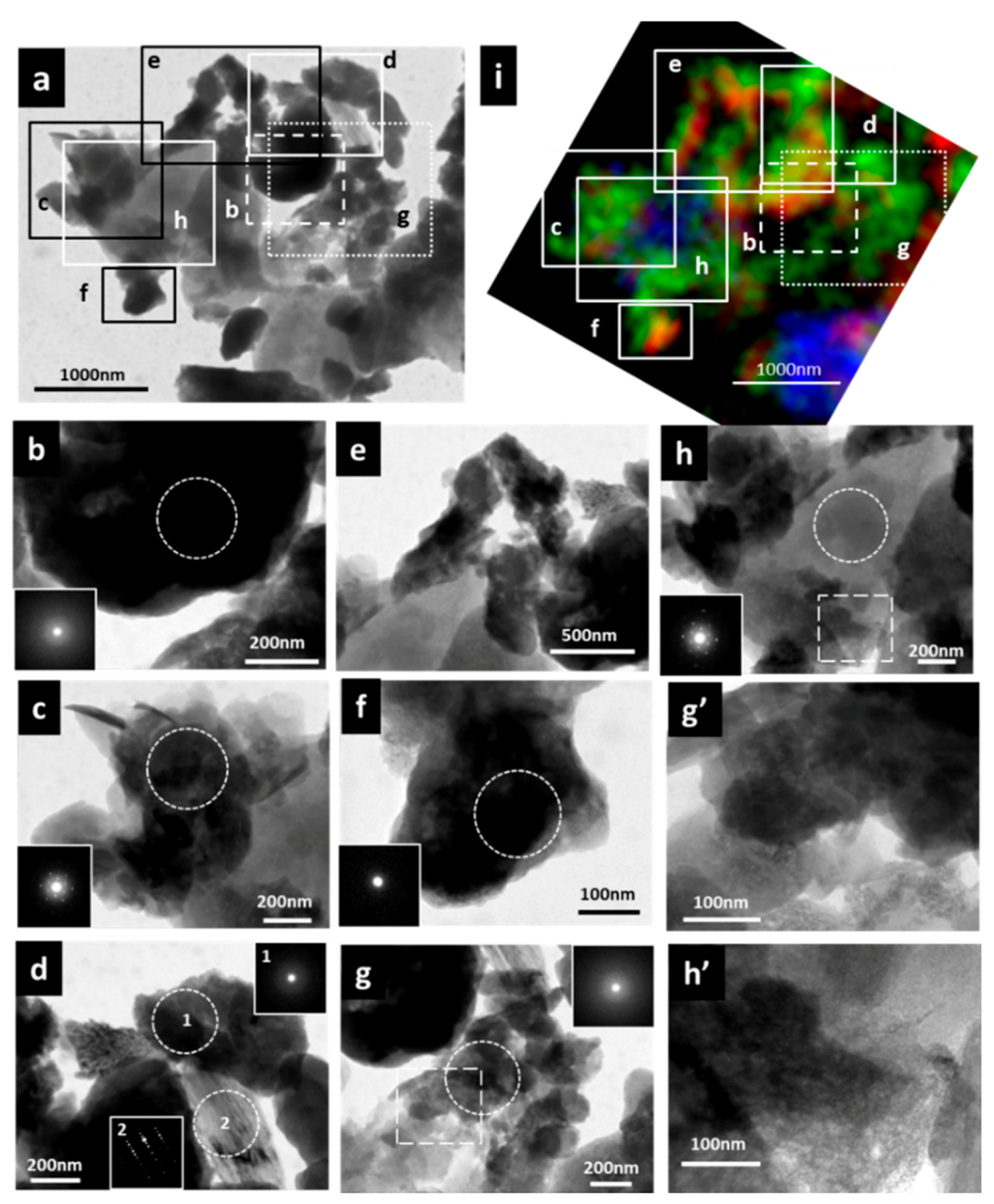
3.3. Micro-Morphological Observation and Electron Diffraction Analysis by TEM and NEXAFS Spectrum within Specific Area
4. Discussion
4.1. Reactive Inorganic (RI) Phase Control on OM Distribution within Particle
4.2.Carbon Forms of the Organo-Mineral/Metal Nanocomposit
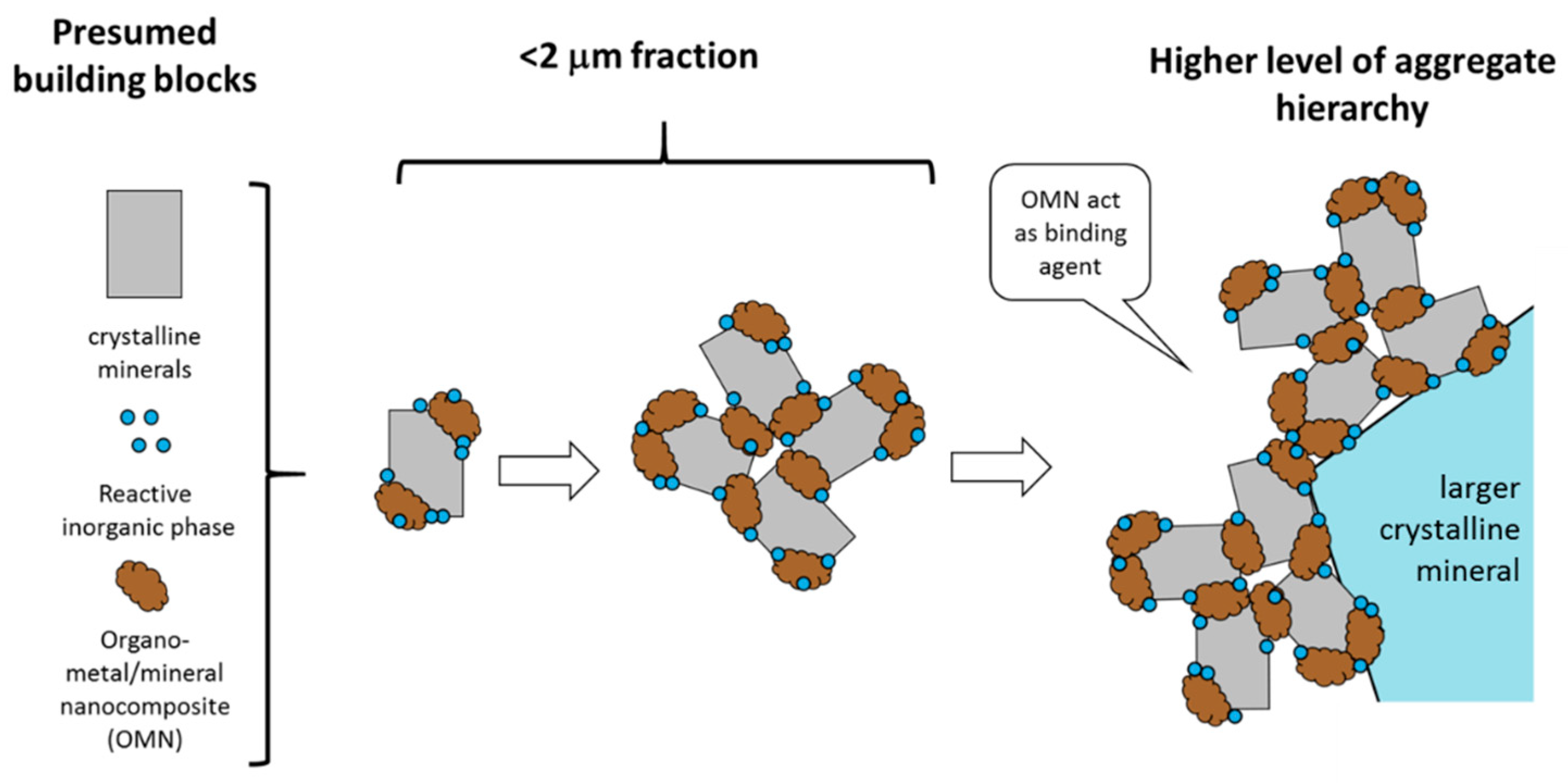
4.3. Organo Mineral/Metal Nanocomposite Act as Binding Agents
4.4. Concluding Remarks
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Schmidt, M.W.I.; Torn, M.S.; Abiven, S.; Dittmar, T.; Guggenberger, G.; Janssens, I.A.; Kleber, M.; Kogel-Knabner, I.; Lehmann, J.; Manning, D.A.C.; et al. Persistence of soil organic matter as an ecosystem property. Nature 2011, 478, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Wieder, W.R.; Boehnert, J.; Bonan, G.B. Evaluating soil biogeochemistry parameterizations in earth system models with observations. Glob. Biogeochem. Cycle 2014, 28, 211–222. [Google Scholar] [CrossRef]
- Luo, Y.Q.; Keenan, T.F.; Smith, M. Predictability of the terrestrial carbon cycle. Glob. Chang. Biol. 2015, 21, 1737–1751. [Google Scholar] [CrossRef] [PubMed]
- Satoh, T.; Yamane, I. Studies on the separation of naturally occurring organo-mineral complexes. 2. Studies by means of densimetric fractionation. J. Sci. Soil Manure 1972, 43, 63–65. [Google Scholar]
- Turchenek, L.W.; Oades, J.M. Fractionation of organomineral complexes by sedimentation and density techniques. Geoderma 1979, 21, 311–343. [Google Scholar] [CrossRef]
- Sollins, P.; Homann, P.; Caldwell, B.A. Stabilization and destabilization of soil organic matter: Mechanisms and controls. Geoderma 1996, 74, 65–105. [Google Scholar] [CrossRef]
- Baldock, J.A.; Skjemstad, J.O. Role of the soil matrix and minerals in protecting natural organic materials against biological attack. Org. Geochem. 2000, 31, 697–710. [Google Scholar] [CrossRef]
- Lutzow, M.V.; Kogel-Knabner, I.; Ekschmitt, K.; Matzner, E.; Guggenberger, G.; Marschner, B.; Flessa, H. Stabilization of organic matter in temperate soils: Mechanisms and their relevance under different soil conditions—A review. Eur. J. Soil Sci. 2006, 57, 426–445. [Google Scholar] [CrossRef]
- Lehmann, J.; Kleber, M. The contentious nature of soil organic matter. Nature 2015, 528, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Mayer, L.M. Relationships between mineral surfaces and organic-carbon concentrations in soils and sediments. Chem. Geol. 1994, 114, 347–363. [Google Scholar] [CrossRef]
- Mayer, L.M.; Xing, B.S. Organic matter-surface area relationships in acid soils. Soil Sci. Soc. Am. J. 2001, 65, 250–258. [Google Scholar] [CrossRef]
