Distinctive Roles of Two Aggregate Binding Agents in Allophanic Andisols: Young Carbon and Poorly-Crystalline Metal Phases with Old Carbon


Abstract
:
1. Introduction
Terminology
2. Materials and Methods
2.1. Study Site and Soil Properties
2.2. Density Fractionation
2.3. Characterization of Isolated Density Fractions
2.4. Standing Stocks of C and Extractable Metals
2.5. Statistical Analysis
3. Results
3.1. Carbon and Nitrogen
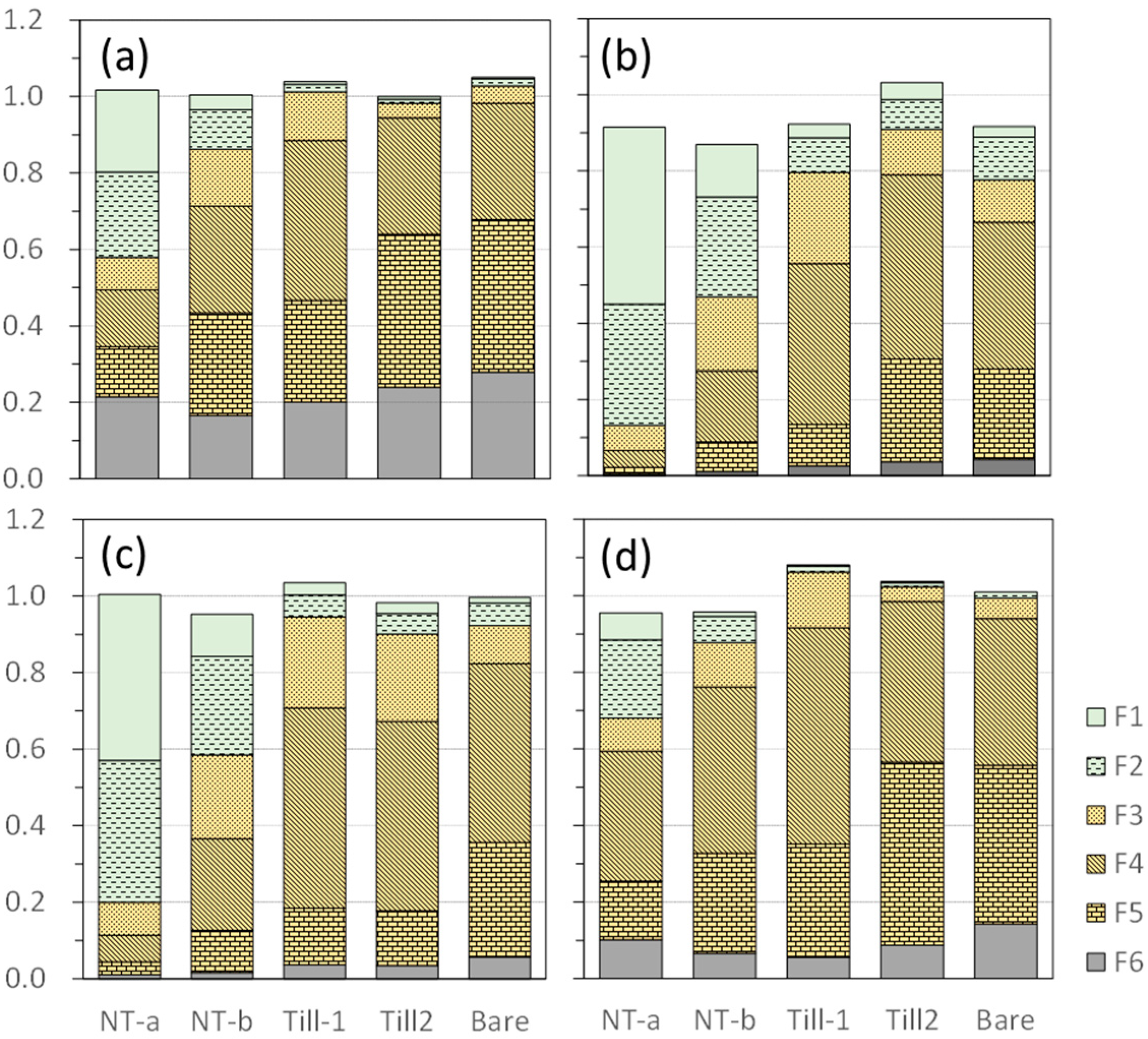
3.1.1. Recovery, Concentration and Distribution
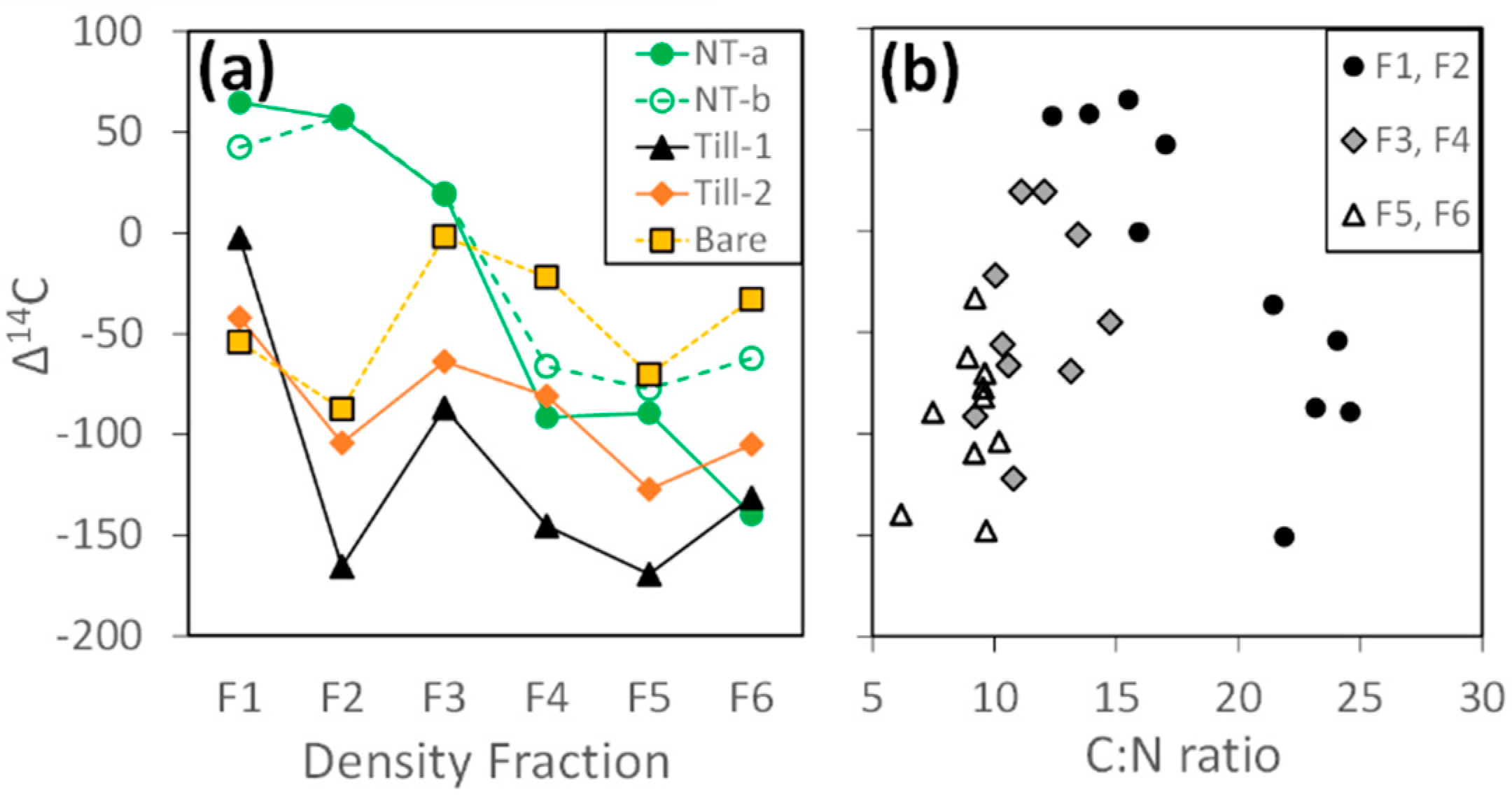
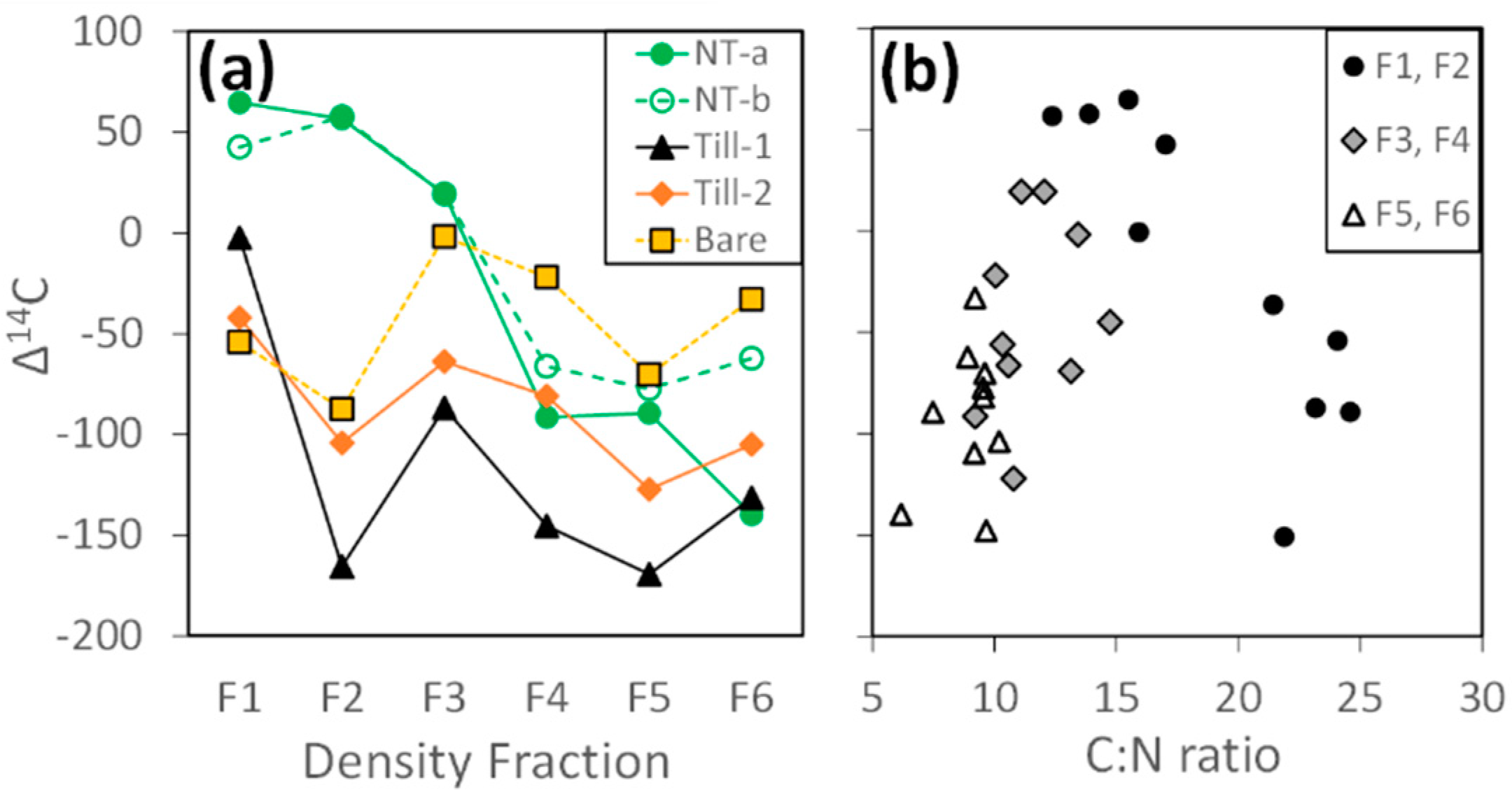
3.1.2. Radiocarbon
3.2. Extractable Iron, Aluminum, Silica, and Manganese
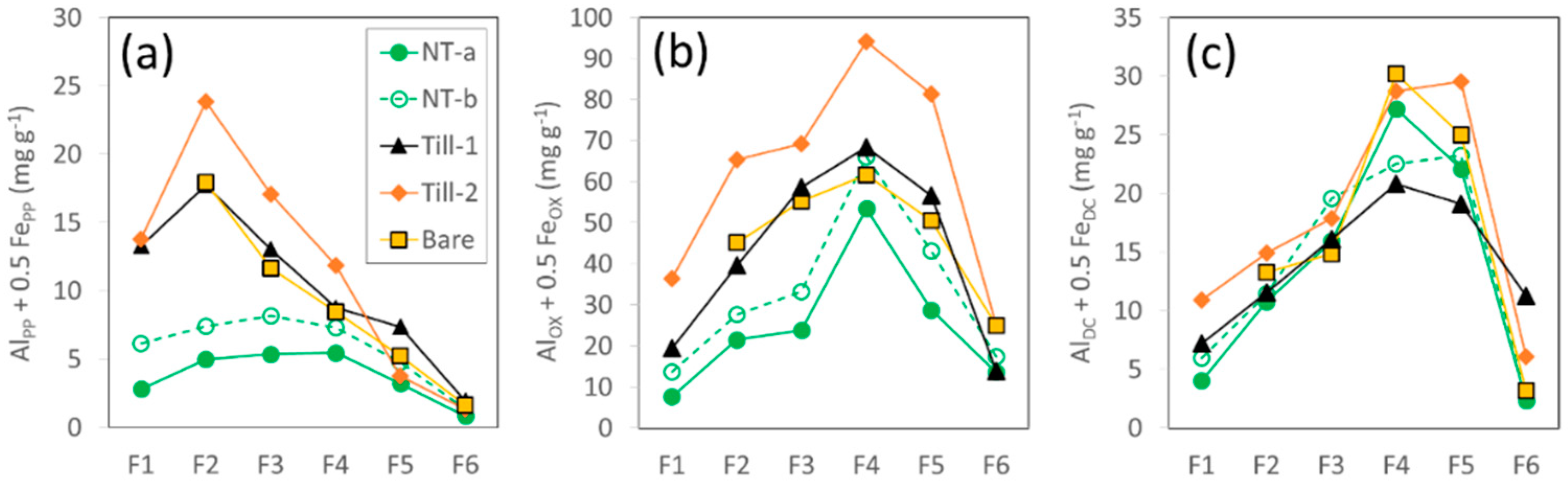
3.2.1. Mineral Composition and Metal Concentration
3.2.2. Recovery and Distribution
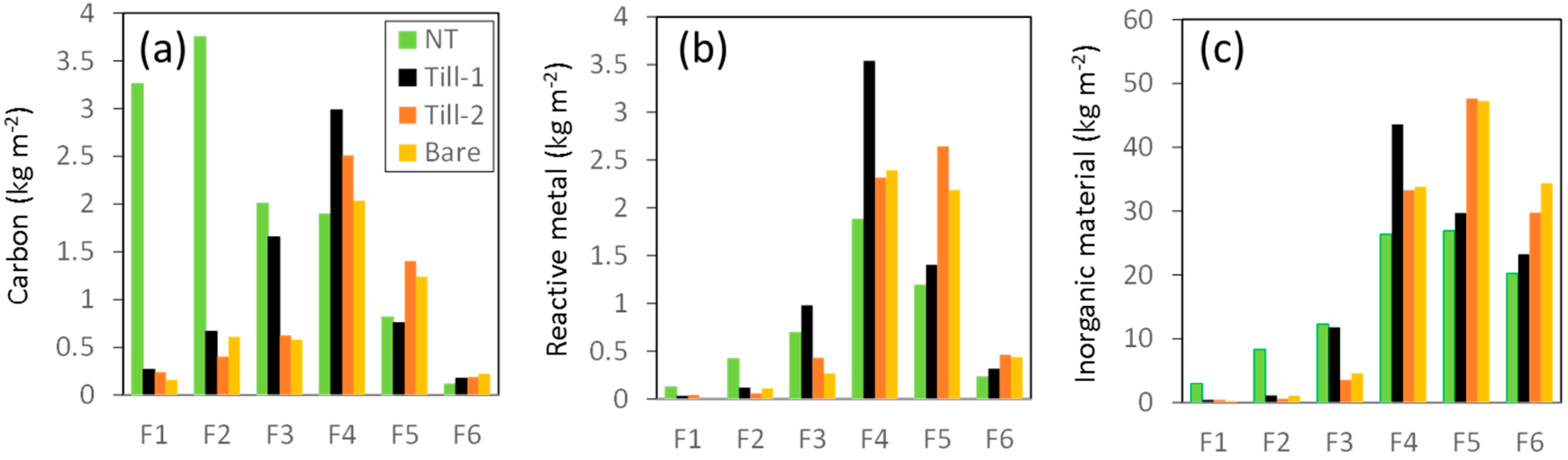
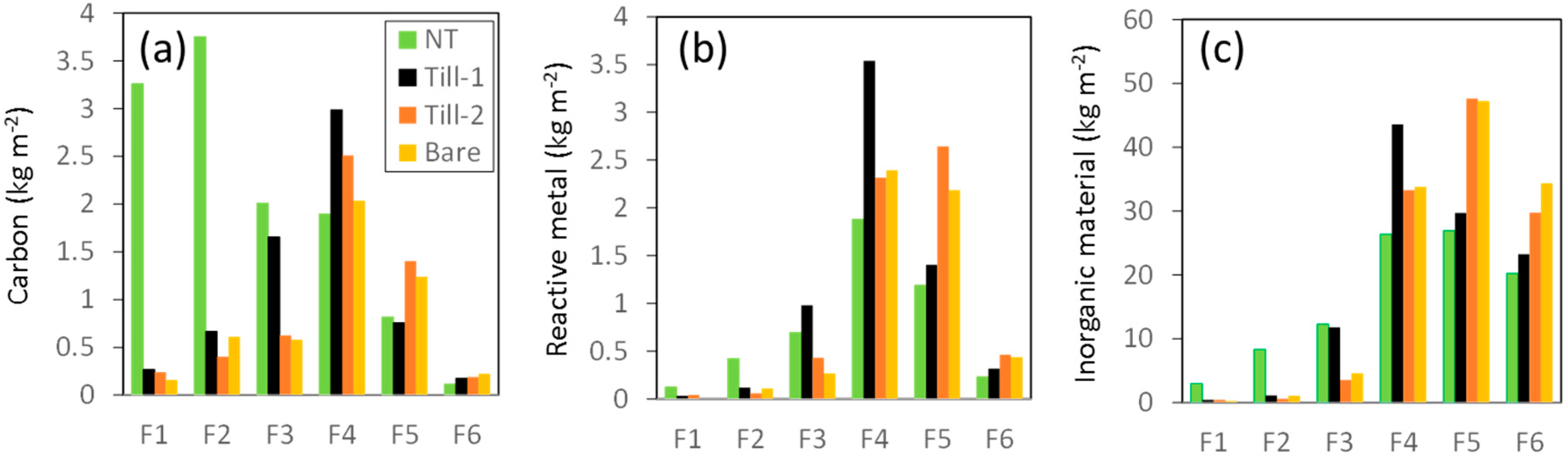
3.3. Standing Stocks of Organic Matter and Reactive Metal Phases
4. Discussion
4.1. Different Roles of Organic Matter and Reactive Metal Phases as Binding Agents
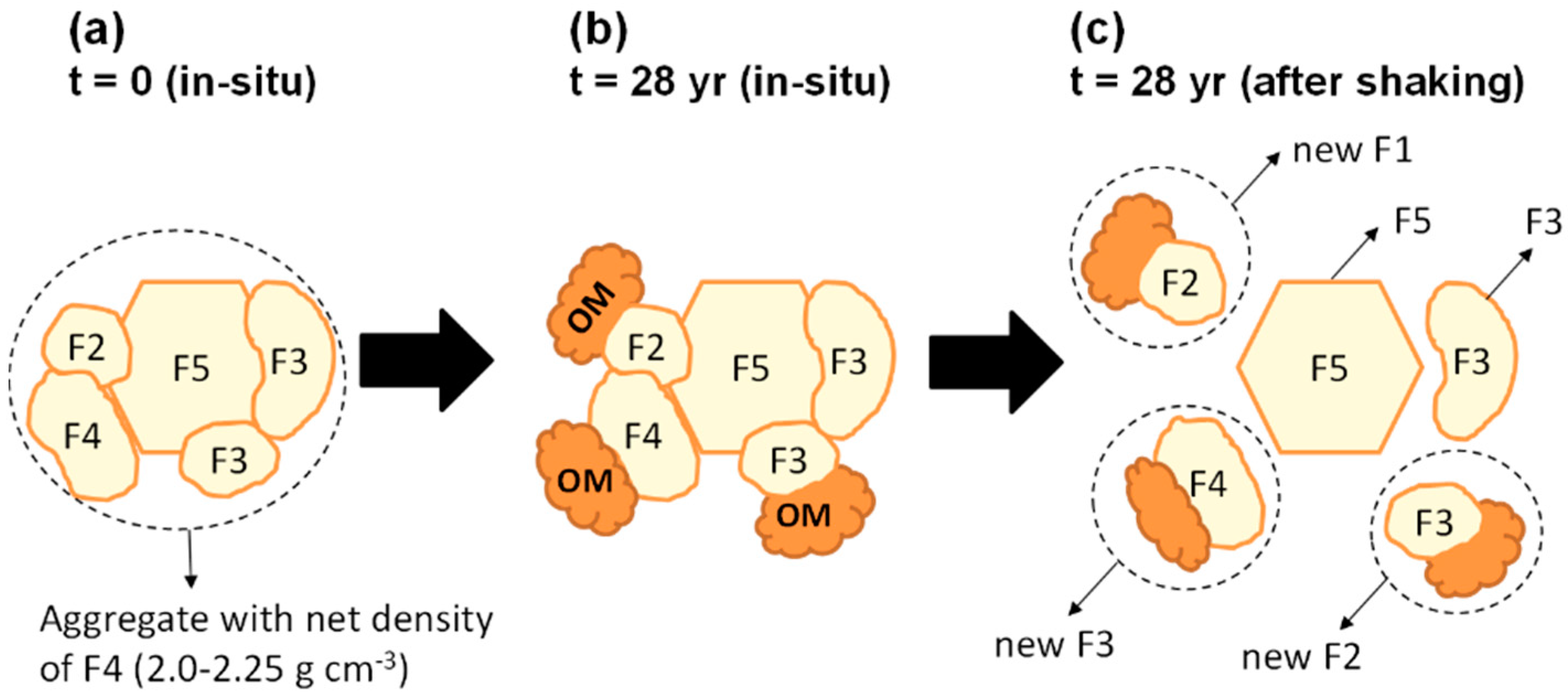
4.2. Dynamic Changes in Organo-Mineral Aggregation Controlled by OM Input
4.3. Insights from Radiocarbon Analysis: Two Pools of Stable C
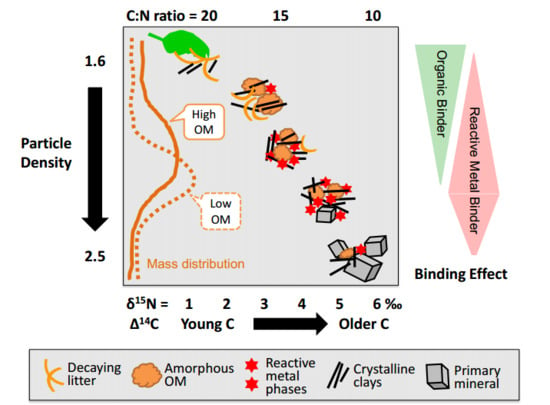
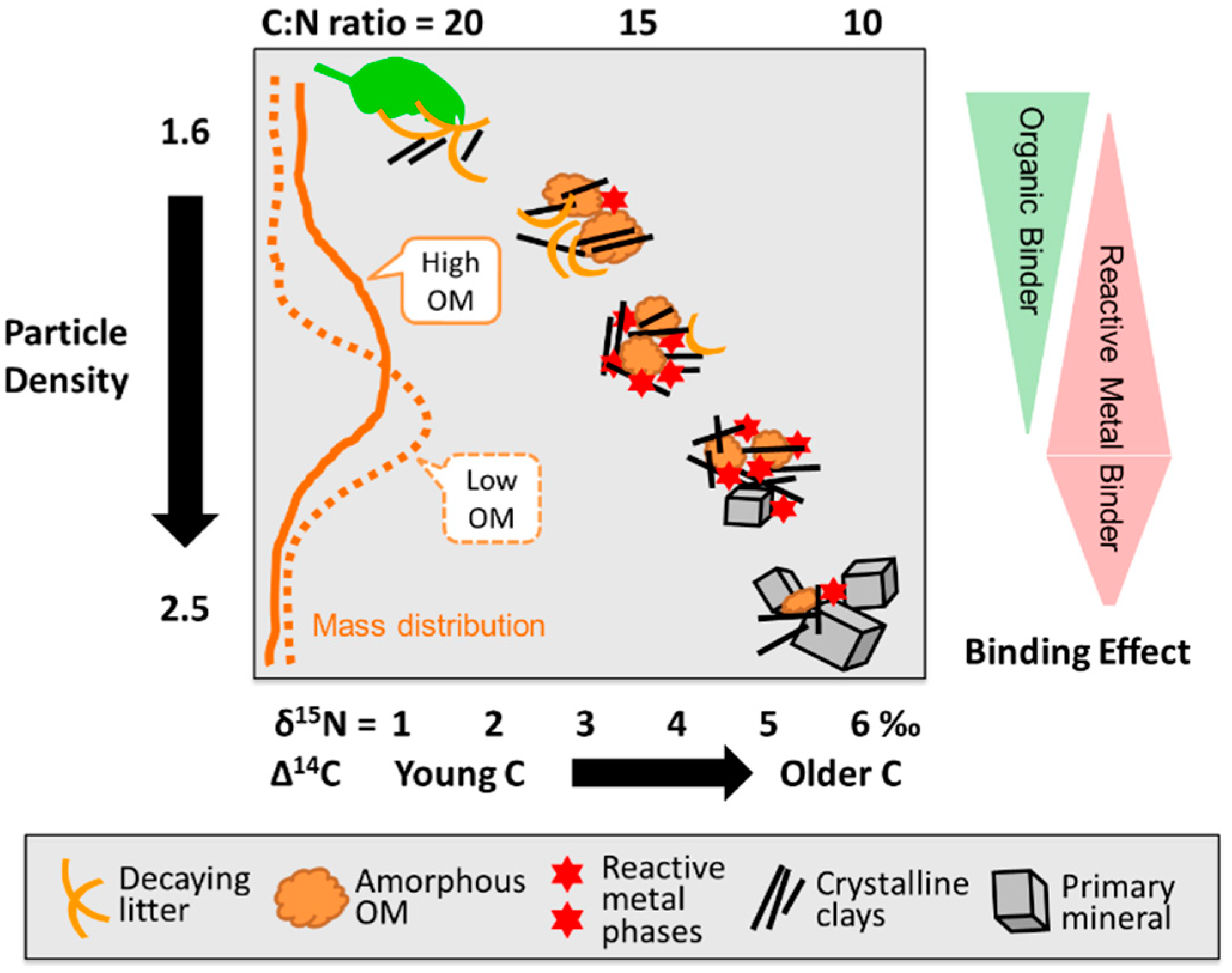
4.4. A Tentative Synthesis: Linking Aggregate Formation, Binding Agents, and OM Dynamics
4.5. Concluding Remarks with Some Implications
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A. Mass Balance (Elemental Budget) and Caution in Pyrophosphate Extraction
Appendix B. Site History Inferred from Radiocarbon Results
References
- Oades, J. Soil organic matter and structural stability: Mechanisms and implications for management. Plant Soil 1984, 76, 319–337. [Google Scholar] [CrossRef]
- Plante, A.F.; Parton, W.J. The dynamics of soil organic matter and nutrient cycling. In Soil Microbiology, Ecology and Biochemistry, 3rd ed.; Academic Press: San Diego, CA, USA, 2007; pp. 433–467. [Google Scholar]
- Chenu, C.; Cosentino, D. Microbial regulation of soil structural dynamics. In The Architecture and Biology of Soils: Life in Inner Space; Ritz, K., Young, I., Eds.; CAB: Wallingford, UK, 2011; pp. 37–70. [Google Scholar]
- Six, J.; Bossuyt, H.; Degryze, S.; Denef, K. A history of research on the link between (micro)aggregates, soil biota, and soil organic matter dynamics. Soil Tillage Res. 2004, 79, 7–31. [Google Scholar] [CrossRef]
- Totsche, K.U.; Amelung, W.; Gerzabek, M.H.; Guggenberger, G.; Klumpp, E.; Knief, C.; Lehndorff, E.; Mikutta, R.; Peth, S.; Prechtel, A.; et al. Microaggregates in soils. J. Plant Nutr. Soil Sci. 2018, 181, 104–136. [Google Scholar] [CrossRef]
- Tisdall, J.M.; Oades, J.M. Organic-matter and water-stable aggregates in soils. J. Soil Sci. 1982, 33, 141–163. [Google Scholar] [CrossRef]
- Abiven, S.; Menasseri, S.; Chenu, C. The effects of organic inputs over time on soil aggregate stability—A literature analysis. Soil Biol. Biochem. 2009, 41, 1–12. [Google Scholar] [CrossRef]
