
Heptanuclear [FeIII6CrIII]3+ Complexes Experimentally Studied by Means of Magnetometry, X-ray Diffraction, XAS, XMCD and Spin-Polarized Electron Spectroscopy in Cross-Comparison with [MnIII6CrIII]3+ Single-Molecule Magnets

Abstract
:1. Introduction
2. Scientific Background
3. Experimental Approach
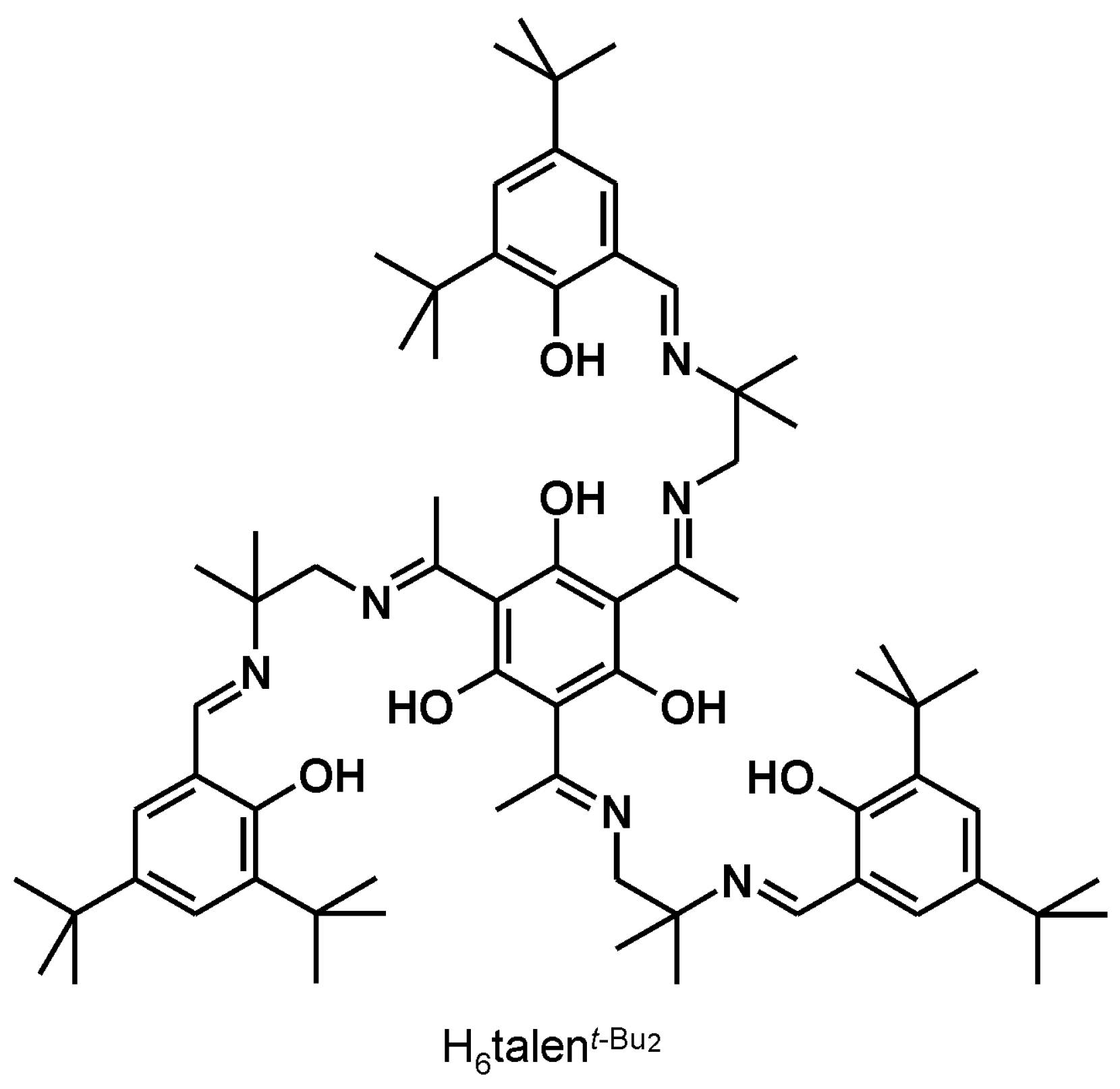
3.1. [{(talent−Bu2)(FeIII(MeOH))3}2{CrIII(CN)6}](ClO4)3 ([FeIII6CrIII](ClO4)3)
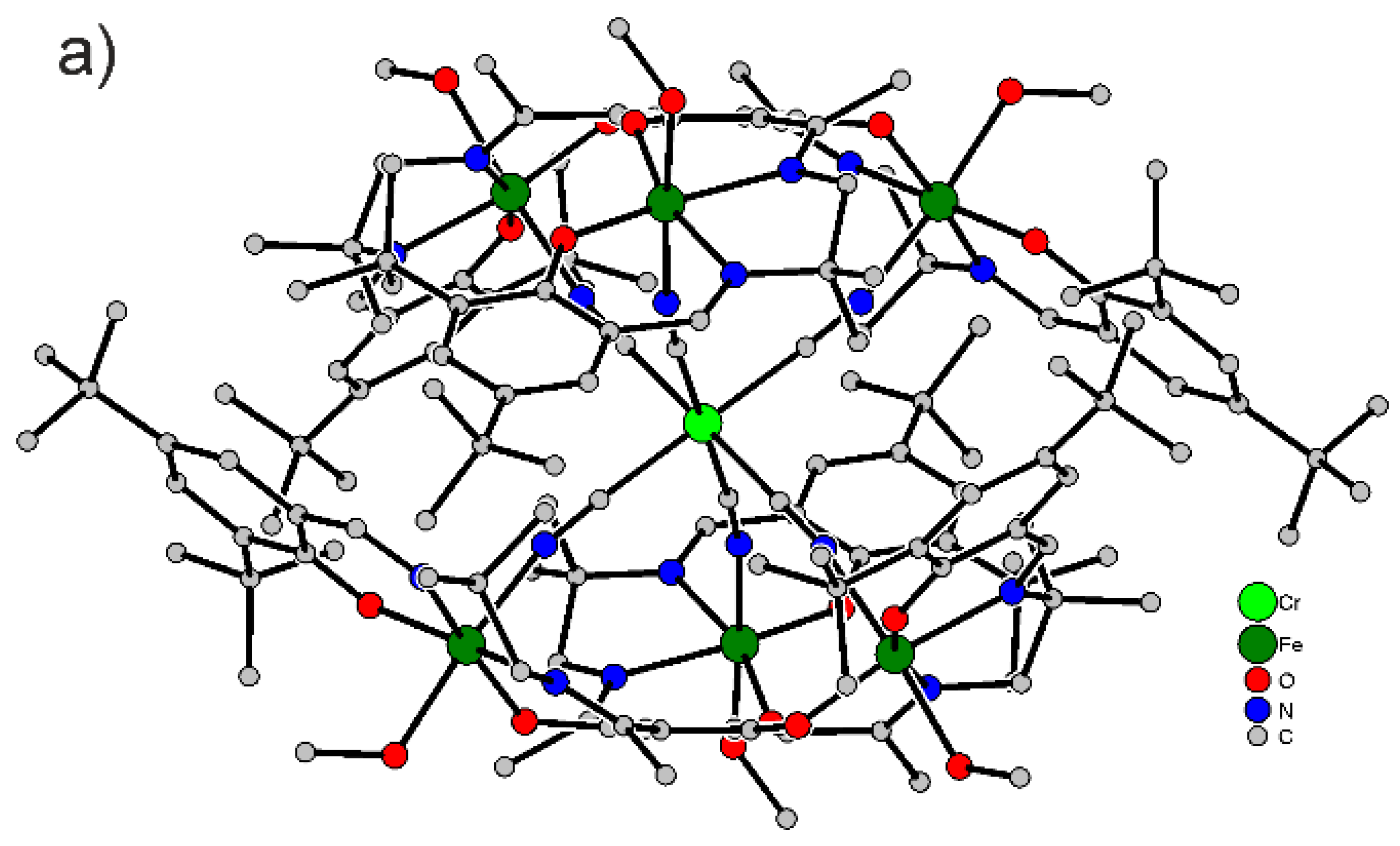
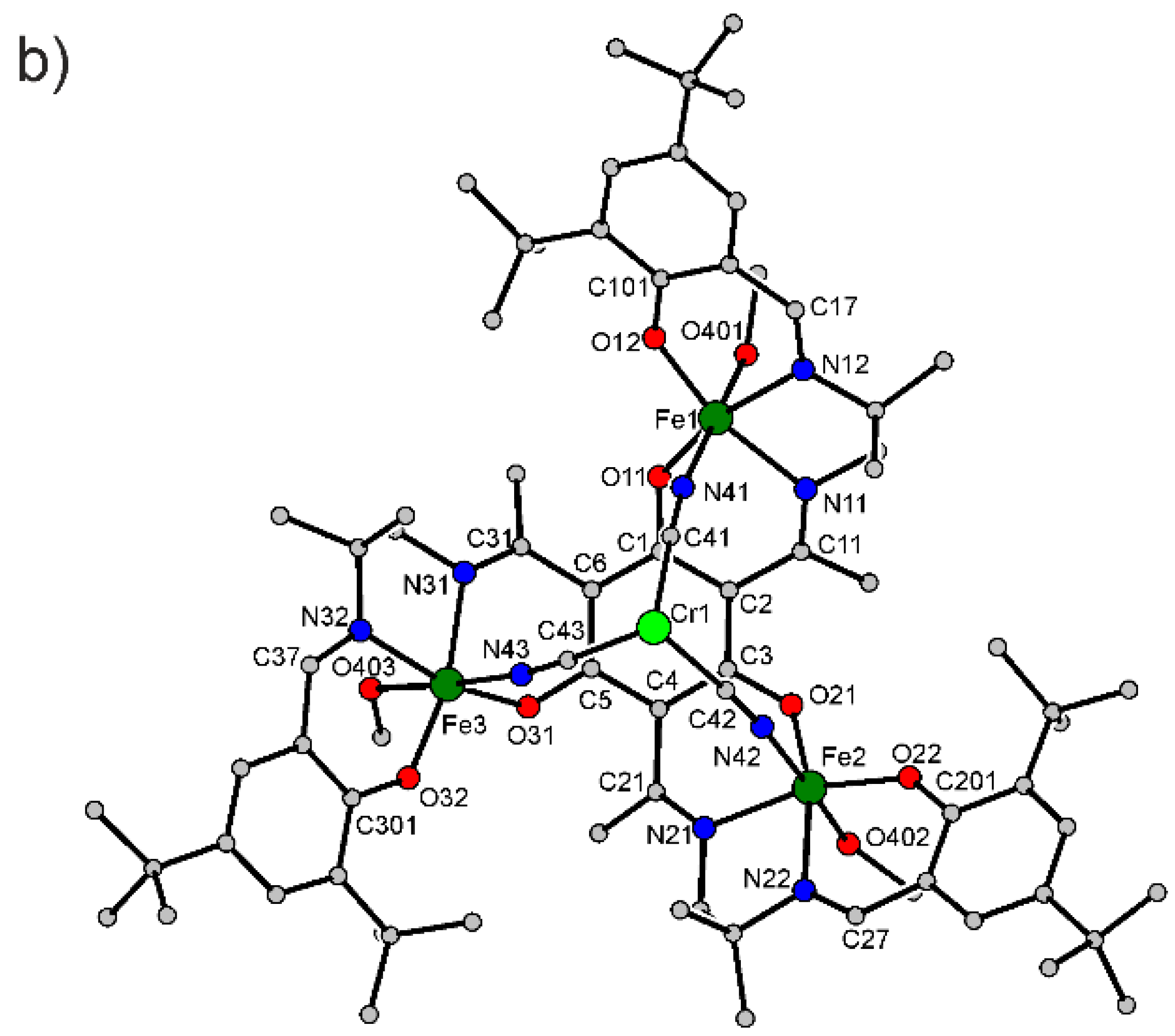
3.2. X-ray Crystallography
3.3. Other Physical Measurements
3.4. Sample Preparation

4. Results and Discussion
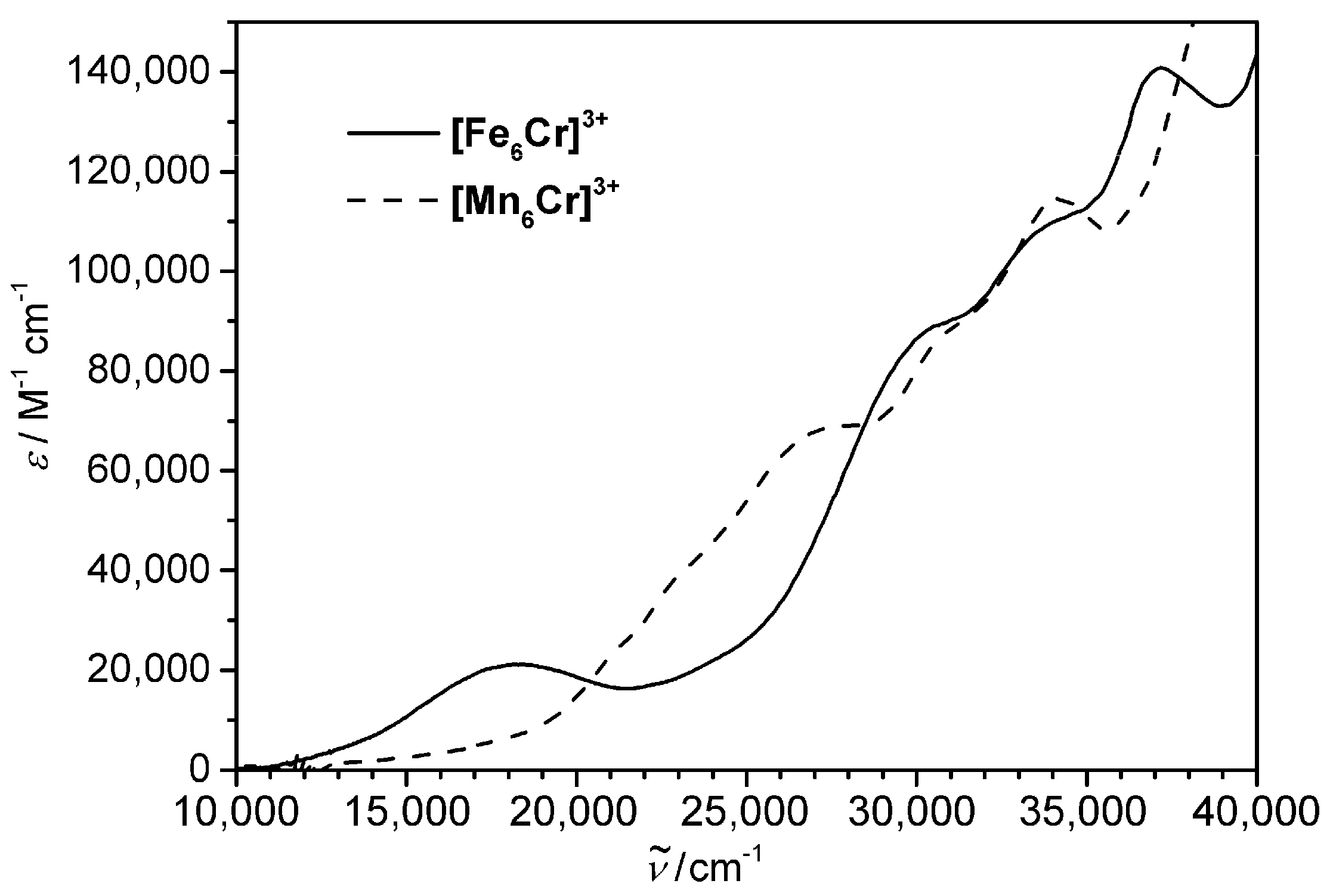
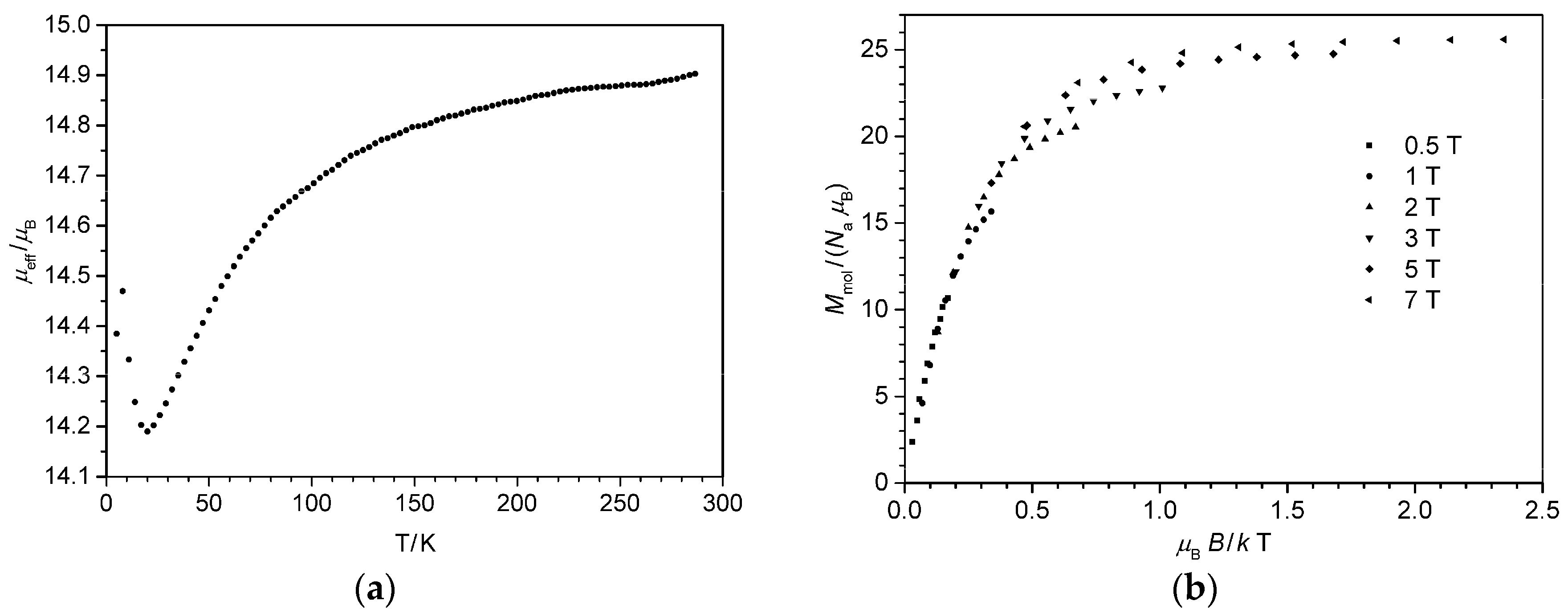
4.1. Synthesis and Characterization of [FeIII6Cr]3+

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
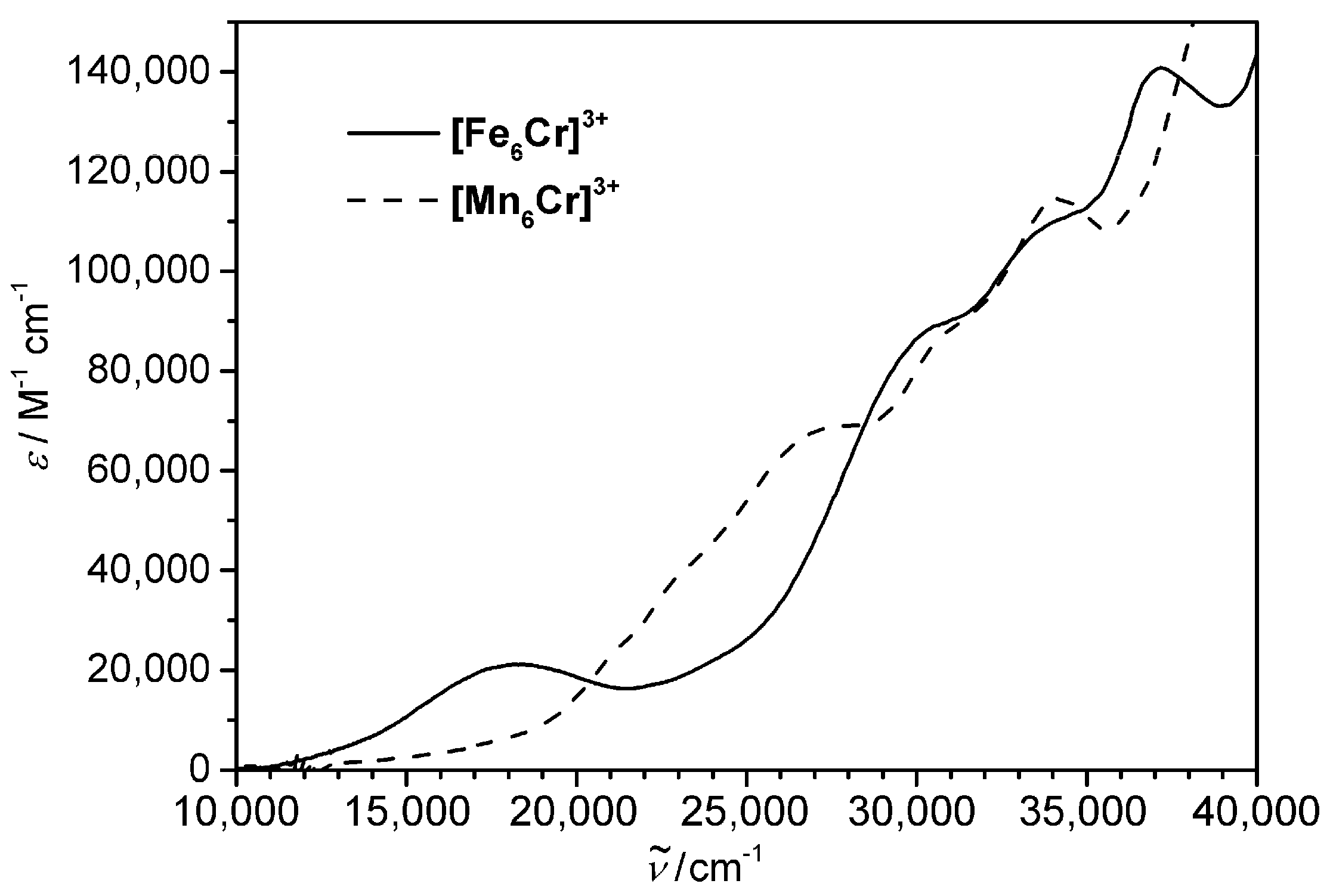
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fe1–O11 | 1.902(2) | O32–C301 | 1.325(4) |
| Fe1–O12 | 1.888(2) | N11–C11 | 1.291(4) |
| Fe1–N11 | 2.110(3) | N12–C17 | 1.298(4) |
| Fe1–N12 | 2.104(3) | N21–C21 | 1.293(4) |
| Fe1–N41 | 2.095(3) | N22–C27 | 1.290(4) |
| Fe1–O401 | 2.141(3) | N31–C31 | 1.293(5) |
| Fe2–O21 | 1.908(2) | N32–C37 | 1.291(5) |
| Fe2–O22 | 1.895(2) | N41–C41 | 1.149(4) |
| Fe2–N21 | 2.111(3) | N42–C42 | 1.147(4) |
| Fe2–N22 | 2.101(3) | N43–C43 | 1.149(4) |
| Fe2–N42 | 2.113(3) | C1–C2 | 1.416(5) |
| Fe2–O402 | 2.103(2) | C2–C3 | 1.422(5) |
| Fe3–O31 | 1.907(2) | C3–C4 | 1.415(5) |