- Vogel, C.; Mueller, C.W.; Hoschen, C.; Buegger, F.; Heister, K.; Schulz, S.; Schloter, M.; Kogel-Knabner, I. Submicron structures provide preferential spots for carbon and nitrogen sequestration in soils. Nat. Commun. 2014, 5, 2947. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, J.; Kinyangi, J.; Solomon, D. Organic matter stabilization in soil microaggregates: Implications from spatial heterogeneity of organic carbon contents and carbon forms. Biogeochemistry 2007, 85, 45–57. [Google Scholar] [CrossRef]
- Kögel-Knabner, I.; Guggenberger, G.; Kleber, M.; Kandeler, E.; Kalbitz, K.; Scheu, S.; Eusterhues, K.; Leinweber, P. Organo-mineral associations in temperate soils: Integrating biology, mineralogy, and organic matter chemistry. J. Plant Nutr. Soil Sci. 2008, 171, 61–82. [Google Scholar] [CrossRef]
- Filimonova, S.; Kaufhold, S.; Wagner, F.E.; Häusler, W.; Kögel-Knabner, I. The role of allophane nano-structure and fe oxide speciation for hosting soil organic matter in an allophanic andosol. Geochim. Cosmochim. Acta 2016, 180, 284–302. [Google Scholar] [CrossRef]
- Kaiser, K.; Guggenberger, G. Mineral surfaces and soil organic matter. Eur. J. Soil Sci. 2003, 54, 219–236. [Google Scholar] [CrossRef]
- Calabi-Floody, M.; Bendall, J.S.; Jara, A.A.; Welland, M.E.; Theng, B.K.G.; Rumpel, C.; Mora, M.d.l.L. Nanoclays from an andisol: Extraction, properties and carbon stabilization. Geoderma 2011, 161, 159–167. [Google Scholar] [CrossRef]
- Barré, P.; Fernandez-Ugalde, O.; Virto, I.; Velde, B.; Chenu, C. Impact of phyllosilicate mineralogy on organic carbon stabilization in soils: Incomplete knowledge and exciting prospects. Geoderma 2014, 235–236, 382–395. [Google Scholar] [CrossRef]
- Eusterhues, K.; Rumpel, C.; Kogel-Knabner, I. Organo-mineral associations in sandy acid forest soils: Importance of specific surface area, iron oxides and micropores. Eur. J. Soil Sci. 2005, 56, 753–763. [Google Scholar] [CrossRef]
- Mikutta, R.; Kaiser, K.; Dörr, N.; Vollmer, A.; Chadwick, O.A.; Chorover, J.; Kramer, M.G.; Guggenberger, G. Mineralogical impact on organic nitrogen across a long-term soil chronosequence (0.3–4100kyr). Geochim. Cosmochim. Acta 2010, 74, 2142–2164. [Google Scholar] [CrossRef]
- Six, J.; Bossuyt, H.; Degryze, S.; Denef, K. A history of research on the link between (micro)aggregates, soil biota, and soil organic matter dynamics. Soil Tillage Res. 2004, 79, 7–31. [Google Scholar] [CrossRef]
- Fukumasu, J.; Shaw, L.J. The role of macro-aggregation in regulating enzymatic depolymerization of soil organic nitrogen. Soil Biol. Biochem. 2017, 115, 100–108. [Google Scholar] [CrossRef]
- Zimmerman, A.R.; Goyne, K.W.; Chorover, J.; Komarneni, S.; Brantley, S.L. Mineral mesopore effects on nitrogenous organic matter adsorption. Org. Geochem. 2004, 35, 355–375. [Google Scholar] [CrossRef]