- Jastrow, J.D. Soil aggregate formation and the accrual of particulate and mineral-associated organic matter. Soil Biol. Biochem. 1996, 28, 665–676. [Google Scholar] [CrossRef]
- Oades, J.M.; Waters, A.G. Aggregate hierarchy in soils. Aust. J. Soil Res. 1991, 29, 815–828. [Google Scholar] [CrossRef]
- Krishna Murti, G.S.R.; Huang, P.M. Influence of constituents on the stability of mechanical separates of soils representing major taxonomic orders. Appl. Clay Sci. 1987, 2, 299–308. [Google Scholar] [CrossRef]
- Duiker, S.W.; Rhoton, F.E.; Torrent, J.; Smeck, N.E.; Lal, R. Iron (hydr)oxide crystallinity effects on soil aggregation. Soil Sci. Soc. Am. J. 2003, 67, 606–611. [Google Scholar] [CrossRef]
- Emerson, W. A classification of soil aggregates based on their coherence in water. Soil Res. 1967, 5, 47–57. [Google Scholar] [CrossRef]
- Boix-Fayos, C.; Calvo-Cases, A.; Imeson, A.C.; Soriano-Soto, M.D. Influence of soil properties on the aggregation of some mediterranean soils and the use of aggregate size and stability as land degradation indicators. Catena 2001, 44, 47–67. [Google Scholar] [CrossRef]
- Chenu, C.; Plante, A.F. Clay-sized organo-mineral complexes in a cultivation chronosequence: Revisiting the concept of the ‘primary organo-mineral complex’. Eur. J. Soil Sci. 2006, 57, 596–607. [Google Scholar] [CrossRef]
- Boudot, J.P.; Bel Hadj Brahim, A.; Steiman, R.; Seigle-Murandi, F. Biodegradation of synthetic organo-metallic complexes of iron and aluminium with selected metal to carbon ratios. Soil Biol. Biochem. 1989, 21, 961–966. [Google Scholar] [CrossRef]
- Kaiser, K.; Guggenberger, G. Sorptive stabilization of organic matter by microporous goethite: Sorption into small pores vs. Surface complexation. Eur. J. Soil Sci. 2007, 58, 45–59. [Google Scholar] [CrossRef]
- Schneider, M.P.W.; Scheel, T.; Mikutta, R.; van Hees, P.; Kaiser, K.; Kalbitz, K. Sorptive stabilization of organic matter by amorphous al hydroxide. Geochim. Cosmochim. Acta 2010, 74, 1606–1619. [Google Scholar] [CrossRef] [Green Version]
- Asano, M.; Wagai, R. Evidence of aggregate hierarchy at micro- to submicron scales in an allophanic andisol. Geoderma 2014, 216, 62–74. [Google Scholar] [CrossRef]
- Field, D.J.; Minasny, B. A description of aggregate liberation and dispersion in a horizons of Australian vertisols by ultrasonic agitation. Geoderma 1999, 91, 11–26. [Google Scholar] [CrossRef]
- Vrdoljak, G.; Sposito, G. Soil aggregate hierarchy in a Brazilian oxisol. Dev. Soil Sci. 2002, 28A, 197–217. [Google Scholar]
- Shoji, S.; Nanzyo, M.; Dahlgren, R. Volcanic Ash Soils: Genesis, Properties, and Utilization; Elsevier: Amsterdam, The Netherlands, 1993; p. 288. [Google Scholar]
- Basile-Doelsch, I.; Amundson, R.; Stone, W.E.E.; Borschneck, D.; Bottero, J.Y.; Moustier, S.; Masin, F.; Colin, F. Mineral control of carbon pools in a volcanic soil horizon. Geoderma 2007, 137, 477–489. [Google Scholar] [CrossRef]
- Percival, H.J.; Parfitt, R.L.; Scott, N.A. Factors controlling soil carbon levels in new zealand grasslands is clay content important? Soil Sci. Soc. Am. J. 2000, 64, 1623–1630. [Google Scholar] [CrossRef]
