New Silver(I) Coordination Polymer with Fe4 Single-Molecule Magnets as Long Spacer
by
 , , , and
, , , and
Luca Rigamonti
1,* ,
,
Manuela Vaccari
1,
Fabrizio Roncaglia
1,
Carlo Baschieri
1 and
Alessandra Forni
2 1
Dipartimento di Scienze Chimiche e Geologiche, Università degli Studi di Modena e Reggio Emilia, via G. Campi 103, 41125 Modena, Italy
2
Istituto di Scienze e Tecnologie Molecolari, Consiglio Nazionale delle Ricerche (ISTM-CNR), via C. Golgi 19, 20133 Milano, Italy
*
Author to whom correspondence should be addressed.
Magnetochemistry 2018, 4(4), 43; https://doi.org/10.3390/magnetochemistry4040043
Submission received: 11 May 2018
/
Revised: 12 September 2018
/
Accepted: 25 September 2018
/
Published: 1 October 2018
Abstract
:In continuation of our work on supramolecular architectures of single-molecule magnets (SMMs) as a promising strategy in developing their magnetic performance, in this paper we report the synthesis and single crystal X-ray structure of the centered triangular tetrairon(III) SMM, [Fe4(PhpPy)2(dpm)6], Fe4 (Hdpm = dipivaloylmethane, H3PhpPy = 2-(hydroxymethyl)-2-(4-(pyridine-4-yl)phenyl)propane-1,3-diol), and its assembly in the coordination polymer {[Fe4(PhpPy)2(dpm)6Ag](ClO4)}n, Fe4Ag, upon reaction with silver(I) perchlorate. Thanks to the presence of the pyridyl rings on the two tripodal ligands, Fe4 behaves as divergent ditopic linker, and due to the Fe4:AgClO4 1:1 ratio, Fe4Ag probably possesses a linear arrangement in which silver(I) ions are linearly coordinated by two nitrogen atoms, forming 1D chains whose positive charge is balanced by the perchlorate anions. The stabilization of such a polymeric structure can be ascribed to the long distance between the two donor nitrogen atoms (23.4 Å) and their donor power. Fe4Ag shows slow relaxation of the magnetization which follows a thermally activated process with Ueff/kB = 11.17(18) K, τ0 = 2.24(17) 10−7 s in zero field, and Ueff/kB = 14.49(5) K, τ0 = 3.88(8) 10−7 s in 1-kOe applied field, in line with what reported for tetrairon(III) SMMs acting as building blocks in polymeric structures.
1. Introduction
Single-molecule magnets (SMMs) [1], thanks to their directionally bistable magnetic moment, have been investigated over the last two decades as nanoscale, chemically tunable entities for encoding binary information. Though SMMs developed so far show slow relaxation of the magnetization only at low temperatures (below liquid nitrogen), preventing their application in spintronics devices [2], a fast increase of their performance is expected, as recently shown through the synthesis of a dysprosocenium complex with opening of magnetic hysteresis at a temperature as high as 60 K [3,4]. Another promising strategy to develop SMM features consists of the assembly into extended coordination networks [5,6,7,8,9,10], which may allow enhanced magnetic remanence due to intramolecular magnetic coupling [11].
Complexes of the family of tetrairon(III) SMMs with general formula [Fe4(L)2(dpm)6] [12,13,14] (H3L = tripodal ligand, Hdpm = dipivaloylmethane) include four coplanar high spin iron(III) ions (s = 5/2) forming a centered triangular geometry. The central metal atom (Fec) is antiferromagnetically coupled to the three peripheral ones (Fep) so as to give an S = 5 spin ground state. In general, clusters of this family feature easy axis anisotropy normal to the metal plane. Despite the low energy barrier of Fe4 SMMs reaching up to 15–17 K, their chemical versatility has been exploited by creating clusters able to sublimate by fluorination of the peripheral β-diketonato ligands [15,16,17] or anchor on gold surfaces by sulfur-rich tripodal ligands [18,19], as well as modulating the spin ground state by selective replacement of the central metal ion with chromium(III) [20,21], vanadium(III) [22], or lanthanide ions [23,24]. This family of SMMs has been also characterized as single-molecule junction in scanning tunneling microscope (STM) experiments at 0.5 K [25] and through theoretical calculations for its behavior in spin-polarized transport [26], revealing how Fe4 SMMs would be excellent candidates for molecular spintronics devices. Recently, the derivative [Fe4(pPy)2(dpm)6] (A) (H3pPy = 2-(hydroxymethyl)-2-(pyridine-4-yl)propane-1,3-diol) [11] has also revealed to behave as efficient ditopic supramolecular synthon due to the presence of two divergent 4-pyridyl substituents on the tripodal ligands, as formerly found in the assembly of 1D chains by using ruthenium paddlewheel dimers as coordination nodes [11]. By modulating the redox state of the Ru2 units (II,II vs. II,III), we could introduce a significant exchange bias with intra-chain coupling, and concomitant enhancement of the remnant magnetization. The ability of A as building block of supramolecular structures has been further confirmed by reaction with silver(I) perchlorate yielding {[Fe4(pPy)2(dpm)6]2Ag}(ClO4) (B), a tridimensional metal–organic framework (3D-MOF) [14]. In this compound, Ag+ ions behave as tetrahedral nodes coordinated by four pyridyl nitrogen atoms of Fe4 units, and the cubic crystals, although with no overall second-order anisotropy, can be persistently magnetized parallel or antiparallel to the four main diagonals of the unit cell.
Compound B also possesses accessible voids (14.3% of unit cell volume) able to trap small solvent molecules, which can be exploited as a means for modulating the magnetic properties. However, due to the presence of two interlocked dia networks (according to IUPAC nomenclature [27]), the space left inside the 3D-MOF is not available for bigger molecules [14]. In the attempt to build-up a new polymeric structure with larger cavities by elongating the SMM spacer, in this work we synthesized the new tripodal ligand H3PhpPy, 2-(hydroxymethyl)-2-(4-(pyridine-4-yl)phenyl) propane-1,3-diol (Scheme 1), where we added a phenyl ring between the triol unit and the pyridyl terminal fragment with respect to H3pPy. This ensures the same divergent ditopic linker behavior once incorporated into an Fe4 SMM, constituting at the same time a longer spacer upon coordination of the pyridyl rings to silver(I) ions. Here we report the synthesis of H3PhpPy, its assembly into [Fe4(PhpPy)2(dpm)6] (Fe4), whose structure was confirmed by single crystal X-ray diffraction studies, and further reaction of Fe4 with AgClO4. The behavior as SMM of the resulting coordination polymer {[Fe4(PhpPy)2(dpm)6Ag](ClO4)}n (Fe4Ag) will be then described, in comparison with the 3D-MOF B and the Fe4 building block.
2. Results and Discussion
2.1. Synthesis of the Tripodal Ligand H3PhpPy
Two possible synthetic paths for H3PhpPy were identified, both starting from commercial 2-(4-bromophenyl)ethanol and involving three synthetic steps. The most convenient method proved to be the one featuring a first Suzuki cross-coupling with pyridin-4-ylboronic acid, followed by oxidation of the homobenzylic alcohol and completed by a Tollens addition with paraformaldehyde (Scheme 1). Another variant was also investigated, featuring the oxidation, Tollens and Suzuki sequence, but its efficacy was lower (Scheme S1 in Supplementary Materials).
Following Scheme 1, the cross-coupling reaction between 2-(4-bromophenyl)ethanol and pyridin-4-ylboronic acid was performed on the basis of a procedure reported in the literature [28] by means of Pd(PPh3)4 as catalyst. The presence of the pyridyl group gave us the opportunity to separate the product 1 from N,N-dimethylformamide (DMF) by precipitating it as hydrochloride salt, avoiding the use of column chromatography and still guaranteeing good yield and optimal purity as detectable with GC-MS and 1H NMR analyses. The oxidation of 1 was secured with a radical method, employing trichloroisocyanuric acid (TICA) and (2,2,6,6-tetramethylpiperidin-1-yl)oxyl (TEMPO) radical [29], while other tested methods caused the partial conversion to the more stable benzaldehyde derivative. The following reaction of 2 with paraformaldehyde and Ca(OH)2 in tetrahydrofuran (THF) [29,30,31] allowed the double aldolic addition and the Cannizzaro reaction to the final tripodal ligand H3PhpPy with a reasonable yield of 41%. The addition of Al(OiPr)3 as additive had not a clear scope in this reaction, but we could observe easier purification of the crude product. The overall yield of the tripodal ligand starting from commercial 2-(4-bromophenyl)ethanol is then 23%.
The alternative pathway reported in Scheme S1 in Supplementary Materials was also attempted in order to move the triol-formation step, characterized by the lowest yield, before the cross-coupling. In this way it would have been possible to save on the amount of used palladium catalyst, which is the most expensive reagent in this synthesis. The first oxidation step of 2-(4-bromophenyl)ethanol with TICA and TEMPO led to 3 in very high yield, and the following reaction with paraformaldehyde allowed the isolation of 4 with the formed triol fragment. Unfortunately, the cross-coupling of 4 with pyridin-4-ylboronic acid in the presence of Pd(PPh3)4 did not yield the desired final tripodal ligand even after several attempts. This might possibly due to an interference of the triol unit that can coordinate the palladium ion, sequestering it from the catalytic cycle, or an interaction between the triol unit and the boronic acid.
2.2. Assembly of Fe4 and Its Molecular Structure
The assembly of the Fe4 cluster with H3PhpPy was performed as previously established [11,12] by reacting a solution of [Fe4(OMe)6(dpm)6] [32] in diethyl ether (Et2O) with the tripodal ligand suspended in ethanol (EtOH). Upon addition of H3PhpPy a darkening of the orange solution could be observed, hint of the exchange of the six bridging methoxido ligands with two tripodal ones. Nevertheless, by vapor diffusion of EtOH with the reaction mixture we could almost always obtain crystals of the dimeric cluster [Fe2(OEt)2(dpm)4] (thermodynamic product), whose formation is known to be favored in basic conditions [33]. Differently from H3pPy, the inductive effect of the added phenyl ring might pushes electron density toward the pyridyl moiety [34] in the tripodal ligand H3PhpPy, with resulting pronounced basicity.