- McCarthy, J.F.; Ilavsky, J.; Jastrow, J.D.; Mayer, L.M.; Perfect, E.; Zhuang, J. Protection of organic carbon in soil microaggregates via restructuring of aggregate porosity and filling of pores with accumulating organic matter. Geochim. Cosmochim. Acta 2008, 72, 4725–4744. [Google Scholar] [CrossRef]
- Totsche, K.U.; Amelung, W.; Gerzabek, M.H.; Guggenberger, G.; Klumpp, E.; Knief, C.; Lehndorff, E.; Mikutta, R.; Peth, S.; Prechtel, A.; et al. Microaggregates in soils. J. Plant Nutr. Soil Sci. 2017, 18, 64–73. [Google Scholar] [CrossRef]
- Solomon, D.; Lehmann, J.; Harden, J.; Wang, J.; Kinyangi, J.; Heymann, K.; Karunakaran, C.; Lu, Y.; Wirick, S.; Jacobsen, C. Micro- and nano-environments of carbon sequestration: Multi-element stxm–nexafs spectromicroscopy assessment of microbial carbon and mineral associations. Chem. Geol. 2012, 329, 53–73. [Google Scholar] [CrossRef]
- Oades, J.M.; Waters, A.G. Aggregate hierarchy in soils. Soil Res. 1991, 29, 815–828. [Google Scholar] [CrossRef]
- Tisdall, J.M.; Oades, J.M. Organic-matter and water-stable aggregates in soils. J. Soil Sci. 1982, 33, 141–163. [Google Scholar] [CrossRef]
- Dahlgren, R.A.; Saigusa, M.; Ugolini, F.C. The nature, properties and management of volcanic soils. Adv. Agron. 2004, 82, 113–182. [Google Scholar]
- Kitagawa, Y. Unit particle of Allophane. Am. Miner 1971, 56, 465–475. [Google Scholar]
- Basile-Doelsch, I.; Amundson, R.; Stone, W.E.E.; Borschneck, D.; Bottero, J.Y.; Moustier, S.; Masin, F.; Colin, F. Mineral control of carbon pools in a volcanic soil horizon. Geoderma 2007, 137, 477–489. [Google Scholar] [CrossRef]
- Levard, C.; Doelsch, E.; Basile-Doelsch, I.; Abidin, Z.; Miche, H.; Masion, A.; Rose, J.; Borschneck, D.; Bottero, J.Y. Structure and distribution of allophanes, imogolite and proto-imogolite in volcanic soils. Geoderma 2012, 183–184, 100–108. [Google Scholar] [CrossRef]
- Thompson, A.; Rancourt, D.G.; Chadwick, O.A.; Chorover, J. Iron solid-phase differentiation along a redox gradient in basaltic soils. Geochim. Cosmochim. Acta 2011, 75, 119–133. [Google Scholar] [CrossRef]
- Nanzyo, M.; Dahlgren, R.; Shoji, S. Chemical characteristics of volcanic ash soils. In Volcanic Ash Soils. Development in Soil Sciences; Shoji, S., Nanzyo, M., Dahlgren, R., Eds.; Elsevier Science Publishers: Amsterdam, The Netherlands, 1993; pp. 145–187. [Google Scholar]
- Takahashi, T.; Dahlgren, R.A. Nature, properties and function of aluminum–humus complexes in volcanic soils. Geoderma 2016, 263, 110–121. [Google Scholar] [CrossRef]
- Wada, K.; Higashi, T. Categories of aluminum-humus and iron-humus complexes in ando soils determined by selective dissolution. J. Soil Sci. 1976, 27, 357–368. [Google Scholar] [CrossRef]
- Shang, C.; Tiessen, H. Organic matter stabilization in two semiarid tropical soils: Size, density, and magnetic separations. Soil Sci. Soc. Am. J. 1998, 62, 1247–1257. [Google Scholar] [CrossRef]