| Fe3–O32 | 1.890(2) | C4–C5 | 1.416(5) |
| Fe3–N31 | 2.102(3) | C5–C6 | 1.410(5) |
| Fe3–N32 | 2.101(3) | C6–C1 | 1.411(5) |
| Fe3–N43 | 2.098(3) | C2–C11 | 1.470(5) |
| Fe3–O403 | 2.159(2) | C4–C21 | 1.482(4) |
| Cr1–C41 | 2.060(4) | C6–C31 | 1.487(5) |
| Cr1–C43 | 2.059(3) | ||
| Cr1–C42 | 2.068(4) | Cr1···Fe1 | 5.2582(5) |
| O11–C1 | 1.333(4) | Cr1···Fe3 | 5.2602(5) |
| O21–C3 | 1.312(4) | Cr1···Fe2 | 5.2758(5) |
| O31–C5 | 1.326(4) | Fe1···Fe3 | 6.7678(7) |
| O12–C101 | 1.322(4) | Fe1···Fe2 | 6.8200(7) |
| O22–C201 | 1.319(4) | Fe2···Fe3 | 6.7415(7) |

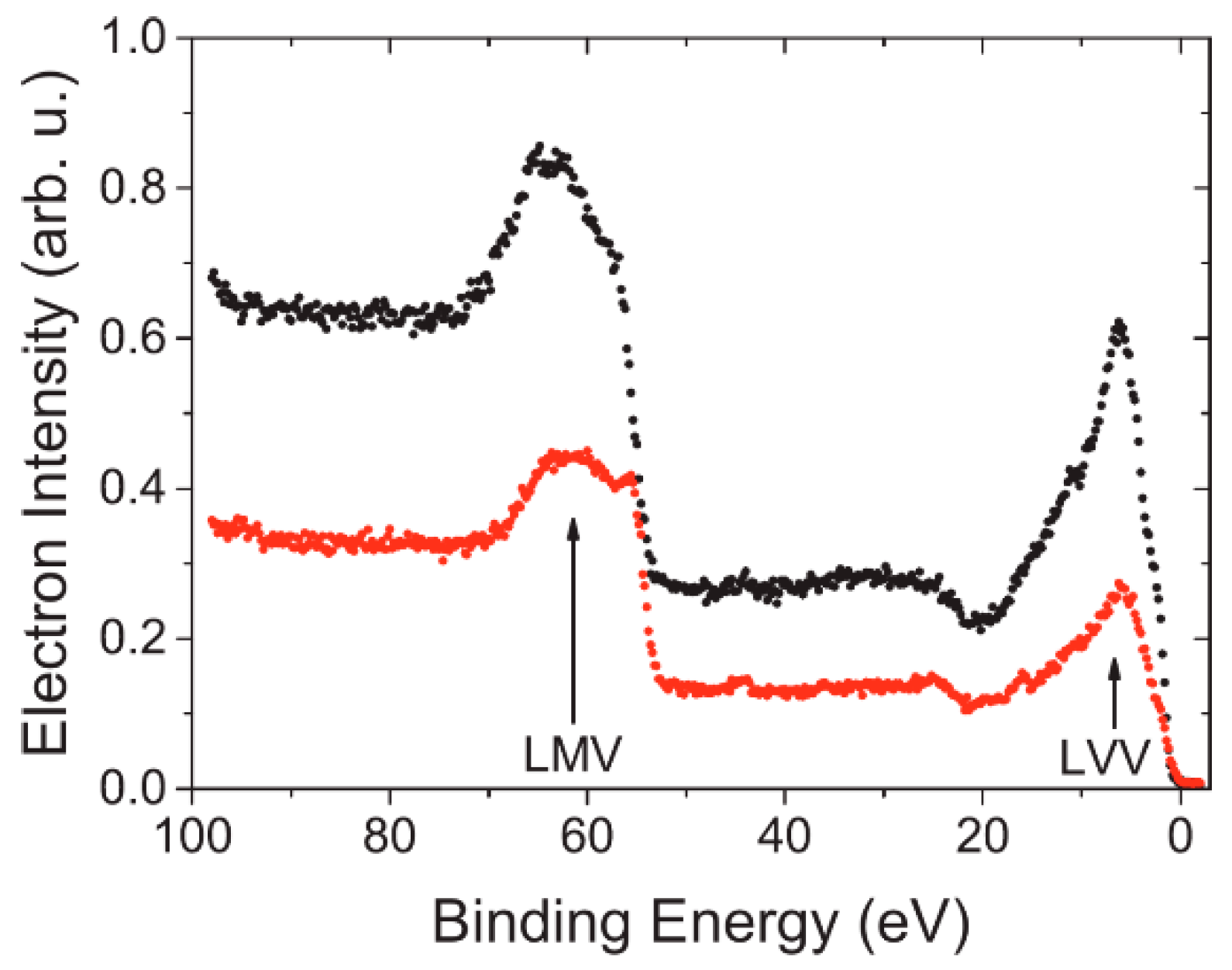
4.2. Radiation Sensitivity
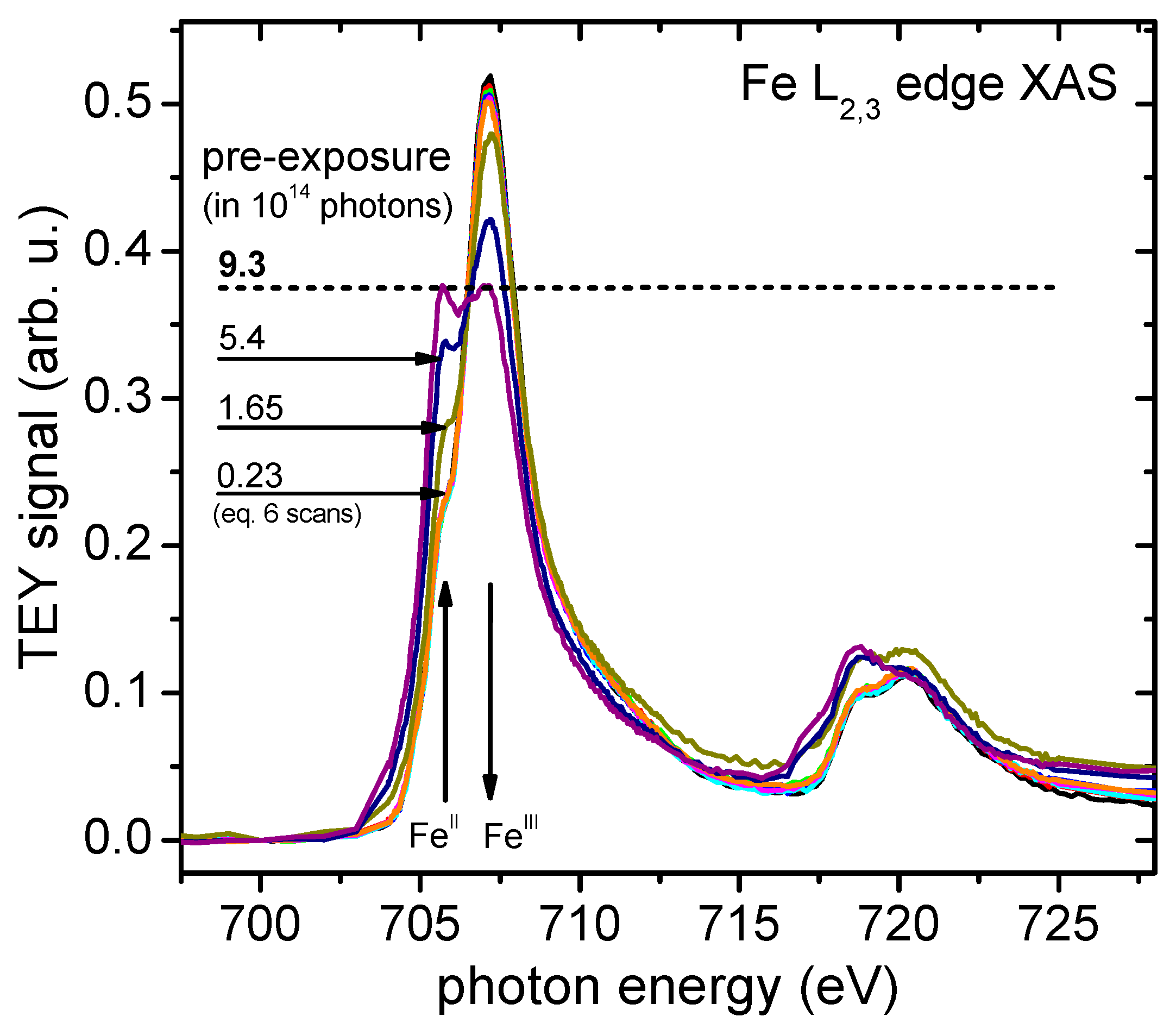
4.3. Spin-Resolved Electron Spectroscopy


4.4. XMCD Measurements
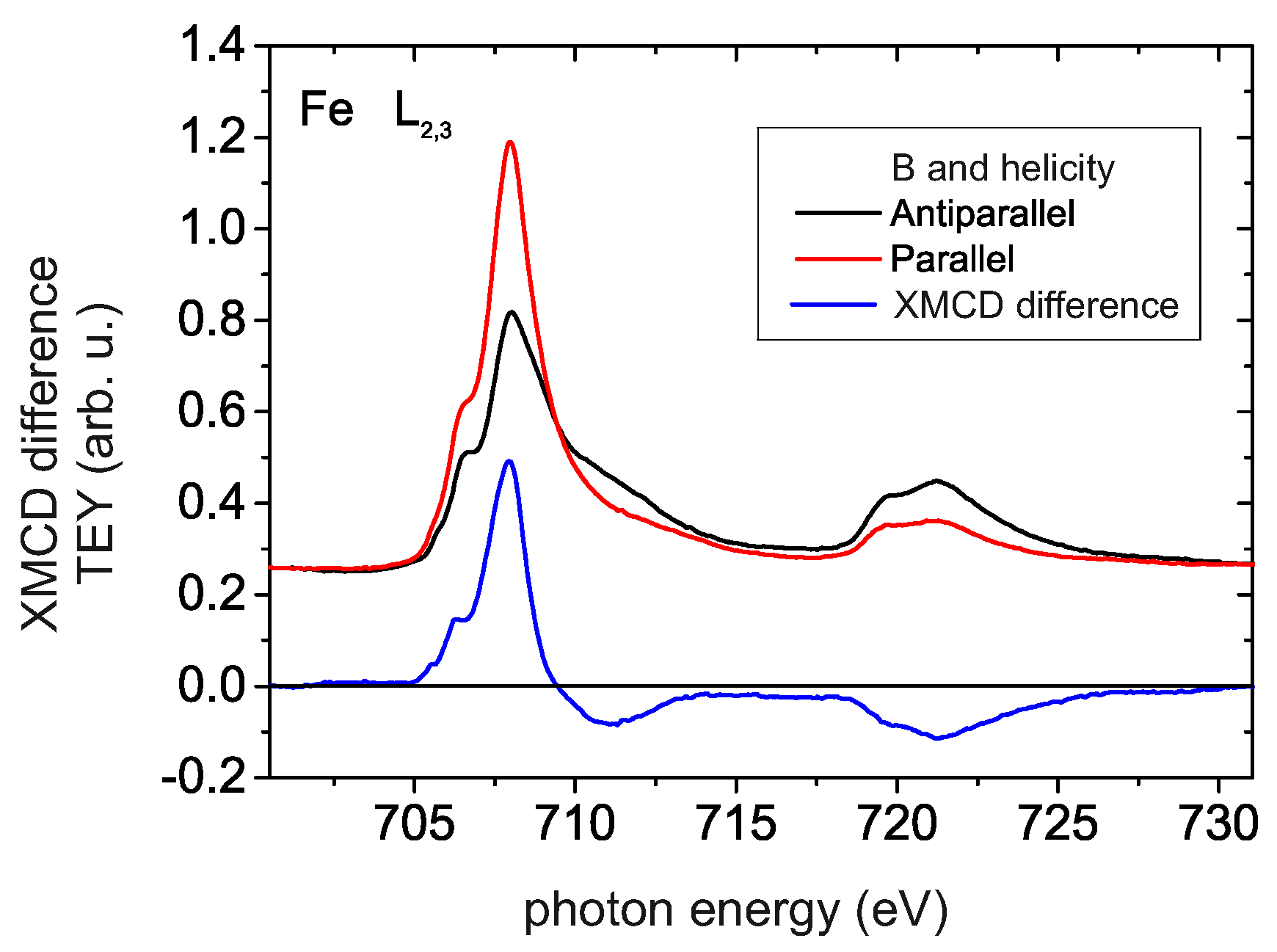
4.5. Electron Spin Polarization and XMCD Results of [FeIII6CrIII](ClO4)3 Adlayers in the Cross-Comparison with the Corresponding Results of [MnIII6CrIII]3+ SMM
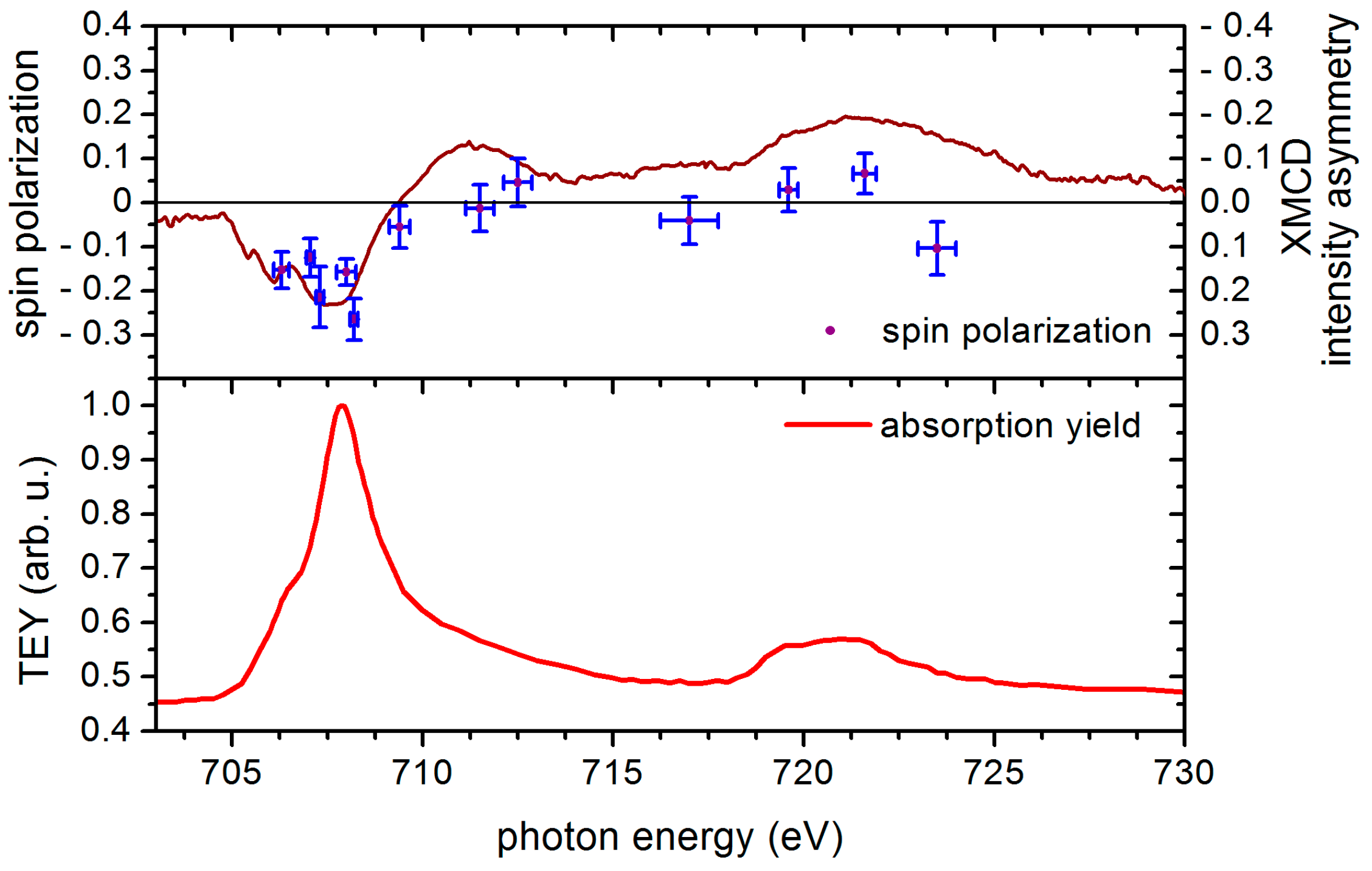
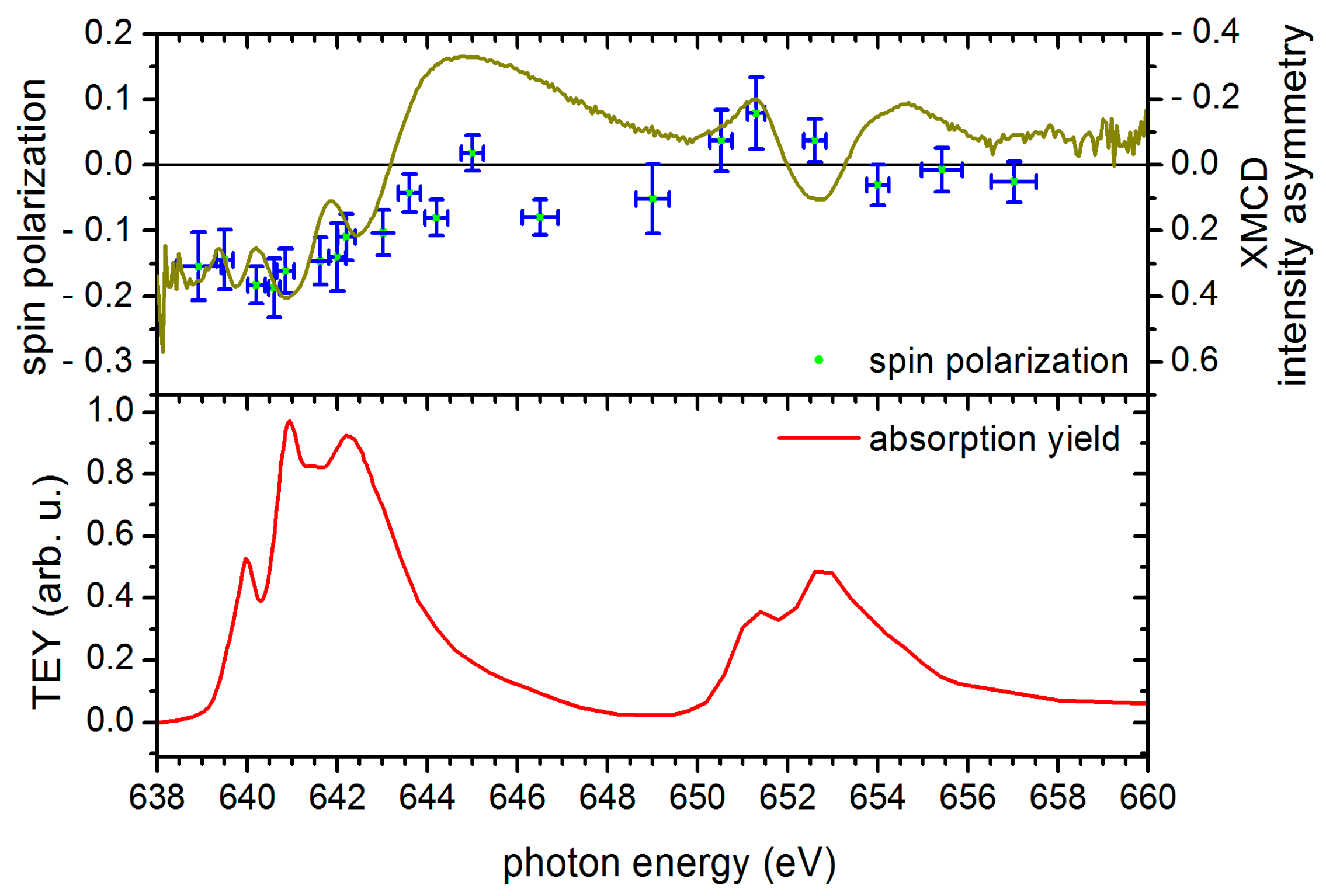
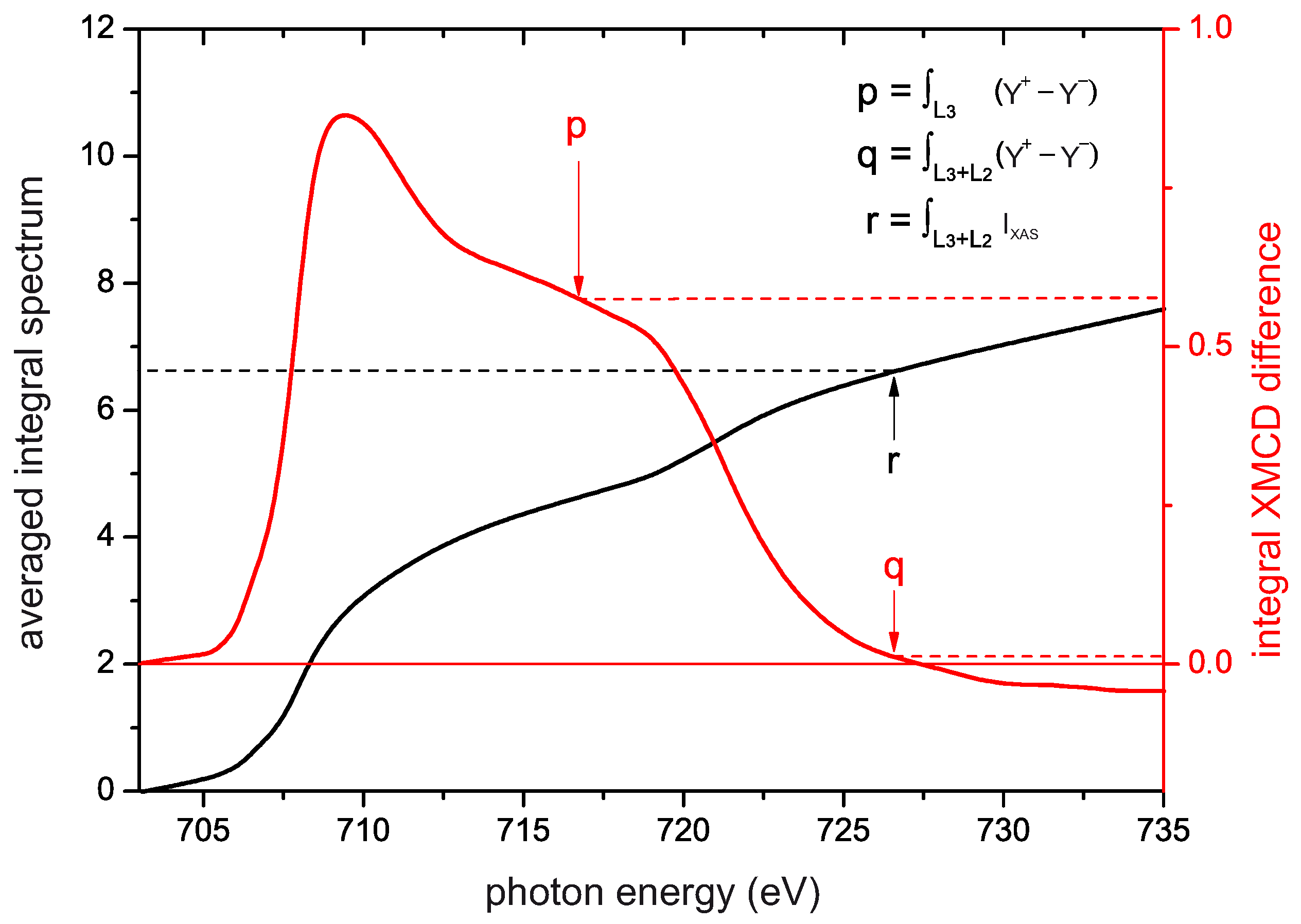
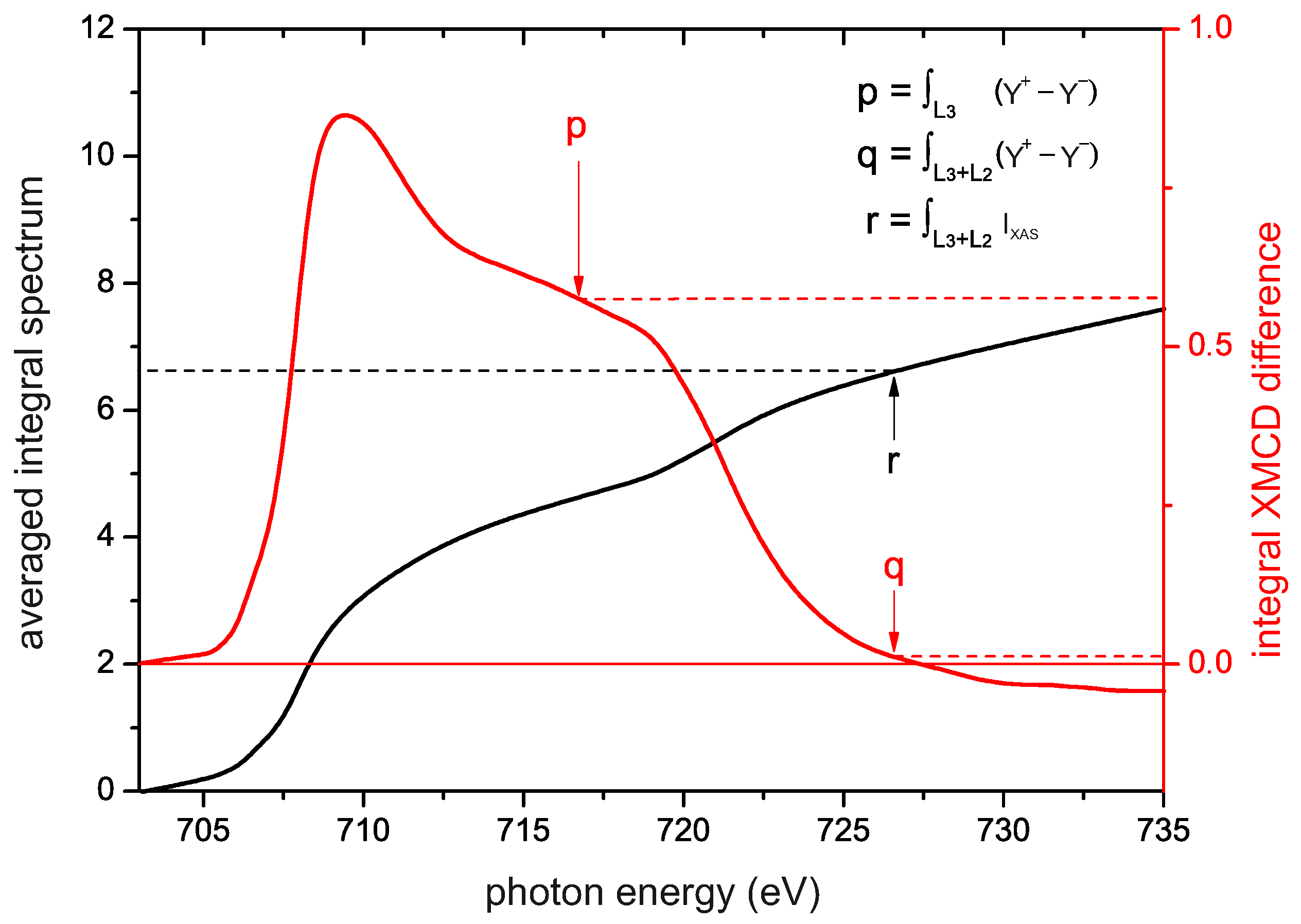
4.6. Sum Rule Evaluation of SPES and XMCD Data

5. Summary and Conclusions
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Helmstedt, A.; Dohmeier, N.; Müller, N.; Gryzia, A.; Brechling, A.; Heinzmann, U.; Hoeke, V.; Krickemeyer, E.; Glaser, T.; Leicht, P.; et al. Probing the magnetic moments of [MnIII6CrIII]3+ single-molecule magnets—A cross comparison of XMCD and spin-resolved electron spectroscopy. J. Electron Spectrosc. Relat. Phenom. 2015, 198, 12–19. [Google Scholar] [CrossRef]