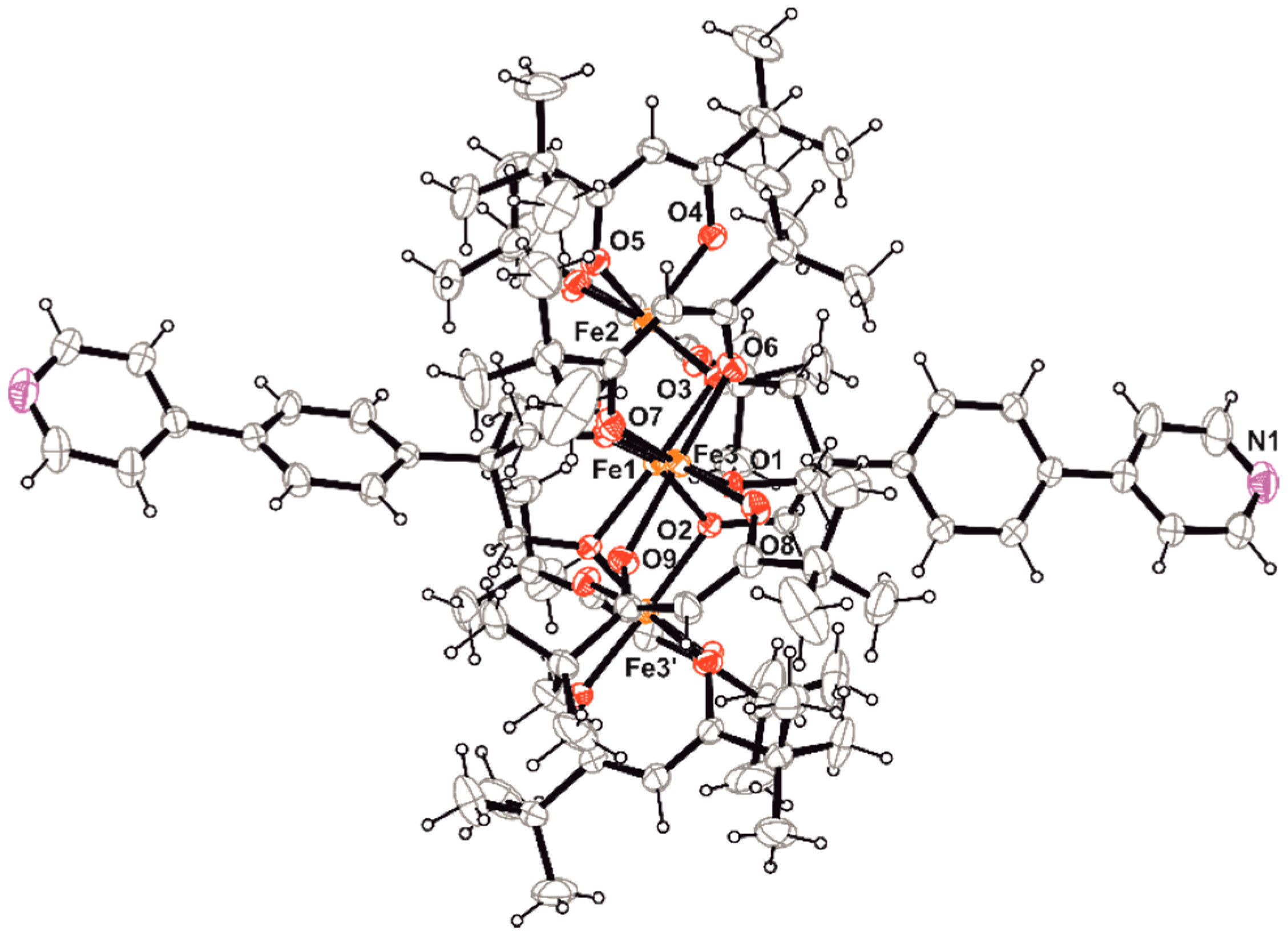
Nevertheless, we could isolate single crystals of Fe4 suitable for X-ray diffraction from the concentrated mother liquor of one synthetic attempt. They belong to the monoclinic space group C2/c as most of the reported homologous tetrairon(III) clusters [12,13,29], and in particular its shorter analogue A⋅2EtOH [11]. Selected geometrical parameters at 293(2) K are reported in Table 1, and a view of the molecular structure is shown in Figure 1.
The four iron(III) ions define a metal-centered triangle that develops around the crystallographic two-fold axis passing through Fe1 and Fe2. The molecular structure is held together by two tri-deprotonated tripodal ligands that bridge the central ion (Fe1) to the peripheral ones (Fe2, Fe3, Fe3’; primed atom is generated by the two-fold rotation). The coordination sphere of each peripheral metal ion is completed by two bidentate dpm− ligands to form the classical propeller-like structure with helical pitch, calculated as dihedral angle between Fe4 and FeO2Fe planes, of about 67–69°. Thus, all metal centers achieve a distorted octahedral environment formed by six oxygen atoms with Fe–O distances falling in the 1.97–2.03 Å narrow range. The isosceles triangle given by the peripheral iron(III) ions possesses two long sides (5.3903(11) Å) and a short one (5.2063(15) Å), and no ethanol molecules are involved in hydrogen bonds with the nitrogen atom of the pyridyl ring in the solid state, differently from A⋅2EtOH [11]. Phenyl and pyridyl rings of the tripodal ligand are not co-planar, being rotated each other of 6.47(5)°. The last ones are involved in weak π⋅⋅⋅π stacking interactions (the shortest C–C distance is 3.67 Å) that, together with weak hydrogen bonds, contribute to stabilize the overall crystal structure (Figure S1 in Supplementary Materials).
Due to the difficulty to isolate the desired cluster as pure crystalline phase, we decided to investigate whether or not Fe4 was already formed in solution after 5 h of stirring between [Fe4(OMe)6(dpm)6] and H3PypPy. The reaction mixture was analyzed via ESI-MS spectrometry and we could positively identify the main peaks at m/z = 1874.7 and 1836.8, belonging to the +K and +H positive ions of Fe4 (Figure S2 in Supplementary Materials). The only other detected peak appears at m/z = 644.4, which corresponds to [Fe(dpm)3 + K]+, a decomposition product of [Fe4(OMe)6(dpm)6] upon standing in solution [12]. Proved that the desired cluster is formed in solution (kinetic product), the reaction mixture was then taken to dryness under reduced pressure without heating and the infrared spectrum of the obtained orange solid was compared with the ones of the dimeric species [Fe2(OEt)2(dpm)4] and the starting cluster [Fe4(OMe)6(dpm)6] (Figure S3 in Supplementary Materials). Even if very similar, the three spectra contain substantial differences in the position and relative intensities of several bands, which all hint to confirm the formation of Fe4. In particular, the coordination of the tripodal ligand is supported by the alkoxido C–O stretching band shifted to 1096 from 1050 cm−1 in the progenitor tetranuclear cluster [Fe4(OMe)6(dpm)6].
Furthermore, the orange solid was analyzed via 1H NMR spectroscopy dissolving the product in toluene-d8: the spectrum (Figure S4 in Supplementary Materials) clearly entails the intense broad signal at 10.9 ppm characteristic of the paramagnetically shifted tBu substituents in this kind of Fe4 complexes [12,15,16]. Other four broad signals at 12.0 (shoulder) 8.8, 8.5 and 6.5 ppm can be observed, ascribable to the corresponding four different aromatic protons of the tripodal ligand; in particular the shoulder at 12.0 ppm can be attributed to H5 (Scheme 1 for atom numbering), in analogy to the known Fe4 complexes with phenyl substituent [12,16], while the narrower peak at 8.8 ppm should be generated by H9, close to the nitrogen atom of the pyridyl ring and farther from the paramagnetic core. An additional shoulder at 13.1 ppm due to decomposition to [Fe(dpm)3] is present [12], in agreement with what observed in the ESI spectrum; anyway, this signal is weaker than the ones of the desired main product Fe4. Most important, the 1H NMR spectrum remained almost unchanged after 16 h, which allow us to conclude that the desired cluster is stable in toluene and suitable for its subsequent use.
2.3. Assembly of Fe4Ag
The reaction of Fe4 with AgClO4 was performed by liquid/liquid diffusion of a toluene solution containing the tetrairon(III) cluster into a THF solution of AgClO4. Regardless of the relative concentration, we could always isolate needle-like microcrystals of Fe4Ag with a 1:1 ratio between the two components. This was confirmed by comparing the experimental percentages with the calculated elemental analysis of different stoichiometric combinations, always fitting with the [Fe4AgClO4] minimal formula. When using a Fe4:AgClO4 2:1 ratio, the formation of microcrystalline Fe4Ag resulted in a darker orange solution containing the excess of unreacted Fe4.
The infrared spectrum of Fe4Ag fully resembles the one of Fe4 (Figure S5 in Supplementary Materials), which proves the intactness of the starting tetrairon(III) cluster. The presence of AgClO4 is confirmed by the appearance of a unique perchlorate stretching band at 1122 cm−1, distinctive of an anion in tetrahedral symmetry and purely ionic character [35], as previously observed for B [14]. It is also possible to detect a weak band at 479 cm−1 tentatively ascribable to the Ag–N bond stretching [36], confirming the coordination of silver(I) by the nitrogen atoms of the pyridyl rings. Another proof can be found in the ESI-MS spectrum of Fe4Ag in THF/CH3CN with the peak at m/z = 2014.7 for the [Fe4Ag(THF)]+ ion, in addition to the peaks at m/z = 1836.4 and 1874.8, as previously observed for Fe4 (Figure S6 in Supplementary Materials). Further confirmation is given by the X-ray fluorescence (XRF) spectrum, where Fe, Ag, and Cl are clearly detected (Figure S7 in Supplementary Materials).
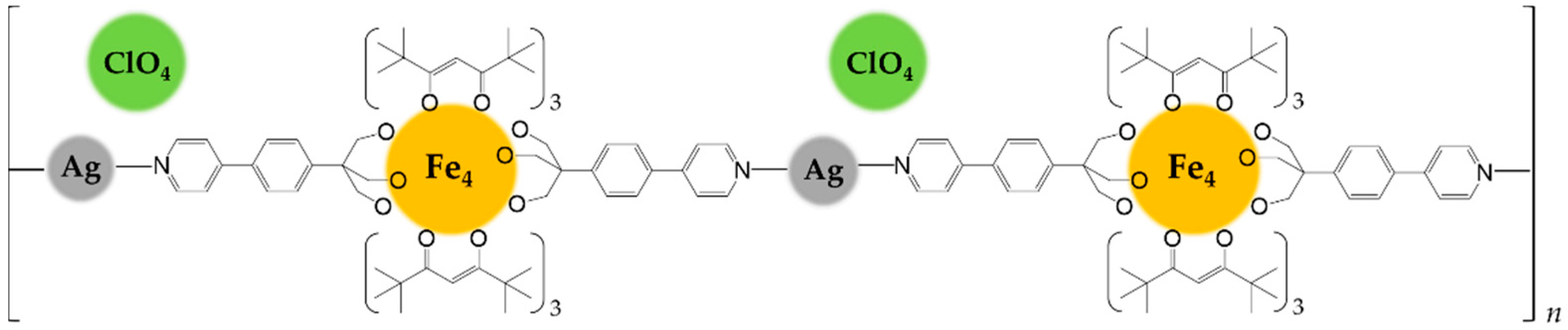
The orange microcrystals of Fe4Ag are practically insoluble in all common solvents, clearly suggesting the polymeric nature of the product. It revealed to be soluble only in pyridine, which coordinates silver(I) ions replacing Fe4. The Fe4:AgClO4 1:1 ratio, the needle-like crystal habit which resemble the 1D chains with ruthenium paddlewheel dimers [11], the insolubility of the product and the ionic Td symmetry of the perchlorate anions as given by the infrared spectrum led to identify Fe4Ag as a probable linear coordination polymer of silver(I) ions linked together by intact Fe4 units acting as long spacer (N1⋅⋅⋅N1’ = 23.413(11) Å), whose positive charge is balanced by ClO4− ions occupying voids in between adjacent 1D chains (Scheme 2). The propensity of silver(I) to give linear coordination environment with pyridine [37,38,39] or nitrogen ligands [40,41,42,43] is reported, and in this case the high basicity of the pyridyl ring given by the releasing electronic effect of the attached phenyl ring [34] might convey enough donor power to only two nitrogen atoms in order to satisfy the coordinative electronic demand of silver(I).