- Parfitt, R.L.; Childs, C.W. Estimation of forms of fe and al—A review, and analysis of contrasting soils by dissolution and mossbauer methods. Aust. J. Soil Res. 1988, 26, 121–144. [Google Scholar] [CrossRef]
- Garrido, E.; Matus, F. Are organo-mineral complexes and allophane content determinant factors for the carbon level in chilean volcanic soils? Catena 2012, 92, 106–112. [Google Scholar] [CrossRef]
- Percival, H.J.; Parfitt, R.L.; Scott, N.A. Factors controlling soil carbon levels in New Zealand grasslands: Is clay content important? Soil Sci. Soc. Am. J. 2000, 64, 1623–1630. [Google Scholar] [CrossRef]
- Matus, F.; Garrido, E.; Sepúlveda, N.; Cárcamo, I.; Panichini, M.; Zagal, E. Relationship between extractable al and organic c in volcanic soils of chile. Geoderma 2008, 148, 180–188. [Google Scholar] [CrossRef]
- Asano, M.; Wagai, R. Evidence of aggregate hierarchy at micro- to submicron scales in an allophanic andisol. Geoderma 2014, 216, 62–74. [Google Scholar] [CrossRef]
- Asano, M.; Wagai, R. Distinctive organic matter pools among particle-size fractions detected by solid-state13C-NMR, δ13C and δ15N analyses only after strong dispersion in an allophanic andisol. Soil Sci. Plant Nutr. 2014, 61, 242–248. [Google Scholar] [CrossRef]
- Baldock, J.A.; Oades, J.M.; Waters, A.G.; Peng, X.; Vassallo, A.M.; Wilson, M.A. Aspects of the chemical-structure of soil organic materials as revealed by solid-state c-13 nmr-spectroscopy. Biogeochemistry 1992, 16, 1–42. [Google Scholar] [CrossRef]
- Schumacher, M.; Christl, I.; Scheinost, A.C.; Jacobsen, C.; Kretzschmar, R. Chemical heterogeneity of organic soil colloids investigated by scanning transmission X-ray microscopy and c-1s nexafs microspectroscopy. Environ. Sci. Technol. 2005, 39, 9094–9100. [Google Scholar] [CrossRef] [PubMed]
- Kinyangi, J.; Solomon, D.; Liang, B.; Lerotic, M.; Wirick, S.; Lehmann, J. Nanoscale biogeocomplexity of the organomineral assemblage in soil. Soil Sci. Soc. Am. J. 2006, 70, 1708–1718. [Google Scholar] [CrossRef]
- Xiao, J.; He, X.; Hao, J.; Zhou, Y.; Zheng, L.; Ran, W.; Shen, Q.; Yu, G. New strategies for submicron characterization the carbon binding of reactive minerals in long-term contrasting fertilized soils: Implications for soil carbon storage. Biogeosciences 2016, 13, 3607–3618. [Google Scholar] [CrossRef]
- Stuckey, J.W.; Yang, J.; Wang, J.; Sparks, D.L. Advances in scanning transmission X-ray microscopy for elucidating soil biogeochemical processes at the submicron scale. J. Environ. Q. 2017, 46, 1166–1174. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Dynes, J.J.; Wang, J.; Karunakaran, C.; Sparks, D.L. Soft X-ray spectromicroscopy study of mineral-organic matter associations in pasture soil clay fractions. Environ. Sci. Technol. 2014, 48, 6678–6686. [Google Scholar] [CrossRef] [PubMed]