- Wood, R. Future hard disk drive systems. J. Magn. Magn. Mater. 2009, 321, 555–561. [Google Scholar] [CrossRef]
- Wood, R. The feasibility of magnetic recording at 1 Terabit per square inch. IEEE Trans. Magn. 2000, 36, 36. [Google Scholar] [CrossRef]
- Stefanita, C.G. From bulk to nano. In The Many Sides of Magnetism; Springer Verlag: Berlin, Heidelberg, Germany, 2008; p. 131. [Google Scholar]
- Charap, S.H.; Lu, P.L.; He, Y. Thermal Stability of Recorded Information at High Densities. IEEE Trans. Magn. 1997, 33, 978. [Google Scholar] [CrossRef]
- Shiroishi, Y.; Fukuda, K.; Tagawa, I.; Iwasaki, H.; Takenoiri, S.; Tanaka, H.; Mutoh, H.; Yoshikawa, N. Future Options for HDD Storage. IEEE Trans. Magn. 2009, 45, 3816. [Google Scholar] [CrossRef]
- Nickel, J.; Gibson, G. Beyond magnetic storage: Atoms and molecules. In Magnetic Storage Systems beyond 2000; Hadjipanayis, G.C., Ed.; Kluwer Academic Publishers: Dordrecht, The Netherlands, 2001; p. 599. [Google Scholar]
- Hush, N.S. An overview of the first half-century of molecular electronics. Ann. N. Y. Acad. Sci. 2003, 1006, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Tsutsui, M.; Taniguchi, M. Single Molecule Electronics and Devices. Sensors 2012, 12, 7259–7298. [Google Scholar] [CrossRef] [PubMed]