2.4. Magnetic Characterization
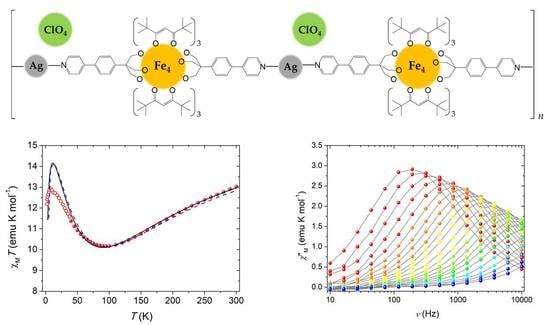
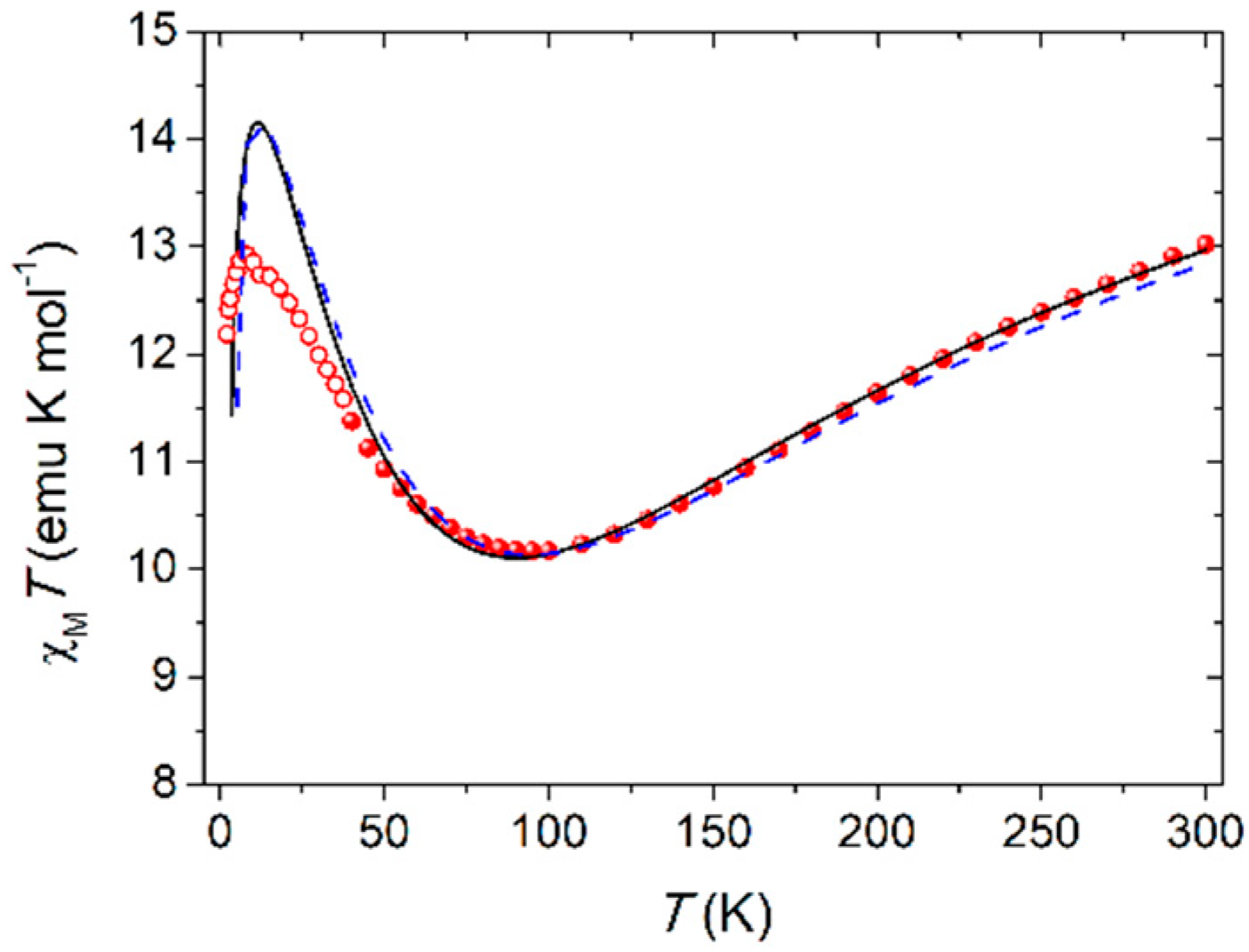
Thanks to the intactness of Fe4 units within the polymer and the purity of the crystalline phase as given by elemental analysis and optical microscope inspection, we performed direct current (dc) magnetic measurements on Fe4Ag. The temperature dependence of the molar magnetic susceptibility, χM, in low fields (1–10 kOe) was measured between 1.9 and 300 K, and the data are reported in Figure 2 as χMT vs. T plot. The shape is characteristic for triangular-centered Fe4 clusters, where the dominant antiferromagnetic interactions between Fec and Fep centers lead to an S = 5 ground state [12,14,15,32]. The χMT value at room temperature is about 13.0 emu K mol−1, hence lower than the Curie constant for four s = 5/2 spins (17.51 emu K mol−1 with g = 2.00); upon cooling, it first decreases towards a minimum at 95 K (ca. 10.2 emu K mol−1) and then increases again reaching 12.9 emu K mol−1 at 8 K, before a further drop at the lowest temperatures. The χMT value at low T is then rising toward the Curie constant for the expected S = 5 ground state (15.00 emu K mol−1 with g = 2.00). Magnetic exchange interactions among the four iron(III) centers are usually obtained by fitting the χMT vs. T curve to a Heisenberg plus Zeeman spin Hamiltonian that assumes threefold molecular symmetry to avoid over-parametrization [15]:
where S1 is the spin vector for Fec; S2, S3, and S4 are the spin vectors for Fep ions; S is the total spin vector; H is the applied magnetic field; and J and J’ coupling constants are the nearest-neighbor (n.n.) and next-nearest-neighbor (n.n.n.) interactions, respectively. Due to the lower increase of the χMT value at low T with respect to the expected Curie constant for the S = 5 ground state reported above and the fact that n.n. interactions are most effective in the high-temperature regime, the best-fit procedure was conducted in the 40–300 K range with data obtained with a 10-kOe applied field (filled circles in Figure 2) using PHI program [44], yielding a satisfactorily reproduction of the experimental data with g = 1.9840(12), J = 13.98(8) cm−1 and J’ = 0.86(2) cm−1 (black curve in Figure 2).
Ĥ = J (Ŝ1⋅Ŝ2 + Ŝ1⋅Ŝ3 + Ŝ1⋅Ŝ4) + J’ (Ŝ2⋅Ŝ3 + Ŝ2Ŝ4 + Ŝ3Ŝ4) + gμBŜ⋅Ĥ
These parameters are in line with what reported for the Fe4 SMM family with isotropic g close to 2.00, as expected for high spin iron(III) ions, and the presence of dominant n.n. antiferromagnetic coupling and J’ of one order of magnitude smaller than J [11,12,14,15]. This last coupling constant is slightly lower than expected (15–19 cm−1 [11,13]), but this can be easily envisaged by the lower value of the minimum of the χMT curve, which usually falls between 110 and 120 K in other Fe4 derivatives [12,13,14]. Furthermore, the shift to about 90 K of the reproduced minimum from the experimental value of 95 K suggests that the real J might be slightly higher. Correction of this shift was carried out by simulating different curves with J ranging from 12 to 16 cm−1, leaving g and J’ unaltered. The best prevision of the minimum in the χMT curve is obtained with J = 14.6 cm−1 (dashed blue curve in Figure 2), still maintaining a satisfactory reproduction of the experimental data at T > 40 K. Upon further cooling below 40 K, the χMT product (empty circles in Figure 2 obtained with a 1-kOe applied field) follows the expected growing trend toward the Curie constant of the S = 5 ground state, but it is lower than the expected values from the fitting curve, and this can be probably ascribed to the presence of anisotropic effects or weak intermolecular interactions, which are more effective at low temperature.
Even if not isolated as crystalline phase, dc magnetic data were also collected for a powder sample of Fe4, whose elemental analysis closely reproduced the one of the pure compound, in order to perform a direct comparison with Fe4Ag. The temperature dependence of χM in low fields (1–10 kOe) was measured between 5 and 250 K, and the data are reported in Figure S8 in Supplementary Materials as χMT vs. T plot. As before, data were well-fitted by means of Equation (1) in the whole temperature range obtaining g = 1.9832(9), J = 15.10(6) cm−1, J’ = 0.80(2) cm−1. Differently from Fe4Ag, in the case of Fe4 the anisotropic effects at low temperature seem to be negligible; nevertheless, both n.n. and n.n.n. interactions in the polymeric 1D chain are fully comparable with the ones in the progenitor cluster.