- Wada, K.; Higashi, T. The categories of aluminum- and iron-humus complexes in ando soils determined by selective dissolution. J. Soil Sci. 1976, 27, 357–368. [Google Scholar] [CrossRef]
- Huygens, D.; Boeckx, P.; Van Cleemput, O.; Oyarzun, C.; Godoy, R. Aggregate and soil organic carbon dynamics in south chilean andisols. Biogeosciences 2005, 2, 159–174. [Google Scholar] [CrossRef] [Green Version]
- Rasmussen, C.; Torn, M.S.; Southard, R.J. Mineral assemblage and aggregates control carbon dynamics in a California conifer forest. Soil Sci. Soc. Am. J. 2005, 69, 1711–1721. [Google Scholar] [CrossRef]
- Wagai, R.; Mayer, L.M. Sorptive stabilization of organic matter in soils by hydrous iron oxides. Geochim. Cosmochim. Acta 2007, 71, 25–35. [Google Scholar] [CrossRef]
- Kramer, M.G.; Sanderman, J.; Chadwick, O.A.; Chorover, J.; Vitousek, P.M. Long-term carbon storage through retention of dissolved aromatic acids by reactive particles in soil. Glob. Chang. Biol. 2012, 18, 2594–2605. [Google Scholar] [CrossRef]
- Rasmussen, C.; Heckman, K.; Wieder, W.R.; Keiluweit, M.; Lawrence, C.R.; Berhe, A.A.; Blankinship, J.C.; Crow, S.E.; Druhan, J.L.; Pries, C.E.H.; et al. Beyond clay: Towards an improved set of variables for predicting soil organic matter content. Biogeochemistry 2018, 137, 297–306. [Google Scholar] [CrossRef]
- Christensen, B. Physical fractionation of soil and organic matter in primary particle size and density separates. In Advances in Soil Science; Stewart, B.A., Ed.; Springer: New York, NY, USA, 1992; Volume 20, pp. 1–90. [Google Scholar]
- Von Lützow, M.; Kögel-Knabner, I.; Ekschmitt, K.; Flessa, H.; Guggenberger, G.; Matzner, E.; Marschner, B. Som fractionation methods: Relevance to functional pools and to stabilization mechanisms. Soil Biol. Biochem. 2007, 39, 2183–2207. [Google Scholar] [CrossRef]
- Hatton, P.-J.; Bodé, S.; Angeli, N.; Boeckx, P.; Zeller, B.; Boiry, S.; Gelhaye, L.; Derrien, D. Assimilation and accumulation of c by fungi and bacteria attached to soil density fractions. Soil Biol. Biochem. 2014, 79, 132–139. [Google Scholar] [CrossRef]
- Kondo, M.; Uchida, M.; Shibata, Y. Radiocarbon-based residence time estimates of soil organic carbon in a temperate forest: Case study for the density fractionation for Japanese volcanic ash soil. Nucl. Instrum. Methods Phys. Res. Sect. B Beam Interact. Mater. At. 2010, 268, 1073–1076. [Google Scholar] [CrossRef]
- Lajtha, K.; Townsend, K.L.; Kramer, M.G.; Swanston, C.; Bowden, R.D.; Nadelhoffer, K. Changes to particulate versus mineral-associated soil carbon after 50 years of litter manipulation in forest and prairie experimental ecosystems. Biogeochemistry 2014, 119, 341–360. [Google Scholar] [CrossRef]
- Sollins, P.; Kramer, M.G.; Swanston, C.; Lajtha, K.; Filley, T.; Aufdenkampe, A.K.; Wagai, R.; Bowden, R.D. Sequential density fractionation across soils of contrasting mineralogy: Evidence for both microbial- and mineral-controlled soil organic matter stabilization. Biogeochemistry 2009, 96, 209–231. [Google Scholar] [CrossRef]