- Renaud, N.; Hliwa, M.; Joachim, C. Single molecule logical devices. In Unimolecular and Supramolecular Electronics II - Chemistry and Physics Meet at Metal-Molecule Interfaces; Metzger, R.M., Ed.; Springer Verlag: Berlin, Germany; Heidelberg, Germany, 2012; p. 217. [Google Scholar]
- Christou, G.; Gatteschi, D.; Hendickson, D.; Sessoli, R. Single-molecule magnets. MRS Bull. 2000, 25, 66–71. [Google Scholar] [CrossRef]
- Gatteschi, D.; Sessoli, R. Quantum Tunneling of Magnetization and Related Phenomena in Molecular Materials. Angew. Chem. Int. Ed. 2003, 42, 268–297. [Google Scholar] [CrossRef] [PubMed]
- Gatteschi, D.; Sessoli, R.; Villain, J. Molecular Nanomagnets; Oxford University Press: Oxford, UK, 2006. [Google Scholar]
- Tejada, J.; Chudnovsky, E.M.; del Barco, E.; Hernandez, J.M.; Spiller, T.P. Magnetic qubits as hardware for quantum computers. Nanotechnology 2001, 12, 181–186. [Google Scholar] [CrossRef]
- Awschalom, D.D.; DiVincenzo, D.P.; Smyth, J.F. Macroscopic Quantum Effects in Nanometer-Scale Magnets. Science 1992, 258, 414. [Google Scholar] [CrossRef] [PubMed]
- Leuenberger, M.N.; Loss, D. Quantum computing in molecular magnets. Nature 2001, 410, 789. [Google Scholar] [CrossRef] [PubMed]
- Bogani, L.; Wernsdorfer, W. Molecular spintronics using single-molecule magnetsle. Nat. Mater. 2008, 7, 179. [Google Scholar] [CrossRef] [PubMed]