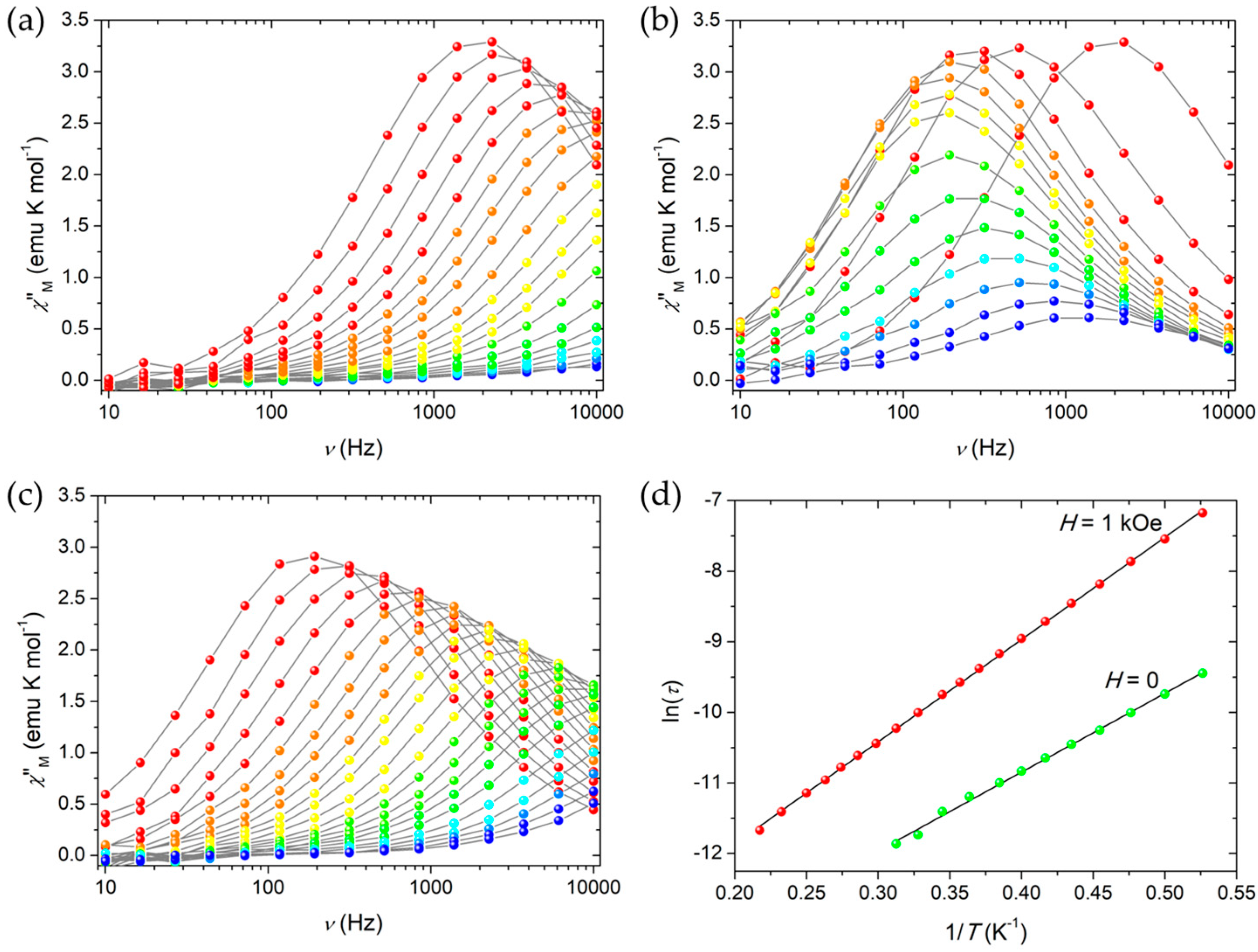
The magnetization dynamics of both Fe4Ag and Fe4 was then probed with alternating current (ac) susceptibility measurements in zero static field as a function of both temperature (down to 1.9 K) and frequency of the oscillating field (ν = 10–10,000 Hz). Characteristic frequency-dependent maxima in χ”M vs. T plots were detected, which indicate that Fe4Ag and Fe4 behave as SMMs (Figure 3a and Figure S9a in Supplementary Materials). Measurements were then repeated at 1.9 K by applying a static field H in the 0–50 kOe range for Fe4Ag in order to evaluate the most effective field in slowing down relaxation by cutting quantum tunneling (QT) pathways (Figure 3b), and the static field H = 1 kOe was found to be the best option to repeat ac measurements at variable T (Figure 3c for Fe4Ag and Figure S9b in Supplementary Materials for Fe4). Quantitative data treatment was based on a generalized Debye model [45,46] to extract the temperature-dependent relaxation time τ and the width of its distribution α. The model was found to be fully satisfactory and afforded α values that approach 0.20 and 0.23 at H = 0 and 1 kOe, respectively, at the lowest T, while they decrease toward zero at high T for both compounds.
By plotting ln(τ) vs. 1/T, linear trends can be clearly recognized at both static fields (Figure 3d for Fe4Ag and Figure S9c,d in Supplementary Materials for Fe4), which indicate that reversal of the magnetization proceeds via a thermally activated process in the temperature range analyzed. Data can be suitably fitted by the Arrhenius law τ = τ0 exp (Ueff/kBT) obtaining Ueff/kB = 11.17(18) K and τ0 = 2.24(17) 10−7 s in zero field, and Ueff/kB = 14.49(5) K and τ0 = 3.88(8) 10−7 s in 1-kOe applied field for Fe4Ag, and Ueff/kB = 10.69(16) K and τ0 = 7.0(7) 10−7 s in zero static field, and Ueff/kB = 14.9(2) K and τ0 = 5.1(3) 10−7 s at 1 kOe for Fe4.
These thermally activated relaxation parameters fully resemble the ones found for the 3D-MOF B (τ0 = 1.65(8) 10−7 s and Ueff/kB = 11.46(10) K in zero static field, and τ0 = 3.21(11) 10−7 s and Ueff/kB = 14.25(8) K at 1 kOe) [14], which are similar but lower compared to Fe4 clusters not involved in coordination networks [12], and especially with A⋅2EtOH [11] as building block of B [14]. This seems to confirm the different spin-phonon interactions, which affect the magnetic relaxation [47], between an organized 1D/3D framework and a molecular crystal held together only by van der Waals forces. The values for Fe4, similar to the ones of Fe4Ag and B and lower to the ones of A⋅2EtOH [11] and the tetrairon SMM with phenyl rings on the tripodal ligands [12], seems also to suggest that the weak but extended π⋅⋅⋅π stacking interactions between the phenylpyridine fragments of adjacent molecules in the crystal packing of Fe4 are sufficient to mimic the spin-phonon effect in a coordination polymer.
3. Materials and Methods
3.1. General Information
Reagent-grade 2-(4-bromophenyl)ethanol, pyridin-4-ylboronic acid, TICA, TEMPO radical, paraformaldehyde, Pd(PPh3)4, solvents and inorganic reagents for organic synthesis, AgClO4, anhydrous toluene (VWR, max. 20 ppm H2O) and anhydrous THF (VWR, max. 30 ppm H2O) were used as received. Petroleum ether (PE) used was the 40–60 °C boiling fraction. EtOH was distilled over magnesium ethoxide and stored over 3 Å molecular sieves, while Et2O was pre-dried over CaCl2 overnight and distilled from sodium and benzophenone under N2 before use. Elemental analyses were performed with a Carlo Erba EA1110 CHNS-O automatic analyzer. IR spectra were recorded as KBr discs by using a Jasco FTIR-4700 LE spectrophotometer with 2 cm−1 resolution; bands are reported as wavenumbers (cm−1) together with the assignment and relative intensity (vs = very strong, s = strong, m = medium, w = weak, br = broad). Thin layer chromatography (TLC) was performed on silica gel and retention factors (r.f.) are reported for the given eluent. GC-MS analyses were performed with an HP GCD G1800C gas-chromatograph with injector and detector temperatures of 250 and 280 °C, respectively; the heating procedure started at 100 °C for 5 min, heated to 240 °C with a rate of 10 °C min−1, and then kept at 240 °C for 16 min; retention times (r.t.) are given in minutes. 1H and 13C NMR spectra at 400 and 100 MHz, respectively, were recorded at room temperature with a Bruker Advance 400 spectrometer with chemical shifts given in parts per million (ppm) versus external TMS. The chemical shifts reported were determined by reference to the solvent residual signal (proton: 7.26, 2.50 and 2.08 ppm and carbon: 77.2, 39.5, and 20.4 ppm for CDCl3, DMSO-d6, and toluene-d8, respectively); coupling constants are given in Hz. [Fe2(OEt)2(dpm)4] [33] and [Fe4(OMe)6(dpm)6] [12,32] were synthesized as previously reported in the literature. Synthesis of 3 and 4 and attempted synthesis of H3PhpPy from 4 are reported in the Supplementary Materials.
3.2. Synthesis of 2-(4-(pyridin-4-yl)phenyl)ethanol (1)
This compound was obtained with a modification of a published procedure [28], especially in the final purification step. 2-(4-bromophenyl)ethanol (1.00 g, 4.97 mmol), pyridin-4-ylboronic acid (0.552 g, 4.49 mmol), and Na2CO3 (0.526 g, 4.97 mmol) were introduced in a Schlenk tube under Ar. Then, Pd(PPh3)4 (0.262 g, 0.23 mmol) was added together with the solvent mixture (DMF:H2O 3:1, 24 mL overall). The yellow-green solution was kept under stirring at 110 °C for 24 h, and then it was cooled to room temperature and open to air to decompose any unreacted catalyst. The mixture was transferred in a round-bottom flask with the help of CH2Cl2 (2 × 5 mL). The volatile organic phase was removed under reduced pressure yielding a yellow-green residue still containing DMF, H2O, or both. It was then transferred in a separating funnel and extracted with CH2Cl2 (40 mL) and H2O (20 mL). The aqueous layer was further extracted with CH2Cl2 (2 × 10 mL), and the organic phases were washed with H2O (10 mL). The aqueous phase was extracted with Et2O (3 × 10 mL) and all the organic phases were unified and concentrated under reduced pressure, yielding a yellow-orange concentrated solution still containing some DMF. Both TLC (PE:AcOEt 1:4) and GC-MS analysis showed the presence of the desired product as main component of the mixture. This was then diluted with Et2O (anhydrous over molecular sieves, 10 mL) and the resulting solution was treated with HCl (1.32 mol L−1 solution in Et2O, 5.0 mL, 6.6 mmol), obtaining the efficient precipitation of the product as light yellow hydrochloride salt. After 5 min under stirring the solid was filtered on a Gooch G3 and washed with Et2O (anhydrous over molecular sieves, 2 × 5 mL). The solid was then suspended in CH2Cl2 (20 mL) and aqueous NaOH (5% w/w solution, 4.5 mL, 5.62 mmol) was slowly added under stirring. The mixture was transferred in a separating funnel, the aqueous solution was extracted with further CH2Cl2 (5 mL) and the organic phases were unified, dried over MgSO4 and taken to dryness under reduced pressure. The dense oil was dissolved in CH2Cl2 (4 mL) and Et2O (15 mL) yielding a turbid yellow solution. This was refluxed for 5 min obtaining the precipitation of a brown solid, which was filtered off and discharged. The solution was then concentrated under reduced pressure till formation of a solid, then n-hexane (20 mL) was added and the mixture again concentrated under reduced pressure and cooled with an ice bath in order to favor the precipitation. The title compound as light yellow solid was filtered on a Gooch G3, washed with n-hexane (5 mL) and dried under vacuum (0.57 g, 58%). TLC (PE:AcOEt 1:4) r.f. = 0.14. GC-MS: r.t. = 18.55 min, m/z = 199 [M]+. Elemental analysis calcd (%) for C13H13NO (199.25): C 78.36, H 6.58, N 7.03; found: C 78.05, H 6.68, N 7.10. IR (KBr): ṽ = 3189 (br, ν O–H), 3043, 2997, 2943, 2919, and 2858 (m–s, ν C–H), 1603 (vs, ν C=N), 1495, 1424, 1406, and 1365 (s, ν aromatic C=C), 1236, 1207, and 1164 (s, ν aliphatic C–C), 1064 (vs, ν alkoxido C–O). 1H NMR (400 MHz, 298 K, CDCl3): δ 2.30 (br, 1H, OH), 2.95 (t, J = 6.49 Hz, 2H, H2), 3.93 (t, J = 6.49 Hz, 2H, H1), 7.37 (m, 2H, H4), 7.53 (m, 2H, H5), 7.60 (m, 2H, H8), 8.64 (m, 2H, H9) ppm. 13C NMR (100 MHz, 298 K, CDCl3): δ 38.9 (C2), 63.4 (C1), 121.7 (C8), 127.2 (C5), 129.9 (C4), 135.9 (C3), 140.3 (C6), 148.9 (C7), 149.4 (C9) ppm (see Scheme 1 for atom numbering).