- Sollins, P.; Swanston, C.; Kleber, M.; Filley, T.; Kramer, M.; Crow, S.; Caldwell, B.A.; Lajtha, K.; Bowden, R. Organic c and n stabilization in a forest soil: Evidence from sequential density fractionation. Soil Biol. Biochem. 2006, 38, 3313–3324. [Google Scholar] [CrossRef]
- Baisden, W.T.; Amundson, R.; Cook, A.C.; Brenner, D.L. Turnover and storage of c and n in five density fractions from california annual grassland surface soils. Glob. Biogeochem. Cycle 2002, 16, 1117. [Google Scholar] [CrossRef]
- Castanha, C.; Trumbore, S.; Amundson, R. Methods of separating soil carbon pools affect the chemistry and turnover time of isolated fractions. Radiocarbon 2008, 50, 83–97. [Google Scholar] [CrossRef]
- Golchin, A.; Baldock, J.A.; Oades, J.M. A model linking organic matter decomposition, chemistry, and aggregate dynamics. In Soil Processes and the Carbon Cycle; Lal, R., Kimble, J.M., Follett, R.F., Stewart, B.A., Eds.; CRC Press: Boca Raton, FL, USA, 1997; pp. 245–266. [Google Scholar]
- Gunina, A.; Kuzyakov, Y. Pathways of litter c by formation of aggregates and som density fractions: Implications from 13c natural abundance. Soil Biol. Biochem. 2014, 71, 95–104. [Google Scholar] [CrossRef]
- Crow, S.; Reeves, M.; Schubert, O.; Sierra, C. Optimization of method to quantify soil organic matter dynamics and carbon sequestration potential in volcanic ash soils. Biogeochemistry 2014, 123, 1–21. [Google Scholar]
- Wagai, R.; Kajiura, M.; Asano, M.; Hiradate, S. Nature of soil organo-mineral assemblage examined by sequential density fractionation with and without sonication: Is allophanic soil different? Geoderma 2015, 241–242, 295–305. [Google Scholar] [CrossRef]
- Kaiser, K.; Guggenberger, G. Distribution of hydrous aluminium and iron over density fractions depends on organic matter load and ultrasonic dispersion. Geoderma 2007, 140, 140–146. [Google Scholar] [CrossRef]
- Mayer, L.M.; Schick, L.L.; Hardy, K.R.; Wagai, R.; McCarthy, J. Organic matter in small mesopores in sediments and soils. Geochim. Cosmochim. Acta 2004, 68, 3863–3872. [Google Scholar] [CrossRef]
- Nickel, E.H.; Nichols, M.C. Mineral Reference Mannual; Springer: Boston, MA, USA, 1991. [Google Scholar]
- Sposito, G. The Chemistry of Soils, 2nd ed.; Oxford University Press: New York, NY, USA, 2008. [Google Scholar]
- Parfitt, R.; Childs, C. Estimation of forms of Fe and Al—A review, and analysis of contrasting soils by dissolution and mossbauer methods. Soil Res. 1988, 26, 121–144. [Google Scholar] [CrossRef]
- Levard, C.; Doelsch, E.; Basile-Doelsch, I.; Abidin, Z.; Miche, H.; Masion, A.; Rose, J.; Borschneck, D.; Bottero, J.Y. Structure and distribution of allophanes, imogolite and proto-imogolite in volcanic soils. Geoderma 2012, 183, 100–108. [Google Scholar] [CrossRef]
- Thompson, A.; Rancourt, D.G.; Chadwick, O.A.; Chorover, J. Iron solid-phase differentiation along a redox gradient in basaltic soils. Geochim. Cosmochim. Acta 2011, 75, 119–133. [Google Scholar] [CrossRef]
- Misra, S.G.; Mishra, P.C. Forms of manganese as influenced by organic matter and iron oxide. Plant Soil 1969, 30, 62–70. [Google Scholar] [CrossRef]
- Chorover, J.; Amistadi, M.K. Reaction of forest floor organic matter at goethite, birnessite and smectite surfaces. Geochim. Cosmochim. Acta 2001, 65, 95–109. [Google Scholar] [CrossRef]