- Keiluweit, M.; Bougoure, J.J.; Zeglin, L.H.; Myrold, D.D.; Weber, P.K.; Pett-Ridge, J.; Kleber, M.; Nico, P.S. Nano-scale investigation of the association of microbial nitrogen residues with iron (hydr)oxides in a forest soil o-horizon. Geochim. Cosmochim. Acta 2012, 95, 213–226. [Google Scholar] [CrossRef]
- Wan, J.; Tyliszczak, T.; Tokunaga, T.K. Organic carbon distribution, speciation, and elemental correlations within soil microaggregates: Applications of stxm and nexafs spectroscopy. Geochim. Cosmochim. Acta 2007, 71, 5439–5449. [Google Scholar] [CrossRef]
- IUSS Working Group WRB. World Reference Base for Soil Resources 2006; World Soil Resources Reports No. 103; FAO: Rome, Italy, 2006. [Google Scholar]
- North, P.F. Toward an absolute measurement of soil structural stability using ultrasound. Eur. J. Soil Sci. 1976, 27, 451–459. [Google Scholar] [CrossRef]
- Wagai, R.; Kishimoto-Mo, A.W.; Yonemura, S.; Shirato, Y.; Hiradate, S.; Yagasaki, Y. Linking temperature sensitivity of soil organic matter decomposition to its molecular structure, accessibility, and microbial physiology. Glob. Chang. Biol. 2013, 19, 1114–1125. [Google Scholar] [CrossRef] [PubMed]
- Wagai, R.; Mayer, L.M.; Kitayama, K. Extent and nature of organic coverage of soil mineral surfaces assessed by a gas sorption approach. Geoderma 2009, 149, 152–160. [Google Scholar] [CrossRef]
- Hiradate, S. Structural changes of allophane during purification procedures as determined by solid-state al-27 and si-29 nmr. Clays Clay Miner. 2005, 53, 653–658. [Google Scholar] [CrossRef]
- Tashiro, Y.; Nakao, A.; Wagai, R.; Yanai, J.; Kosaki, T. Inhibition of radiocesium adsorption on 2:1 clay minerals under acidic soil environment: Effect of organic matter vs. Hydroxy aluminum polymer. Geoderma 2018, 319, 52–60. [Google Scholar] [CrossRef]
- Stuiver, M.; Polach, H.A. Reporting of c-14 data—Discussion. Radiocarbon 1977, 19, 355–363. [Google Scholar] [CrossRef]
- Takeichi, Y.; Inami, N.; Suga, H.; Miyamoto, C.; Ueno, T.; Mase, K.; Takahashi, Y.; Ono, K. Design and performance of a compact scanning transmission x-ray microscope at the photon factory. Rev. Sci. Instrum. 2016, 87, 013704. [Google Scholar] [CrossRef] [PubMed]
- Hitchcock, A.P. Hitchcock Group aXis 2000. Available online: http://unicorn.mcmaster.ca/axis/aXis2000-windows-pre-IDL8.3.html (accessed on 21 May 2018).
- Mitsunobu, S.; Zhu, M.; Takeichi, Y.; Ohigashi, T.; Suga, H.; Makita, H.; Sakata, M.; Ono, K.; Mase, K.; Takahashi, Y. Nanoscale identification of extracellular organic substances at the microbe–mineral interface by scanning transmission x-ray microscopy. Chem. Lett. 2015, 44, 91–93. [Google Scholar] [CrossRef]