3.3. Synthesis of 2-(4-(Pyridin-4-yl)phenyl)acetaldehyde (2)
This compound was obtained with a modification of a published procedure [29]. 1 (0.50 g, 2.51 mmol) was dissolved in CH2Cl2 (13 mL, anhydrous over molecular sieves) and the resulting solution was cooled with an ice bath. After 10 min, TICA (0.50 g, 2.15 mmol) was added yielding a white suspension, which was further stirred for 20 min at low temperature. TEMPO (0.073 mol L−1 solution in CH2Cl2, 0.30 mL, 0.022 mmol) was added, the reaction mixture left at 0 °C for 15 min, and then diluted with CH2Cl2 (7 mL). Aqueous Na2CO3 (1.5 mL of a saturated solution) was added observing a color change from orange to yellow and formation of a white solid. The liquid phase was separated, the solid residue was washed with CH2Cl2 (2 × 5 mL) and the organic phases taken to dryness under reduced pressure. The orange residue was dissolved in CH2Cl2 (8 mL) and Et2O (10 mL), heated to reflux for 5 min, and the yellow liquid phase was filtered on a Gooch G3 to eliminate the solid and taken to dryness, yielding the title compound as yellow solid (0.47 g, 95%). TLC (PE:AcOEt 1:4) r.f. = 0.26. GC-MS: r.t. = 17.10 min, m/z = 197 [M]+. Elemental analysis calcd (%) for C13H11NO·⅓H2O (203.24): C 76.83, H 5.79, N 6.89; found: C 76.94, H 5.86, N 6.91. IR (KBr): ṽ = 1701 (s, ν C=O), 1596 (s, ν C=N), 1540, 1488 and 1403 (m, ν aromatic C=C). 1H NMR (400 MHz, 298 K, CDCl3): δ 3.81 (d, J = 1.99 Hz, 2H, H2), 7.38 (m, 2H, H4), 7.66 (m, 2H, H8), 7.71 (m, 2H, H5), 8.70 (m, 2H, H9), 9.82 (t, J = 1.99 Hz, 1H, H1) ppm. 13C NMR (100 MHz, 298 K, CDCl3): δ 50.2 (C2), 122.3 (C8), 127.8 (C5), 130.7 (C4), 134.1 (C3), 136.1 (C6), 147.4 (C7), 150.5 (C9), 198.4 (C1) ppm (see Scheme 1 for atom numbering).
3.4. Synthesis of 2-(Hydroxymethyl)-2-(4-(pyridin-4-yl)phenyl)propane-1,3-diol (H3PhpPy)
2 (0.50 g, 2.54 mmol), paraformaldehyde (0.50 g, 16.7 mmol), Ca(OH)2 (0.25 g, 3.37 mmol) and Al(OiPr)3 (0.05 g, 0.24 mmol) were dissolved in THF (5.5 mL, anhydrous over molecular sieves) in a Schlenk tube under Ar atmosphere. The mixture was stirred at 60 °C for 3 days, then cooled to room temperature, diluted with fresh THF (20 mL) and filtered on a Gooch G3 and Celite, washing with THF (2 × 10 mL). The obtained yellow solution was taken to dryness under reduced pressure, and the crude orange dense oil was purified with column chromatography (h = 17.0 cm, Ø = 2.5 cm) over silica gel and PE:AcOEt:THF (10:85:5, 100 mL), AcOEt:THF (95:5, 100 mL), AcOEt:THF:EtOH (95:2.5:2.5, 100 mL), AcOEt:EtOH (95:5, 200 mL) and AcOEt:EtOH (90:10, 200 mL) as eluent. The title compound was obtained as light yellow solid by removing of the solvent under reduced pressure of the fractions containing the desired product, washing it with CHCl3 (1.5 mL) and drying under vacuum (0.27 g, 41%). TLC (AcOEt:EtOH 9:1) r.f. = 0.11. GC-MS: not performed due to the high affinity of the triol unit for the stationary phase of the column. Elemental analysis calcd (%) for C15H17NO3·0.6H2O (270.11): C 66.70, H 6.79, N 5.19; found: C 66.66, H 6.67, N 4.91. IR (KBr): ṽ = 3354 and 3255 (br, ν O–H), 2946 and 2887 (m, ν C–H), 1602 (vs, ν C=N), 1491 and 1406 (m–s, ν C=C), 1025 (vs, ν alkoxido C–O). 1H NMR (400 MHz, 298 K, DMSO-d6): δ 3.77 (s, 6H, H1), 4.48 (br, 3H, OH), 7.58 (m, 2H, H5), 7.69 (m, 2H, H8), 7.71 (m, 2H, H4), 8.62 (m, 2H, H9). 13C NMR (100 MHz, 298 K, DMSO-d6): δ 49.3 (C2), 63.3 (C1), 121.2 (C8), 125.9 (C4), 128.6 (C5), 134.2 (C6), 144.7 (C7), 147.3 (C3), 150.0 (C9) (see Scheme 1 for atom numbering).
3.5. Synthesis of [Fe4(PhpPy)2(dpm)6] (Fe4)
A suspension of H3PhpPy (21.5 mg, 0.0829 mmol) in distilled EtOH (3.5 mL) was added dropwise to an orange solution of [Fe4(OMe)6(dpm)6] (50.0 mg, 0.0331 mmol) in distilled Et2O (21.0 mL). The reaction mixture became turbid and darker orange, and after 5 h under stirring at room temperature it was filtered to eliminate the undissolved material. The clear orange solution was taken to dryness yielding an orange powder, which appeared homogeneous under visual inspection at the microscope. IR (KBr): ṽ = 2962 (s, ν C–H), 1564, 1546, 1536, and 1505 (vs, ν C=N, C=O), 1401, 1385 and 1357 (s, ν aliphatic C–C), 1096 (m, ν alkoxido C–O), 566 (w, ν Fe–O). 1H NMR (400 MHz, 298 K, toluene-d8): δ 6.5, 8.8, 10.9, 12.0, 13.1 ppm. MS ESI (CH3CN): 644.4 ([Fe(dpm)3 + K]+, 50%), 1835.8 ([Fe4 + H]+, 10), 1874.7 ([Fe4 + K]+, 100). Elemental analysis calcd (%) for C96H142Fe4N2O18 (1835.54): C 62.82, H 7.80, N 1.53; found for powder: C 62.94, H 8.07, N 1.81. Different attempts to crystallize the title cluster by vapor diffusion of EtOH or evaporation of the mother liquor always yielded the dimer [Fe2(OEt)2(dpm)4] as main component; in one case we could also isolate single crystals of Fe4 suitable for X-ray diffraction. Elemental analysis calcd (%) for C96H142Fe4N2O18 (1835.54): C 62.82, H 7.80, N 1.53; found for manually-selected crystals: C 62.98, H 7.86, N 1.56.
3.6. Synthesis of {[Fe4(PhpPy)2(dpm)6Ag](ClO4)}n (Fe4Ag)
A solution of Fe4 (obtained from 25.0 mg of [Fe4(OMe)6(dpm)6], 0.0165 mol) in anhydrous toluene (4.0 mL), was put in liquid/liquid diffusion with a solution of AgClO4 in anhydrous THF (1.8 mL of a 9.12 × 10−3 mol L−1 solution, 0.0164 mmol). After one week the diffusion was complete and the orange needle-like microcrystals formed at the interface were filtered, washed with a toluene:THF 2:1 mixture and dried under vacuum (23.9 mg, 71.3%). Elemental analysis calcd (%) for C96H142AgClFe4N2O22 (2042.86): C 56.44, H 7.01, N 1.37; found: C 56.81, H 6.92, N 1.41. IR (KBr): ṽ = 3434 (br), 2963 (s, ν C–H), 1564, 1548, and 1505 (vs, ν C=N and C=O), 1400, 1385, and 1357 (s, ν aliphatic C–C), 1122 (m, ν ClO4), 1092 (m, ν alkoxido C–O), 567 (w, ν Fe–O), 479 (w, ν Ag–N). MS ESI (THF/CH3CN): m/z 1230.9 ([Fe3(PhpPy)(dpm)4(CH3CN) + H + Na]+, 100%), 1836.4 ([Fe4 + H]+, 10), 1837.8 ([Fe4 + K]+, 55), 2014.7 ([Fe4Ag(THF)]+, 5).
3.7. X-ray Data Collection and Structure Determination
Crystals of Fe4 suitable for X-ray diffraction experiments were selected from the mother liquor in EtOH/Et2O. Intensity data were collected on a Bruker Apex CCD area detector using graphite monochromatic Mo-Kα radiation. Crystal structure data for Fe4: C96H142Fe4N2O18, Mr = 1835.54, crystal dimensions 0.37 × 0.15 × 0.15 mm, monoclinic, space group: C2/c (No. 15), a = 19.328(3), b = 23.163(3), c = 24.483(4) Å, β = 108.155(2), V = 10,416(3) Å3, Z = 4, ρcalc = 1.171 g cm−3, 2θmax = 46.78°, T = 293(2) K, collected/independent reflections = 55,040/7575, R(int) = 0.0644, parameters/restraints = 651/108, final R indices: R1 = 0.0561, wR2 = 0.1384 [on 5266 reflections with I > 2σ(I)], R1 = 0.0859, wR2 = 0.1690 [all data], goodness-of-fit = 1.074.
During data collection, no crystal decay was observed, so that no time-decay correction was needed. Data reductions were performed with SAINT, and absorption corrections based on multiscan were obtained with SADABS [48]. The structure was solved by direct methods and refined with SHELXL-2016/6 [49] implemented in WinGX—Version 2014.1 system [50]. The program ORTEPIII was used for graphics [51]. Three over six tBu groups were refined for their disorder by splitting them into two positions, A and B, with complementary occupancy factors of 0.624(15) and 0.376(15); restraint ISOR (sigma = 0.1) was also applied to the disordered terminal carbon atoms to ensure reasonable thermal ellipsoids. Anisotropic thermal parameters were used for all non-H atoms. The isotropic thermal parameters of H atoms were fixed at 1.2 (1.5 for methyl groups) times those of the atom to which they were attached. All H atoms were placed in calculated positions and refined by a riding model.
CCDC 1842624 contains the supplementary crystallographic data for Fe4. These data can be obtained free of charge via http://www.ccdc.cam.ac.uk/conts/retrieving.html, or from the Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: (+44) 1223-336-033, or e-mail: [email protected].
3.8. Magnetic Measurements
Dc magnetic data were recorded using a Quantum Design MPMS SQUID magnetometer. Magnetic susceptibilities were measured on 7.54 and 8.10 mg powder samples of Fe4 and Fe4Ag, respectively, packed in a Teflon pellet with applied fields of 1 kOe from 1.9 (Fe4Ag) or 5 (Fe4) to 40 K and of 10 kOe from 40 to 300 (Fe4Ag) or 250 (Fe4) K. Data reduction was carried out by using 1835.54 and 2042.86 as molecular weights and −1054.9 and −1114.9 10−6 emu mol−1 as diamagnetic contributions, estimated from the Pascal’s constants [52], for Fe4 and Fe4Ag, respectively. Data fitting was performed by using program PHI v3.1.1 [44]. Ac susceptibility was recorded on a Quantum Design PPMS susceptometer for Fe4Ag at zero static applied field and for both Fe4 and Fe4Ag samples at 1 kOe static applied field, while the previously reported Quantum Design MPMS SQUID was employed for ac measurements on Fe4 at zero static applied field.