- Suda, A.; Makino, T. Functional effects of manganese and iron oxides on the dynamics of trace elements in soils with a special focus on arsenic and cadmium: A review. Geoderma 2016, 270, 68–75. [Google Scholar] [CrossRef]
- Wagai, R.; Kishimoto-Mo, A.W.; Yonemura, S.; Shirato, Y.; Hiradate, S.; Yagasaki, Y. Linking temperature sensitivity of soil organic matter decomposition to its molecular structure, accessibility, and microbial physiology. Glob. Chang. Biol. 2013, 19, 1114–1125. [Google Scholar] [CrossRef] [PubMed]
- Dahlgren, R.; Shoji, S.; Nanzyo, M. Mineralogical characteristics of volcanic ash soils. In Volcanic Ash Soils: Genesis, Properties, and Utilization; Shoji, S., Nanzyo, M., Dahlgren, R., Eds.; Developments in Soil Science 21; Elsevier: Amsterdam, The Netherlands, 1993; Volume 21, pp. 101–143. [Google Scholar]
- Wagai, R.; Mayer, L.M.; Kitayama, K.; Shirato, Y. Association of organic matter with iron and aluminum across a range of soils determined via selective dissolution techniques coupled with dissolved nitrogen analysis. Biogeochemistry 2013, 112, 95–109. [Google Scholar] [CrossRef]
- Shoji, S.; Dahlgren, R.; Nanzyo, M. Chemical characteristics of volcanic ash soils. In Volcanic Ash Soils: Genesis, Properties, and Utilization; Shoji, S., Nanzyo, M., Dahlgren, R., Eds.; Developments in Soil Science 21; Elsevier: Amsterdam, The Netherlands; 1993; Volume 21, pp. 145–187. [Google Scholar]
- Uchida, M.; Shibata, Y.; Yoneda, M.; Kobayashi, T.; Morita, M. Technical progress in AMS microscale radiocarbon analysis. Nucl. Instrum. Methods Phys. Res. Sect. B Beam Interact. Mater. At. 2004, 223–224, 313–317. [Google Scholar] [CrossRef]
- Uchida, M.; Mantoku, K.; Kobayashi, T. Recent improvements for ultra-microscale radiocarbon measurements at NIES-TERRA. Nucl. Instrum. Methods Phys. Res. B 2018, in press. [Google Scholar]
- Uchida, M.; Shibata, Y.; Ohkushi, K.; Yoneda, M.; Kawamura, K.; Morita, M. Age discrepancy between molecular biomarkers and calcareous foraminifera isolated from the same horizons of northwest pacific sediments. Chem. Geol. 2005, 218, 73–89. [Google Scholar] [CrossRef]
- Crow, S.E.; Swanston, C.W.; Lajtha, K.; Brooks, J.R.; Keirstead, H. Density fractionation of forest soils: Methodological questions and interpretation of incubation results and turnover time in an ecosystem context. Biogeochemistry 2007, 85, 69–90. [Google Scholar] [CrossRef]
- Kaiser, K.; Guggenberger, G. Mineral surfaces and soil organic matter. Eur. J. Soil Sci. 2003, 54, 219–236. [Google Scholar] [CrossRef]
- Kajiura, M.; Wagai, R.; Hayashi, K. Optimal thermolysis conditions for soil carbon storage on plant residue burning: Modeling the trade-off between thermal decomposition and subsequent biodegradation. J. Environ. Qual. 2015, 44, 228–235. [Google Scholar] [CrossRef] [PubMed]
- Keiluweit, M.; Nico, P.S.; Johnson, M.G.; Kleber, M. Dynamic molecular structure of plant biomass-derived black carbon (biochar). Environ. Sci. Technol. 2010, 44, 1247–1253. [Google Scholar] [CrossRef] [PubMed]