- Hall, P.; Churchman, G.J.; Theng, B.K.G. Size distribution of allophane unit particles in aqueous suspension. Clays Clay Miner. 1985, 33, 345–349. [Google Scholar] [CrossRef]
- Karube, J. Water retention, adsorption, and microstructure of allophane and imogolite. J. Jpn. Soc. Soil Phys. 2004, 96, 27–33. [Google Scholar]
- Egashira, K.; Aomine, S. Effects of drying and and heating on the surface area of allophane and imogolite. Clay Sci. 1974, 4, 231–242. [Google Scholar]
- Mayer, L.M. Extent of coverage of mineral surfaces by organic matter in marine sediments. Geochim. Cosmochim. Acta 1999, 63, 207–215. [Google Scholar] [CrossRef]
- Kaiser, K.; Guggenberger, G. Sorptive stabilization of organic matter by microporous goethite: Sorption into small pores vs. Surface complexation. Eur. J. Soil Sci. 2007, 58, 45–59. [Google Scholar] [CrossRef]
- Basile-Doelsch, I.; Balesdent, J.; Rose, J. Are interactions between organic compounds and nanoscale weathering minerals the key drivers of carbon storage in soils? Environ. Sci. Technol. 2015, 49, 3997–3998. [Google Scholar] [CrossRef] [PubMed]
- Shindo, H.; Higashi, T.; Matsui, Y. Comparison of humic acids from charred residues of susuki (eulalia, miscanthus sinensisa.) and from the a horizons of volcanic ash soils. Soil Sci. Plant Nutr. 1986, 32, 579–586. [Google Scholar] [CrossRef]
- Kramer, M.G.; Sanderman, J.; Chadwick, O.A.; Chorover, J.; Vitousek, P.M. Long-term carbon storage through retention of dissolved aromatic acids by reactive particles in soil. Glob. Chang. Biol. 2012, 18, 2594–2605. [Google Scholar] [CrossRef]
- Lehmann, J.; Liang, B.Q.; Solomon, D.; Lerotic, M.; Luizao, F.; Kinyangi, J.; Schafer, T.; Wirick, S.; Jacobsen, C. Near-edge X-ray absorption fine structure (nexafs) spectroscopy for mapping nano-scale distribution of organic carbon forms in soil: Application to black carbon particles. Glob. Biogeochem. Cycles 2005, 19. [Google Scholar] [CrossRef]
- Liang, B.; Lehmann, J.; Solomon, D.; Kinyangi, J.; Grossman, J.; O’Neill, B.; Skjemstad, J.O.; Thies, J.; Luizão, F.J.; Petersen, J.; et al. Black carbon increases cation exchange capacity in soils. Soil Sci. Soc. Am. J. 2006, 70, 1719–1730. [Google Scholar] [CrossRef]
- Abe, T.; Watanabe, A. X-ray photoelectron spectroscopy of nitrogen functional groups in soil humic acids. Soil Sci. 2004, 169, 35–43. [Google Scholar] [CrossRef]
- Ikeya, K.; Hikage, T.; Arai, S.; Watanabe, A. Size distribution of condensed aromatic rings in various soil humic acids. Org. Geochem. 2011, 42, 55–61. [Google Scholar] [CrossRef]
- Kleber, M.; Nico, P.S.; Plante, A.; Filley, T.; Kramer, M.; Swanston, C.; Sollins, P. Old and stable soil organic matter is not necessarily chemically recalcitrant: Implications for modeling concepts and temperature sensitivity. Glob. Chang. Biol. 2011, 17, 1097–1107. [Google Scholar] [CrossRef]