4. Conclusions
As a promising strategy in developing the performance of SMMs by their assembly into coordination networks, in this paper we have described the behavior of a tetrairon(III) cluster, Fe4, as longer spacer (23.4 Å) when coordinating to silver(I) ions, in comparison with what previously observed with a shorter spacer (14.8 Å) of the same family, A [14]. As crucial point when engineering novel compounds with desired properties [53], the chemical design of the suitable tripodal ligand and its synthesis revealed fundamental for this project. 1D polymer chains were achieved in Fe4Ag with Fe4:AgClO4 1:1 ratio, in contrast with the aimed 3D arrangement similar to B, which contains an A:AgClO4 2:1 ratio [14]. The enormous cavities that would have formed with a 3D-MOF structure given by an Fe4:AgClO4 2:1 ratio might probably suffer of high instability in the absence of any supplementary big molecules acting as sustain within the pores, and this point will be pursued in our future studies.
Despite the synthesized coordination polymer Fe4Ag does not possess the desired 3D structure, it still revealed invariant relaxation dynamics compared to B, in which Fe4 SMMs behave as building blocks of polymeric structures. Further work to address 1D vs. 3D issue will be perceived also by testing other metal ions or coordination units like ruthenium dimers, which hold minor flexibility in their coordination behavior with respect to silver(I). In fact, this ion can assume different coordination environments only by subtle changes in the surrounding environment or the donor power of the coordinated atoms [43,54,55], as we could clearly observe here by exchanging H3pPy with H3PhpPy and that highlights how small molecular modifications can direct the fate of the final supramolecular assembly.
Supplementary Materials
The following are available online at https://www.mdpi.com/2312-7481/4/4/43/s1, Experimental Section (cont.): Synthesis of 3 and 4, Scheme S1: synthetic path of H3PhpPy through 3 and 4; Figure S1: Crystal packing in Fe4 with highlighted weak π⋅⋅⋅π stacking interactions; Figure S2: ESI-MS spectrum in THF/CH3CN of the reaction mixture for the synthesis of Fe4 after 5 h of stirring in EtOH at room temperature; Figure S3: Comparison among the infrared spectra of [Fe2(OEt)2(dpm)4], [Fe4(OMe)6(dpm)6] and Fe4 as KBr disks; Figure S4: 1H NMR spectrum of Fe4 in toluene-d8 at room temperature; Figure S5: ESI-MS spectrum in THF/CH3CN of Fe4Ag; Figure S6: Comparison among the infrared spectra of Fe4 and Fe4Ag as KBr disks; Figure S7: XRF spectrum of Fe4Ag with a view of the orange microcrystals; Figure S8: Temperature dependence of χMT for Fe4; Figure S9: Imaginary component of the ac susceptibility, χ’’M, of Fe4 measured at zero and 1-kOe applied static fields together with the corresponding ln(τ)-vs.-1/T Arrhenius plots.
Author Contributions
Conceptualization, L.R.; Data curation, L.R., C.B., and A.F.; Formal analysis, M.V. and C.B.; Investigation, L.R., M.V., F.R., and A.F.; Methodology, M.V. and F.R.; Project administration, L.R.; Resources, L.R., F.R., and A.F.; Supervision, L.R.; Writing–review & editing, L.R.
Funding
This project was founded by the European Research Council and the Italian Ministero dell’Istruzione, dell’Università e della Ricerca (MIUR) through the Advanced Grant MolNanoMaS no. 267746 and the FIRB project no. RBAP117RWN, respectively.
Acknowledgments
Authors would like to thank Carri Cotton (Erasmus student at Università degli Studi di Modena e Reggio Emilia from Warwick University, UK) for some preliminary synthetic work, Giorgia Ferrari for helping in XRF analysis, Andrea Nava for helping in preliminary single-crystal analyses, Andrea Cornia (Dipartimento di Scienze Chimiche e Geologiche, Università degli Studi di Modena e Reggio Emilia) for the access to the facilities of his research laboratory, and Roberta Sessoli and the Laboratory of Molecular Magnetism (Dipartimento di Chimica ‘Ugo Schiff’, Università degli Studi di Firenze) for the access to the facilities for magnetic measurements. L.R. also thanks Valentina Nicolini (Università degli Studi di Modena e Reggio Emilia) for helpful hints while writing the paper.
Conflicts of Interest
The authors declare no conflict of interest. The founding sponsors had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, and in the decision to publish the results.
References
- Gatteschi, D.; Sessoli, R.; Villain, J. Molecular Nanomagnets; Oxford University Press: Oxford, UK, 2006; ISBN 978-0-19-856753-0. [Google Scholar]
- Natterer, F.D.; Yang, K.; Paul, W.; Willke, P.; Choi, T.; Greber, T.; Heinrich, A.J.; Lutz, C.P. Reading and writing single-atom magnets. Nature 2017, 543, 226–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodwin, C.A.P.; Ortu, F.; Reta, D.; Chilton, N.F.; Mills, D.P. Molecular magnetic hysteresis at 60 kelvin in dysprosocenium. Nature 2017, 548, 439–442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, F.-S.; Day, B.M.; Chen, Y.-C.; Tong, M.-L.; Mansikkamäki, A.; Layfield, R.A. A Dysprosium Metallocene Single-Molecule Magnet Functioning at the Axial Limit. Angew. Chem. Int. Ed. 2017, 56, 11445–11449. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Zhang, X.; Meng, X.; Shi, W.; Cheng, P.; Powell, A.K. Constraining the coordination geometries of lanthanide centers and magnetic building blocks in frameworks: A new strategy for molecular nanomagnets. Chem. Soc. Rev. 2016, 45, 2423–2439. [Google Scholar] [CrossRef] [PubMed]
- Whitehead, G.F.S.; Moro, F.; Timco, G.A.; Wernsdorfer, W.; Teat, S.J.; Winpenny, R.E.P. A Ring of Rings and Other Multicomponent Assemblies of Cages. Angew. Chem. Int. Ed. 2013, 52, 9932–9935. [Google Scholar] [CrossRef] [PubMed]
- Jeon, I.-R.; Clérac, R. Controlled association of single-molecule magnets (SMMs) into coordination networks: Towards a new generation of magnetic materials. Dalton Trans. 2012, 41, 9569–9586. [Google Scholar] [CrossRef] [PubMed]
- Kostakis, G.E.; Hewitt, I.J.; Ako, A.M.; Mereacre, V.; Powell, A.K. Magnetic coordination clusters and networks: Synthesis and topological description. Philos. Trans. R. Soc. Math. Phys. Eng. Sci. 2010, 368, 1509–1536. [Google Scholar] [CrossRef] [PubMed]
- Roubeau, O.; Clérac, R. Rational Assembly of High-Spin Polynuclear Magnetic Complexes into Coordination Networks: The Case of a [Mn4] Single-Molecule Magnet Building Block. Eur. J. Inorg. Chem. 2008, 2008, 4325–4342. [Google Scholar] [CrossRef]
- Abstracts from the X INSTM National Conference on Materials Science and Technology. J. Appl. Biomater. Funct. Mater. 2016, 14, e84–e128. [CrossRef]
- Nava, A.; Rigamonti, L.; Zangrando, E.; Sessoli, R.; Wernsdorfer, W.; Cornia, A. Redox-Controlled Exchange Bias in a Supramolecular Chain of Fe4 Single-Molecule Magnets. Angew. Chem. Int. Ed. 2015, 54, 8777–8782. [Google Scholar] [CrossRef] [PubMed]
- Accorsi, S.; Barra, A.-L.; Caneschi, A.; Chastanet, G.; Cornia, A.; Fabretti, A.C.; Gatteschi, D.; Mortalò, C.; Olivieri, E.; Parenti, F.; et al. Tuning Anisotropy Barriers in a Family of Tetrairon(III) Single-Molecule Magnets with an S = 5 Ground State. J. Am. Chem. Soc. 2006, 128, 4742–4755. [Google Scholar] [CrossRef] [PubMed]
- Gregoli, L.; Danieli, C.; Barra, A.-L.; Neugebauer, P.; Pellegrino, G.; Poneti, G.; Sessoli, R.; Cornia, A. Magnetostructural Correlations in Tetrairon(III) Single-Molecule Magnets. Chem. Eur. J. 2009, 15, 6456–6467. [Google Scholar] [CrossRef] [PubMed]
- Rigamonti, L.; Cotton, C.; Nava, A.; Lang, H.; Rüffer, T.; Perfetti, M.; Sorace, L.; Barra, A.-L.; Lan, Y.; Wernsdorfer, W.; et al. Diamondoid Structure in a Metal-Organic Framework of Fe4 Single-Molecule Magnets. Chem. Eur. J. 2016, 22, 13705–13714. [Google Scholar] [CrossRef] [PubMed]
- Rigamonti, L.; Piccioli, M.; Malavolti, L.; Poggini, L.; Mannini, M.; Totti, F.; Cortigiani, B.; Magnani, A.; Sessoli, R.; Cornia, A. Enhanced Vapor-Phase Processing in Fluorinated Fe4 Single-Molecule Magnets. Inorg. Chem. 2013, 52, 5897–5905. [Google Scholar] [CrossRef] [PubMed]
- Rigamonti, L.; Piccioli, M.; Nava, A.; Malavolti, L.; Cortigiani, B.; Sessoli, R.; Cornia, A. Structure, magnetic properties and thermal sublimation of fluorinated Fe4 Single-Molecule Magnets. Polyhedron 2017, 128, 9–17. [Google Scholar] [CrossRef]
- Ninova, S.; Lanzilotto, V.; Malavolti, L.; Rigamonti, L.; Cortigiani, B.; Mannini, M.; Totti, F.; Sessoli, R. Valence electronic structure of sublimated Fe4 single-molecule magnets: An experimental and theoretical characterization. J. Mater. Chem. C 2014, 2, 9599–9608. [Google Scholar] [CrossRef]
- Mannini, M.; Pineider, F.; Danieli, C.; Totti, F.; Sorace, L.; Sainctavit, P.; Arrio, M.-A.; Otero, E.; Joly, L.; Cezar, J.C.; et al. Quantum tunnelling of the magnetization in a monolayer of oriented single-molecule magnets. Nature 2010, 468, 417–421. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Douton, M.J.; Mannini, M.; Armelao, L.; Barra, A.-L.; Tancini, E.; Sessoli, R.; Cornia, A. One-step covalent grafting of Fe4 single-molecule magnet monolayers on gold. Chem. Commun. 2011, 47, 1467–1469. [Google Scholar] [CrossRef] [PubMed]