- McBeath, A.V.; Smernik, R.J.; Schneider, M.P.W.; Schmidt, M.W.I.; Plant, E.L. Determination of the aromaticity and the degree of aromatic condensation of a thermosequence of wood charcoal using NMR. Org. Geochem. 2011, 42, 1194–1202. [Google Scholar] [CrossRef]
- Golchin, A.; Baldock, J.A.; Clarke, P.; Higashi, T.; Oades, J.M. The effects of vegetation and burning on the chemical composition of soil organic matter of a volcanic ash soil as shown by 13C NMR spectroscopy. II. Density fractions. Geoderma 1997, 76, 175–192. [Google Scholar] [CrossRef]
- Inoue, Y.; Hiradate, S.; Sase, T.; Hosono, M.; Morita, S.; Matsuzaki, H. Using C-14 dating of stable humin fractions to assess upbuilding pedogenesis of a buried holocene humic soil horizon, towada volcano, Japan. Geoderma 2011, 167–168, 85–90. [Google Scholar] [CrossRef]
- Nishimura, S.; Hirota, T.; Hirahara, O.; Shindo, H. Contribution of charred and buried plant fragments to humic and fulvic acids in Japanese volcanic ash soils. Soil Sci. Plant Nutr. 2006, 52, 686–690. [Google Scholar] [CrossRef]
- Shindo, H.; Honna, T.; Yamamoto, S.; Honma, H. Contribution of charred plant fragments to soil organic carbon in japanese volcanic ash soils containing black humic acids. Org. Geochem. 2004, 35, 235–241. [Google Scholar] [CrossRef]
- Brodowski, S.; John, B.; Flessa, H.; Amelung, W. Aggregate-occluded black carbon in soil. Eur. J. Soil Sci. 2006, 57, 539–546. [Google Scholar] [CrossRef]
- Heckman, K.; Throckmorton, H.; Clingensmith, C.; González Vila, F.J.; Horwath, W.R.; Knicker, H.; Rasmussen, C. Factors affecting the molecular structure and mean residence time of occluded organics in a lithosequence of soils under ponderosa pine. Soil Biol. Biochem. 2014, 77, 1–11. [Google Scholar] [CrossRef]
- Mikutta, R.; Schaumann, G.E.; Gildemeister, D.; Bonneville, S.; Kramer, M.G.; Chorover, J.; Chadwick, O.A.; Guggenberger, G. Biogeochemistry of mineral–organic associations across a long-term mineralogical soil gradient (0.3–4100 kyr), hawaiian islands. Geochim. Cosmochim. Acta 2009, 73, 2034–2060. [Google Scholar] [CrossRef]
- Chevallier, T.; Woignier, T.; Toucet, J.; Blanchart, E. Organic carbon stabilization in the fractal pore structure of andosols. Geoderma 2010, 159, 182–188. [Google Scholar] [CrossRef]
- Filimonova, S.; Kaufhold, S.; Wagner, F.E.; Häusler, W.; Kögel-Knabner, I. The role of allophane nano-structure and Fe oxide speciation for hosting soil organic matter in an allophanic andosol. Geochim. Cosmochim. Acta 2016, 180, 284–302. [Google Scholar] [CrossRef]
- Cotrufo, M.F.; Soong, J.L.; Horton, A.J.; Campbell, E.E.; Haddix, M.L.; Wall, D.H.; Parton, W.J. Formation of soil organic matter via biochemical and physical pathways of litter mass loss. Nat. Geosci. 2015, 8, 776–779. [Google Scholar] [CrossRef]
- Swanston, C.W.; Torn, M.S.; Hanson, P.J.; Southon, J.R.; Garten, C.T.; Hanlon, E.M.; Ganio, L. Initial characterization of processes of soil carbon stabilization using forest stand-level radiocarbon enrichment. Geoderma 2005, 128, 52–62. [Google Scholar] [CrossRef]