- Mikutta, R.; Schaumann, G.E.; Gildemeister, D.; Bonneville, S.; Kramer, M.G.; Chorover, J.; Chadwick, O.A.; Guggenberger, G. Biogeochemistry of mineral-organic associations across a long-term mineralogical soil gradient (0.3-4100 kyr), hawaiian islands. Geochim. Cosmochim. Acta 2009, 73, 2034–2060. [Google Scholar] [CrossRef]
- Dahlgren, R.A.; Dragoo, J.P.; Ugolini, F.C. Weathering of mt. St. Helens tephra under a cryic-udic climatic regime. Soil Sci. Soc. Am. J. 1997, 61, 1519–1525. [Google Scholar] [CrossRef]
- Wagai, R.; Kajiura, M.; Uchida, M.; Asano, M. Distinctive roles of two aggregate binding agents in allophanic Andisols: Young carbon and poorly-crystalline metal phases with old carbon. Soil Syst. 2018, 2, 29. [Google Scholar] [CrossRef]
- Wagai, R.; Kajiura, M.; Asano, M.; Hiradate, S. Nature of soil organo-mineral assemblage examined by sequential density fractionation with and without sonication: Is allophanic soil different? Geoderma 2015, 241–242, 295–305. [Google Scholar] [CrossRef]
- Heckman, K.; Grandy, A.S.; Gao, X.; Keiluweit, M.; Wickings, K.; Carpenter, K.; Chorover, J.; Rasmussen, C. Sorptive fractionation of organic matter and formation of organo-hydroxy-aluminum complexes during litter biodegradation in the presence of gibbsite. Geochim. Cosmochim. Acta 2013, 121, 667–683. [Google Scholar] [CrossRef]
- Baisden, W.T.; Amundson, R.; Cook, A.C.; Brenner, D.L. Turnover and storage of c and n in five density fractions from california annual grassland surface soils. Glob. Biogeochem. Cycles 2002, 16. [Google Scholar] [CrossRef]
- Hatton, P.J.; Bode, S.; Angeli, N.; Boeckx, P.; Zeller, B.; Boiry, S.; Gelhaye, L.; Derrien, D. Assimilation and accumulation of c by fungi and bacteria attached to soil density fractions. Soil Biol. Biochem. 2014, 79, 132–139. [Google Scholar] [CrossRef]
- Sollins, P.; Kramer, M.G.; Swanston, C.; Lajtha, K.; Filley, T.; Aufdenkampe, A.K.; Wagai, R.; Bowden, R.D. Sequential density fractionation across soils of contrasting mineralogy: Evidence for both microbial- and mineral-controlled soil organic matter stabilization. Biogeochemistry 2009, 96, 209–231. [Google Scholar] [CrossRef]
- Cotrufo, M.F.; Soong, J.L.; Horton, A.J.; Campbell, E.E.; Haddix, M.L.; Wall, D.H.; Parton, A.J. Formation of soil organic matter via biochemical and physical pathways of litter mass loss. Nat. Geosci. 2015, 8, 2520. [Google Scholar] [CrossRef]
- Chenu, C.; Plante, A.F. Clay-sized organo-mineral complexes in a cultivation chronosequence: Revisiting the concept of the ‘primary organo-mineral complex’. Eur. J. Soil Sci. 2006, 57, 596–607. [Google Scholar] [CrossRef]