- Mannini, M.; Pineider, F.; Danieli, C.; Totti, F.; Sorace, L.; Sainctavit, P.; Arrio, M.A.; Otero, E.; Joly, L.; Cezar, J.C.; et al. Quantum tunneling of the magnetization in a monolayer of oriented single-molecule magnets. Nature 2010, 468, 417. [Google Scholar] [CrossRef] [PubMed]
- Rogez, G.; Donnio, B.; Terazzi, E.; Gallani, J.L.; Kappler, J.P.; Bucher, J.P.; Drillon, M. The Quest for Nanoscale Magnets: The example of [Mn12] Single Molecule Magnets. Adv. Mater. 2009, 21, 4323–4333. [Google Scholar] [CrossRef] [PubMed]
- Winpenny, R.E.P. Serendipitous assembly of polynuclear cage compounds. Dalton Trans. 2002, 1–10. [Google Scholar] [CrossRef]
- Lis, T. Preparation, structure and magnetic properties of a dodecanuclear mixed-valence manganese carboxylate. Acta Cryst. 1980, B36, 2042–2046. [Google Scholar] [CrossRef]
- Sessoli, R.; Tasi, H.-L.; Schake, A.R.; Wang, S.; Vincent, J.B.; Folting, K.; Gatteschi, D.; Christou, G.; Hendrickson, D.N. High-Spin Molecules: [Mn12O12(O2CR)16(H2O)4]. J. Am. Chem. Soc. 1993, 115, 1804–1816. [Google Scholar] [CrossRef]
- Bagai, R.; Christou, G. The Drosophila of single-molecule magnetism: [Mn12O12(O2CR)16(H2O)4]. Chem. Soc. Rev. 2009, 38, 1011–1026. [Google Scholar] [CrossRef] [PubMed]
- Glaser, T.; Heidemeier, M.; Weyhermüller, T.; Hoffmann, R.D.; Rupp, H.; Müller, P. Property-oriented rational design of single-molecule magnets: A C3-Symmetric Mn6Cr complex based on three molecular building blocks with a spin ground state of St = 21/2. Angew. Chem. Int. Ed. 2006, 45, 6033–6037. [Google Scholar] [CrossRef] [PubMed]