- Vogel, C.; Mueller, C.W.; Höschen, C.; Buegger, F.; Heister, K.; Schulz, S.; Schloter, M.; Kögel-Knabner, I. Submicron structures provide preferential spots for carbon and nitrogen sequestration in soils. Nat. Commun. 2014, 5, 2947. [Google Scholar] [CrossRef] [PubMed]
- Hoyos, N.; Comerford, N.B. Land use and landscape effects on aggregate stability and total carbon of andisols from the Colombian Andes. Geoderma 2005, 129, 268–278. [Google Scholar] [CrossRef]
- Paul, S.; Martinson, G.O.; Veldkamp, E.; Flessa, H. Sample pretreatment affects the distribution of organic carbon in aggregates of tropical grassland soils. Soil Sci. Soc. Am. J. 2008, 72, 500. [Google Scholar] [CrossRef]
- Asano, M.; Wagai, R. Distinctive organic matter pools among particle-size fractions detected by solid-state 13c-NMR, δ13c and δ15n analyses only after strong dispersion in an allophanic andisol. Soil Sci. Plant Nutr. 2015, 61, 242–248. [Google Scholar] [CrossRef]
- Malamoud, K.; McBratney, A.B.; Minasny, B.; Field, D.J. Modelling how carbon affects soil structure. Geoderma 2009, 149, 19–26. [Google Scholar] [CrossRef]
- Sollins, P.; Gregg, J.W. Soil organic matter accumulation in relation to changing soil volume, mass, and structure: Concepts and calculations. Geoderma 2017, 301, 60–71. [Google Scholar] [CrossRef]
- Schuppli, P.A.; Ross, G.J.; McKeague, J.A. The effective removal of suspended materials from pyrophosphate extracts of soils from tropical and temperate regions 1. Soil Sci. Soc. Am. J. 1983, 47, 1026–1032. [Google Scholar] [CrossRef]
- Yamada, Y. The Characterization of Humus Accumulation in Andosols by 14c Dating; National Institute for Agro-Environmental Sciences: Tsukuba, Ibaraki, Japan, 1986; pp. 23–86. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample ID | Sample Depth cm | Bulk Density g cm−3 | TC | TN | C:N | Δ14C ‰ | pH in H2O | PP 1-Extractable | OX 1-Extractable | DC 1-Extractable | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| mg g−1 | Fe | Al | Fe | Al | Fe | Al | |||||||
| mg g−1 | mg g−1 | mg g−1 | |||||||||||
| NT-a | 0–5 | 0.45 | 149 | 10 | 14.9 | 47.4 | 6.2 | 1.5 | 1.6 | 8.6 | 19.1 | 15.1 | 3.6 |
| NT-b | 5–20 | 0.63 | 80.4 | 5.9 | 13.6 | 24.3 | 6.2 | 1.2 | 3.5 | 14.0 | 35.5 | 26.6 | 5.6 |
| Till-1 | 0–20 | 0.78 | 51.4 | 4.1 | 12.5 | −125.8 | 6.1 | 1.0 | 5.0 | 18.5 | 41.6 | 25.5 | 6.3 |
| Till-2 | 0–20 | 0.73 | 42.3 | 3.6 | 11.8 | −65.6 | 6.7 | 1.3 | 3.8 | 29.8 | 53.4 | 29.9 | 6.2 |
| Bare | 0–20 | 0.74 | 36.9 | 3.0 | 12.3 | −33.1 | 6.5 | 0.5 | 3.6 | 18.5 | 39.6 | 30.1 | 5.8 |
| Sample ID | Fraction ID | Mass Fraction | C 1 | N 1 | C:N | C 2 | N 2 | 14C | Δ14C |
|---|---|---|---|---|---|---|---|---|---|
| mg g fraction−1 | mg g bulk−1 | pMC | ‰ | ||||||
| NT-a | F1 | 0.215 | 33.9 | 2.19 | 15.5 | 74.5 | 4.8 | 105.7 | 74.1 |
| F2 | 0.222 | 22.2 | 1.80 | 12.4 | 51.0 | 4.1 | 104.9 | 61.2 | |
| F3 | 0.086 | 12.1 | 1.09 | 11.1 | 10.7 | 1.0 | 101.2 | 19.0 | |
| F4 | 0.148 | 4.7 | 0.51 | 9.2 | 7.1 | 0.8 | 90.2 | −102.5 | |
| F5 | 0.131 | 2.2 | 0.29 | 7.5 | 2.9 | 0.4 | 90.4 | −99.4 | |
| F6 | 0.215 | 0.5 | 0.07 | 6.2 | 0.9 | 0.1 | 85.4 | −147.8 | |
| NT-b | F1 | 0.039 | 30.9 | 1.82 | 17.0 | 12.0 | 0.7 | 103.5 | 50.5 |