- Christensen, B.T. Physical fractionationof soil and organic matter in primary particle size and density separates. Adv. Soil Sci. 1992, 20, 1–89. [Google Scholar]
- Lawrence, C.R.; Harden, J.W.; Xu, X.; Schulz, M.S.; Trumbore, S.E. Long-term controls on soil organic carbon with depth and time: A case study from the cowlitz river chronosequence, wa USA. Geoderma 2015, 247–248, 73–87. [Google Scholar] [CrossRef]
- Torn, M.S.; Trumbore, S.E.; Chadwick, O.A.; Vitousek, P.M.; Hendricks, D.M. Mineral control of soil organic carbon storage and turnover. Nature 1997, 389, 170–173. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mass | TOC a | TN a | TOC Distribution | PP b | OX b | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Al | Fe | Al | Fe | Si | |||||||
| % | mg g−1 | % | mg g−1 | ||||||||
| bulk soil | 53.2 | (0.1) | 4.5 | (0.0) | - | 3.47 | 1.35 | 37.25 | 17.14 | 16.86 | |
| Size fractions | |||||||||||
| <0.2 | 11.88 | 71.4 | (3.2) | 7.5 | (0.4) | 16 | 7.43 | 2.83 | 71.1 | 35.0 | 33.6 |
| 0.2–2 | 36.49 | 68.0 | (1.6) | 5.4 | (0.1) | 47 | 6.25 | 1.86 | 50.7 | 23.4 | 23.4 |
| 2–53 | 37.65 | 32.5 | (3.4) | 1.8 | (0.3) | 23 | 2.26 | 0.52 | 12.9 | 9.66 | 5.58 |
| 53–4000 | 6.61 | 2.07 | - | tr | - | 0.3 | 0.26 | 0.40 | 0.93 | 21.5 | 0.70 |
| SSA Untreated | SSA Oxidized | Organic Coverage | Total Surface Area Distribution | Δ14C | Radiocarbon Lab. Code No. | ||
|---|---|---|---|---|---|---|---|
| m2 g−1 | % | ‰ | |||||
| bulk soil | 85 | NA | - | - | −145.5 | - | TERRA-112210d21 |
| Size fractions (µm) | |||||||
| <0.2 | 99 | 267 | 63 | 28 | −94.8 | (3.4) | Beta-311744 |
| 0.2–2 | 84 | 172 | 51 | 55 | −114.8 | (3.3) | Beta-311745 |
| 2–53 | 30 | 49 | 39 | 16 | −201.7 | (3.0) | Beta-311746 |
| 53–4000 | 9 | 11 | 14 | 1 | NA | - | - |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Asano, M.; Wagai, R.; Yamaguchi, N.; Takeichi, Y.; Maeda, M.; Suga, H.; Takahashi, Y. In Search of a Binding Agent: Nano-Scale Evidence of Preferential Carbon Associations with Poorly-Crystalline Mineral Phases in Physically-Stable, Clay-Sized Aggregates. Soil Syst. 2018, 2, 32. https://doi.org/10.3390/soilsystems2020032
Asano M, Wagai R, Yamaguchi N, Takeichi Y, Maeda M, Suga H, Takahashi Y. In Search of a Binding Agent: Nano-Scale Evidence of Preferential Carbon Associations with Poorly-Crystalline Mineral Phases in Physically-Stable, Clay-Sized Aggregates. Soil Systems. 2018; 2(2):32. https://doi.org/10.3390/soilsystems2020032
Chicago/Turabian StyleAsano, Maki, Rota Wagai, Noriko Yamaguchi, Yasuo Takeichi, Makoto Maeda, Hiroki Suga, and Yoshio Takahashi. 2018. "In Search of a Binding Agent: Nano-Scale Evidence of Preferential Carbon Associations with Poorly-Crystalline Mineral Phases in Physically-Stable, Clay-Sized Aggregates" Soil Systems 2, no. 2: 32. https://doi.org/10.3390/soilsystems2020032