- Glaser, T. Rational design of single-molecule magnets: A supramolecular approach. Chem. Commun. 2011, 47, 116–130. [Google Scholar] [CrossRef] [PubMed]
- Hoeke, V.; Heidemeier, M.; Krickemeyer, E.; Stammler, A.; Bögge, H.; Schnack, J.; Postnikov, A.; Glaser, T. Environmental influence on the single-molecule magnet behavior of [MnIII6CrIII]3+: Molecular symmetry versus solid-state effects. Inorg. Chem. 2012, 51, 10929–10954. [Google Scholar] [CrossRef] [PubMed]
- Glaser, T.; Heidemeier, M.; Krickemeyer, E.; Bögge, H.; Stammler, A.; Fröhlich, R.; Bill, E.; Schnack, J. Exchange interactions and zero-field splittings in C3-Symmetric MnIII6FeIII: Using molecular recognition for the construction of a series of high spin complexes based on the triplesalen ligand. Inorg. Chem. 2009, 48, 607–620. [Google Scholar] [CrossRef] [PubMed]
- Von Richthofen, C.G.; Stammler, A.; Bögge, H.; de Groot, M.W.; Long, J.R.; Glaser, T. Synthesis, Structure, and Magnetic Characterization of a C-3-Symmetric (Mn3CrIII-Cr-III Assembly: Molecular Recognition Between a Trinuclear Mn-III Triplesalen Complex and a fac-Triscyano Cr-III Complex. Inorg. Chem. 2009, 48, 10165–10176. [Google Scholar] [CrossRef] [PubMed]
- Krickemeyer, E.; Hoeke, V.; Stammler, A.; Bögge, H.; Schnack, J.; Glaser, T. Synthesis and characterization of the heptanuclear [MnIII6CoIII]3+ triplesalen complex: Evidence for exchange pathways involving low-spin CoIII. Z. Naturforsch. 2010, 65, 295–303. [Google Scholar] [CrossRef]