- Mannini, M.; Tancini, E.; Sorace, L.; Sainctavit, P.; Arrio, M.-A.; Qian, Y.; Otero, E.; Chiappe, D.; Margheriti, L.; Cezar, J.C.; et al. Spin Structure of Surface-Supported Single-Molecule Magnets from Isomorphous Replacement and X-ray Magnetic Circular Dichroism. Inorg. Chem. 2011, 50, 2911–2917. [Google Scholar] [CrossRef] [PubMed]
- Tancini, E.; Mannini, M.; Sainctavit, P.; Otero, E.; Sessoli, R.; Cornia, A. On-Surface Magnetometry: The Evaluation of Superexchange Coupling Constants in Surface-Wired Single-Molecule Magnets. Chem. Eur. J. 2013, 19, 16902–16905. [Google Scholar] [CrossRef] [PubMed]
- Westrup, K.C.M.; Boulon, M.-E.; Totaro, P.; Nunes, G.G.; Back, D.F.; Barison, A.; Jackson, M.; Paulsen, C.; Gatteschi, D.; Sorace, L.; et al. Adding Remnant Magnetization and Anisotropic Exchange to Propeller-like Single-Molecule Magnets through Chemical Design. Chem. Eur. J. 2014, 20, 13681–13691. [Google Scholar] [CrossRef] [PubMed]
- Rigamonti, L.; Cornia, A.; Nava, A.; Perfetti, M.; Boulon, M.-E.; Barra, A.-L.; Zhong, X.; Park, K.; Sessoli, R. Mapping of single-site magnetic anisotropy tensors in weakly coupled spin clusters by torque magnetometry. Phys. Chem. Chem. Phys. 2014, 16, 17220–17230. [Google Scholar] [CrossRef] [PubMed]
- Rigamonti, L.; Nava, A.; Boulon, M.-E.; Luzon, J.; Sessoli, R.; Cornia, A. Experimental and Theoretical Studies on the Magnetic Anisotropy in Lanthanide(III)-Centered Fe3Ln Propellers. Chem. Eur. J. 2015, 21, 12171–12180. [Google Scholar] [CrossRef] [PubMed]
- Burgess, J.A.J.; Malavolti, L.; Lanzilotto, V.; Mannini, M.; Yan, S.; Ninova, S.; Totti, F.; Rolf-Pissarczyk, S.; Cornia, A.; Sessoli, R.; et al. Magnetic fingerprint of individual Fe4 molecular magnets under compression by a scanning tunnelling microscope. Nat. Commun. 2015, 6, 8216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zu, F.; Liu, Z.; Yao, K.; Gao, G.; Fu, H.; Zhu, S.; Ni, Y.; Peng, L. Nearly Perfect Spin Filter, Spin Valve and Negative Differential Resistance Effects in a Fe4-based Single-molecule Junction. Sci. Rep. 2015, 4, 4838. [Google Scholar] [CrossRef] [PubMed]
- Batten, S.R.; Champness, N.R.; Chen, X.-M.; Garcia-Martinez, J.; Kitagawa, S.; Öhrström, L.; O’Keeffe, M.; Paik Suh, M.; Reedijk, J. Terminology of metal–organic frameworks and coordination polymers (IUPAC Recommendations 2013). Pure Appl. Chem. 2013, 85, 1715–1724. [Google Scholar] [CrossRef] [Green Version]
- Vukotic, V.N.; Loeb, S.J. One-, Two- and Three-Periodic Metal-Organic Rotaxane Frameworks (MORFs): Linking Cationic Transition-Metal Nodes with an Anionic Rotaxane Ligand. Chem. Eur. J. 2010, 16, 13630–13637. [Google Scholar] [CrossRef] [PubMed]
- Condorelli, G.G.; Motta, A.; Pellegrino, G.; Cornia, A.; Gorini, L.; Fragalà, I.L.; Sangregorio, C.; Sorace, L. Site-Specific Anchoring of Tetrairon(III) Single Molecule Magnets on Functionalized Si(100) Surfaces. Chem. Mater. 2008, 20, 2405–2411. [Google Scholar] [CrossRef]
- Viguier, R.; Serratrice, G.; Dupraz, A.; Dupuy, C. New Polypodal Polycarboxylic Ligands—Complexation of Rare-Earth Ions in Aqueous Solution. Eur. J. Inorg. Chem. 2001, 1789–1795. [Google Scholar] [CrossRef]
- Beaufort, L.; Delaude, L.; Noels, A.F. A new tripodal ligand system based on the iminophosphorane functional group. Part 1: Synthesis and characterization. Tetrahedron 2007, 63, 7003–7008. [Google Scholar] [CrossRef]
- Barra, A.L.; Caneschi, A.; Cornia, A.; Fabrizi de Biani, F.; Gatteschi, D.; Sangregorio, C.; Sessoli, R.; Sorace, L. Single-Molecule Magnet Behavior of a Tetranuclear Iron(III) Complex. The Origin of Slow Magnetic Relaxation in Iron(III) Clusters. J. Am. Chem. Soc. 1999, 121, 5302–5310. [Google Scholar] [CrossRef]
- Wu, C.-H.S.; Rossman, G.R.; Gray, H.B.; Hammond, G.S.; Schugar, H.J. Chelates of β-diketones. VI. Synthesis and characterization of dimeric dialkoxo-bridged iron(III) complexes with acetylacetone and 2,2,6,6-tetramethylheptane-3,5-dione (HDPM). Inorg. Chem. 1972, 11, 990–994. [Google Scholar] [CrossRef]
- Rigamonti, L.; Rusconi, M.; Forni, A.; Pasini, A. The role of the atomic charges on the ligands and platinum(II) in affecting the cis and trans influences in [PtXL(PPh3)2]+ complexes (X = NO3, Cl, Br, I.; L = 4-substituted pyridines, amines, PPh3). A 31P NMR and DFT investigation. Dalton Trans. 2011, 40, 10162. [Google Scholar] [CrossRef] [PubMed]
- Nakamoto, K. Infrared and Raman Spectra of Inorganic and Coordination Compounds, 6th ed.; Wiley: Hoboken, NJ, USA, 2009; ISBN 978-0-471-74339-2. [Google Scholar]
- Geddes, A.L.; Bottger, G.L. Infrared spectra of silver-ammine complexes. Inorg. Chem. 1969, 8, 802–807. [Google Scholar] [CrossRef]
- Kondo, J.; Tada, Y.; Dairaku, T.; Saneyoshi, H.; Okamoto, I.; Tanaka, Y.; Ono, A. High-Resolution Crystal Structure of a Silver(I)-RNA Hybrid Duplex Containing Watson-Crick-like C-Silver(I)-C Metallo-Base Pairs. Angew. Chem. Int. Ed. 2015, 54, 13323–13326. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.G.; Cho, Y.; Lee, H.; Lee, Y.-A.; Jung, O.-S. o-, m-, and p-Pyridyl isomer effects on construction of 1D loop-and-chains: Silver(I) coordination polymers with Y-type tridentate ligands. J. Mol. Struct. 2016, 1122, 186–191. [Google Scholar] [CrossRef]
- Zhu, H.-L.; Zhang, M.; Sun, Z.-Y.; Rong, N.-N.; Zhang, B.; Li, Y.; Fun, H.-K. Crystal structure of di(4-aminopyridine)silver(I) trifluoromethylsulfate, Ag(C5H6N2)2(CF3SO3). Z. Für Krist. New Cryst. Struct. 2003, 218, 521–522. [Google Scholar] [CrossRef]
- Liu, J.-T.; Ng, S.W. catena-Poly[[silver-μ-piperazine-κ2N:N′]perchlorate]. Acta Crystallogr. Sect. E Struct. Rep. Online 2006, 62, m1992–m1993. [Google Scholar] [CrossRef]
- Cong, F.-D.; Yu, F.-Y.; Wei, Z.; Ng, S.W. Bis(2-methyl-1H-imidazole-κN3)silver(I) nitrate dihydrate. Acta Crystallogr. Sect. E Struct. Rep. Online 2009, 65, m1535. [Google Scholar] [CrossRef] [PubMed]
- Massoud, A.A.; Gohar, Y.M.; Langer, V.; Lincoln, P.; Svensson, F.R.; Jänis, J.; Gårdebjer, S.T.; Haukka, M.; Jonsson, F.; Aneheim, E.; et al. Bis 4,5-diazafluoren-9-one silver(I) nitrate: Synthesis, X-ray structures, solution chemistry, hydrogel loading, DNA coupling and anti-bacterial screening. New J. Chem. 2011, 35, 640–648. [Google Scholar] [CrossRef]
- Shimokawa, C.; Itoh, S. The First β-Diketiminate−Ag(I) Complexes. Macrocyclic Dinuclear and Tetranuclear Ag(I)-Complexes and Linear Coordination Polymer Ag(I)-Complex. Inorg. Chem. 2005, 44, 3010–3012. [Google Scholar] [CrossRef] [PubMed]
- Chilton, N.F.; Anderson, R.P.; Turner, L.D.; Soncini, A.; Murray, K.S. PHI: A powerful new program for the analysis of anisotropic monomeric and exchange-coupled polynuclear d- and f-block complexes. J. Comput. Chem. 2013, 34, 1164–1175. [Google Scholar] [CrossRef] [PubMed]
- Cole, K.S.; Cole, R.H. Dispersion and Absorption in Dielectrics I. Alternating Current Characteristics. J. Chem. Phys. 1941, 9, 341–351. [Google Scholar] [CrossRef]
- Dekker, C.; Arts, A.F.M.; de Wijn, H.W.; van Duyneveldt, A.J.; Mydosh, J.A. Activated dynamics in a two-dimensional Ising spin glass: Rb2Cu1−xCoxF4. Phys. Rev. B 1989, 40, 11243–11251. [Google Scholar] [CrossRef]
- Lunghi, A.; Totti, F.; Sessoli, R.; Sanvito, S. The role of anharmonic phonons in under-barrier spin relaxation of single molecule magnets. Nat. Commun. 2017, 8, 14620. [Google Scholar] [CrossRef] [PubMed]
- Bruker. SMART, SAINT and SADABS; Bruker AXS Inc.: Madison, WI, USA, 1997. [Google Scholar]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Farrugia, L.J. WinGX and ORTEP for Windows: An update. J. Appl. Crystallogr. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Burnett, M.N.; Johnson, C.K. ORTEP-III: Oak Ridge Thermal Ellipsoid Plot Program for Crystal Structure Illustrations; U.S. Department of Energy Office of Scientific and Technical Information: Oak Ridge, TN, USA, 1996.
- Bain, G.A.; Berry, J.F. Diamagnetic Corrections and Pascal’s Constants. J. Chem. Educ. 2008, 85, 532–536. [Google Scholar] [CrossRef]
- Rigamonti, L.; Bridonneau, N.; Poneti, G.; Tesi, L.; Sorace, L.; Pinkowicz, D.; Jover, J.; Ruiz, E.; Sessoli, R.; Cornia, A. A Pseudo-Octahedral Cobalt(II) Complex with bis-Pyrazolylpyridine Ligands Acting as a Zero-Field Single-Molecule Magnet with Easy Axis Anisotropy. Chem. Eur. J. 2018, 24, 8857–8868. [Google Scholar] [CrossRef] [PubMed]
- Fox, B.S.; Beyer, M.K.; Bondybey, V.E. Coordination Chemistry of Silver Cations. J. Am. Chem. Soc. 2002, 124, 13613–13623. [Google Scholar] [CrossRef] [PubMed]
- Shoeib, T.; El Aribi, H.; Siu, K.W.M.; Hopkinson, A.C. A Study of Silver(I) Ion−Organonitrile Complexes: Ion Structures, Binding Energies, and Substituent Effects. J. Phys. Chem. A 2001, 105, 710–719. [Google Scholar] [CrossRef]
Scheme 1.