| F2 | 0.104 | 21.8 | 1.57 | 13.9 | 22.9 | 1.6 | 105.0 | 60.2 | |
| F3 | 0.148 | 11.3 | 0.94 | 12.0 | 16.8 | 1.4 | 101.2 | 13.0 | |
| F4 | 0.279 | 5.8 | 0.55 | 10.6 | 16.2 | 1.5 | 92.7 | −81.7 | |
| F5 | 0.269 | 2.6 | 0.28 | 9.5 | 7.1 | 0.7 | 91.6 | −94.1 | |
| F6 | 0.165 | 0.6 | 0.06 | 8.9 | 0.9 | 0.1 | 93.1 | −77.8 | |
| Till-1 | F1 | 0.008 | 26.1 | 1.64 | 15.9 | 2.0 | 0.1 | 99.2 | −2.5 |
| F2 | 0.019 | 26.2 | 1.20 | 21.9 | 5.1 | 0.2 | 84.3 | −165.6 | |
| F3 | 0.127 | 10.5 | 0.80 | 13.1 | 13.3 | 1.0 | 92.4 | −86.9 | |
| F4 | 0.419 | 5.6 | 0.52 | 10.8 | 23.5 | 2.2 | 87.2 | −145.6 | |
| F5 | 0.266 | 2.3 | 0.24 | 9.7 | 6.0 | 0.6 | 84.6 | −169.4 | |
| F6 | 0.201 | 0.7 | 0.08 | 9.2 | 1.4 | 0.2 | 88.4 | −131.6 | |
| Till-2 | F1 | 0.007 | 25.5 | 1.19 | 21.4 | 1.9 | 0.1 | 95.7 | −42.2 |
| F2 | 0.011 | 28.3 | 1.15 | 24.6 | 3.3 | 0.1 | 90.4 | −104.3 | |
| F3 | 0.038 | 13.1 | 0.89 | 14.7 | 5.3 | 0.4 | 94.8 | −64.0 | |
| F4 | 0.304 | 6.5 | 0.63 | 10.3 | 20.9 | 2.0 | 93.7 | −81.0 | |
| F5 | 0.401 | 2.8 | 0.27 | 10.2 | 11.8 | 1.2 | 89.0 | −127.3 | |
| F6 | 0.240 | 0.6 | 0.07 | 9.5 | 1.6 | 0.2 | 91.2 | −105.1 | |
| Bare | F1 | 0.004 | 26.2 | 1.09 | 24.1 | 1.2 | 0.0 | 93.9 | −64.2 |
| F2 | 0.018 | 25.9 | 1.12 | 23.2 | 4.6 | 0.2 | 90.6 | −102.3 | |
| F3 | 0.046 | 9.9 | 0.74 | 13.4 | 4.6 | 0.3 | 99.1 | −22.6 | |
| F4 | 0.303 | 5.2 | 0.52 | 10.0 | 15.9 | 1.6 | 97.1 | −48.5 | |
| F5 | 0.399 | 2.4 | 0.26 | 9.6 | 9.8 | 1.0 | 92.3 | −95.9 | |
| F6 | 0.279 | 0.6 | 0.07 | 9.2 | 1.8 | 0.2 | 96.0 | −58.2 | |
| Fraction & Density | Allophane/Imogolite-Type Phase 1 | Ferrihydrite 2 | Poorly-Crystalline Minerals 3 | Other Minerals 4 | Organic Matter 5 | |
|---|---|---|---|---|---|---|
| (g cm−3) | (% by mass) | |||||
| Bulk | 13 (8–17) | 3 (2–5) | 16 (9–22) | 71 (65–73) | 14 (7–30) | |
| F1 | <1.6 | 5 (2–8) | 1 (1–3) | 6 (3–11) | 36 (29–41) | 58 (51–68) |
| F2 | 1.6–1.8 | 11 (2–8) | 2 (1–4) | 13 (8–19) | 37 (24–47) | 50 (44–56) |
| F3 | 1.8–2.0 | 14 (7–15) | 3 (1–4) | 16 (9–21) | 61 (53–66) | 23 (20–26) |
| F4 | 2.0–2.25 | 21 (7–17) | 4 (3–6) | 25 (22–29) | 64 (58–69) | 11 (9–13) |
| F5 | 2.25–2.5 | 15 (19–23) | 3 (2–5) | 19 (11–26) | 76 (68–84) | 5 (4–6) |
| F6 | >2.5 | 3 (9–21) | 4 (2–4) | 7 (5–9) | 92 (90–94) | 1 (1–1) |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wagai, R.; Kajiura, M.; Uchida, M.; Asano, M. Distinctive Roles of Two Aggregate Binding Agents in Allophanic Andisols: Young Carbon and Poorly-Crystalline Metal Phases with Old Carbon. Soil Syst. 2018, 2, 29. https://doi.org/10.3390/soilsystems2020029
Wagai R, Kajiura M, Uchida M, Asano M. Distinctive Roles of Two Aggregate Binding Agents in Allophanic Andisols: Young Carbon and Poorly-Crystalline Metal Phases with Old Carbon. Soil Systems. 2018; 2(2):29. https://doi.org/10.3390/soilsystems2020029
Chicago/Turabian StyleWagai, Rota, Masako Kajiura, Masao Uchida, and Maki Asano. 2018. "Distinctive Roles of Two Aggregate Binding Agents in Allophanic Andisols: Young Carbon and Poorly-Crystalline Metal Phases with Old Carbon" Soil Systems 2, no. 2: 29. https://doi.org/10.3390/soilsystems2020029