- Hoeke, V.; Gieb, K.; Müller, P.; Ungur, L.; Chibotaru, L.F.; Heidemeier, M.; Krickemeyer, E.; Stammler, A.; Bögge, H.; Schröder, C.; et al. Hysteresis in the ground and excited spin state up to 10 T of a [MnIII6MnIII]3+ triplesalen single-molecule magnet. Chem. Sci. 2012, 3, 2868–2882. [Google Scholar] [CrossRef]
- Hoeke, V.; Heidemeier, M.; Krickemeyer, E.; Stammler, A.; Bögge, H.; Schnack, J.; Glaser, T. Structural influences on the exchange coupling and zero-field splitting in the single-molecule magnet [MnIII6MnIII]3+. Dalton Trans. 2012, 12942. [Google Scholar] [CrossRef] [PubMed]
- Hoeke, V.; Krickemeyer, E.; Heidemeier, M.; Theil, H.; Stammler, A.; Bögge, H.; Weyhermüller, T.; Schnack, J.; Glaser, T. A Comprehensive Study on Triplesalen-Based [MnIII6FeIII]3+ and [MnIII6FeII]2+ Complexes: Redox-Induced Variation of Molecular Magnetic Properties. Eur. J. Inorg. Chem. 2013, 4398–4409. [Google Scholar] [CrossRef]
- Hoeke, V.; Stammler, A.; Bögge, H.; Schnack, J.; Glaser, T. Strong and anisotropic superexchange in the single-molecule magnet (SMM) [MnIII6OsIII]3+: Promoting SMM behavior through 3d-5d transition metal substitution. Inorg. Chem. 2014, 53, 257–268. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, C.; Hoeke, V.; Stammler, A.; Bögge, H.; Schnack, J.; Glaser, T. Switching from antiferromagnetic to ferromagnetic coupling in Heptanuclear [Mt6Mc]n+ complexes by going from an achiral to a chiral triplesalen ligand. Dalton Trans. 2014, 43, 9690–9703. [Google Scholar] [CrossRef] [PubMed]