(a) Na2CO3, Pd(PPh3)4, DMF:H2O 3:1, 110 °C, 24 h (yield: 58%); (b) TICA, TEMPO, CH2Cl2, 0 °C, 15 min (yield: 95%); (c) paraformaldehyde, Ca(OH)2, Al(OiPr)3, THF, 60 °C, 3 days (yield: 41%); overall yield: 23%; atom numbering as used for NMR assignment (see Section 3).
Scheme 1.
(a) Na2CO3, Pd(PPh3)4, DMF:H2O 3:1, 110 °C, 24 h (yield: 58%); (b) TICA, TEMPO, CH2Cl2, 0 °C, 15 min (yield: 95%); (c) paraformaldehyde, Ca(OH)2, Al(OiPr)3, THF, 60 °C, 3 days (yield: 41%); overall yield: 23%; atom numbering as used for NMR assignment (see Section 3).
Figure 1.
Molecular structure of Fe4 with ellipsoids at 20% probability, viewed along the crystallographic c axis with atom labelling scheme used in Table 1. Color code: Fe = orange, O = red, N = violet, C = grey, H = black.
Figure 1.
Molecular structure of Fe4 with ellipsoids at 20% probability, viewed along the crystallographic c axis with atom labelling scheme used in Table 1. Color code: Fe = orange, O = red, N = violet, C = grey, H = black.
Scheme 2.
Representation of Fe4Ag as 1D coordination polymer.
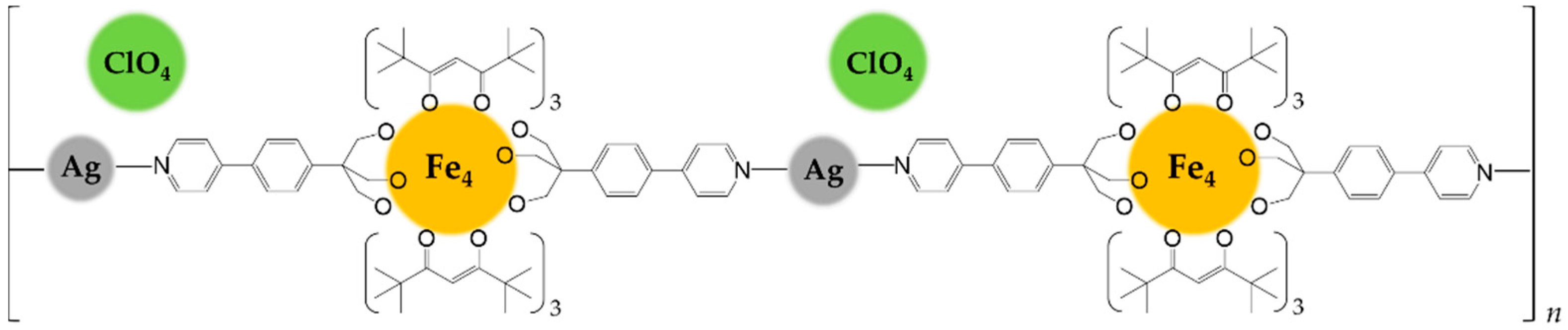
Figure 2.
Temperature dependence of χMT for Fe4Ag; solid and dashed curves are calculated using the best-fit parameters reported in the text.
Figure 2.
Temperature dependence of χMT for Fe4Ag; solid and dashed curves are calculated using the best-fit parameters reported in the text.
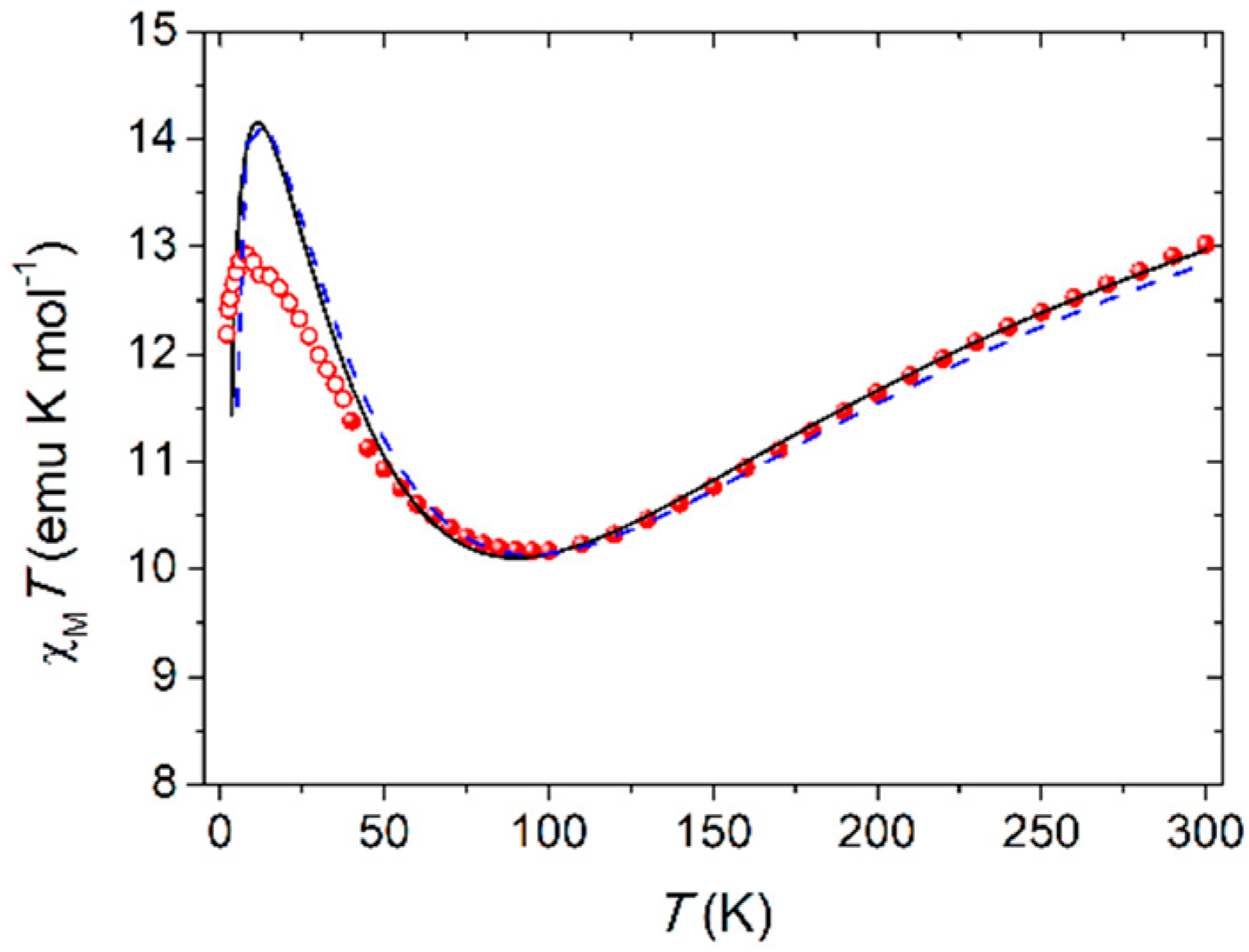
Figure 3.
Imaginary component of the ac susceptibility, χ’’M, of Fe4Ag measured at (a) zero applied static field in the 1.9 (red)–5.5 (blue) K temperature range, (b) 1.9 K and variable applied static field in the 0 (red)–50 (blue) kOe range, (c) 1 kOe applied static field in the 1.9 (red)–5.5 (blue) K temperature range; all measurements were performed in the 10–10,000 Hz frequency range, and grey lines are a guide for the eye. (d) Arrhenius plot for Fe4Ag both at zero and 1-kOe applied static fields; black lines are given by fitting data with the Arrhenius law.
Figure 3.
Imaginary component of the ac susceptibility, χ’’M, of Fe4Ag measured at (a) zero applied static field in the 1.9 (red)–5.5 (blue) K temperature range, (b) 1.9 K and variable applied static field in the 0 (red)–50 (blue) kOe range, (c) 1 kOe applied static field in the 1.9 (red)–5.5 (blue) K temperature range; all measurements were performed in the 10–10,000 Hz frequency range, and grey lines are a guide for the eye. (d) Arrhenius plot for Fe4Ag both at zero and 1-kOe applied static fields; black lines are given by fitting data with the Arrhenius law.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Selected interatomic distances (Å) and angles (°) in Fe4 1 (see Figure 1 for atom labelling).
Table 1.
Selected interatomic distances (Å) and angles (°) in Fe4 1 (see Figure 1 for atom labelling).
| Parameter | Value | Parameter | Value | Parameter | Value |
|---|---|---|---|---|---|
| Fe1–O1 | 1.994(3) | Fe2–O4 | 1.988(3) | Fe1⋅⋅⋅Fe2 | 3.0926(12) |
| Fe1–O2 | 1.971(2) | Fe2–O5 | 2.029(3) | Fe1⋅⋅⋅Fe3 | 3.0700(8) |
| Fe1–O3 | 1.978(2) | Fe3–O6 | 1.994(3) | Fe2⋅⋅⋅Fe3 | 5.3903(11) |
| Fe2–O3 | 1.977(2) | Fe3–O7 | 1.997(3) | Fe3⋅⋅⋅Fe3’ | 5.2063(15) |
| Fe3–O1 | 1.972(2) | Fe3–O8 | 1.998(3) | Fe2⋅⋅⋅Fe1⋅⋅⋅Fe3 | 122.014(17) |
| Fe3–O2 | 1.989(3) | Fe3–O9 | 2.005(3) | Fe3⋅⋅⋅Fe1⋅⋅⋅Fe3’ | 115.970(18) |
| Fe1–O1–Fe3 | 101.46(11) | Fe1–O2–Fe3 | 101.66(11) | Fe1–O3–Fe2 | 102.87(12) |
| γ (Fe2) 2 | 67.16(10) | γ (Fe3) 3 | 69.08(8) | N1⋅⋅⋅N1’ | 23.413(11) |
1 Primed atoms are related to unprimed ones by the two-fold axis passing through Fe1 and Fe2; 2 Dihedral angle between Fe1,Fe2,Fe3 and Fe1,O3,Fe2 planes; 3 Dihedral angle between Fe1,Fe2,Fe3 and Fe1,O1,O2,Fe3 planes.
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Rigamonti, L.; Vaccari, M.; Roncaglia, F.; Baschieri, C.; Forni, A. New Silver(I) Coordination Polymer with Fe4 Single-Molecule Magnets as Long Spacer. Magnetochemistry 2018, 4, 43. https://doi.org/10.3390/magnetochemistry4040043
AMA Style
Rigamonti L, Vaccari M, Roncaglia F, Baschieri C, Forni A. New Silver(I) Coordination Polymer with Fe4 Single-Molecule Magnets as Long Spacer. Magnetochemistry. 2018; 4(4):43. https://doi.org/10.3390/magnetochemistry4040043
Chicago/Turabian StyleRigamonti, Luca, Manuela Vaccari, Fabrizio Roncaglia, Carlo Baschieri, and Alessandra Forni. 2018. "New Silver(I) Coordination Polymer with Fe4 Single-Molecule Magnets as Long Spacer" Magnetochemistry 4, no. 4: 43. https://doi.org/10.3390/magnetochemistry4040043
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.