- Glaser, T.; Hoeke, V.; Gieb, K.; Schnack, J.; Schröder, C.; Müller, P. Quantum tunneling of the magnetization in [MnIII6M]3+ (M = CrIII, MnIII) SMMs: Impact of molecular and crystal symmetry. Coord. Chem. Rev. 2015, 289–290, 261–278. [Google Scholar] [CrossRef]
- Helmstedt, A.; Sacher, M.D.; Gryzia, A.; Harder, A.; Brechling, A.; Müller, N.; Heinzmann, U.; Hoeke, V.; Krickemeyer, E.; Glaser, T.; et al. Exposure of [MnIII6CrIII]3+ single-molecule-magnets to soft X-rays: The effect of the counterions on radiation stability. J. Electron Spectrosc. Relat. Phenom. 2012, 184, 583–588. [Google Scholar] [CrossRef]
- Helmstedt, A.; Müller, N.; Gryzia, A.; Dohmeier, N.; Brechling, A.; Sacher, M.D.; Heinzmann, U.; Hoeke, V.; Krickemeyer, E.; Glaser, T.; et al. Spin-resolved photoelectron spectroscopy of [MnIII6CrIII]3+ single-molecule-magnets and of manganese compounds as reference layers. J. Phys. Condens. Matter 2011, 23, 266001. [Google Scholar] [CrossRef] [PubMed]
- Gryzia, A.; Predatsch, H.; Brechling, A.; Hoeke, V.; Krickemeyer, E.; Derks, C.; Neumann, M.; Glaser, T.; Heinzmann, U. Preparation of monolayers of [Mn6Cr]3+-single-molecule-magnets on HOPG-, mica- and silicon surfaces and characterization by means of nc-AFM. Nanoscale Res. Lett. 2011, 6, 486. [Google Scholar] [CrossRef] [PubMed]
- Gryzia, A.; Volkmann, T.; Brechling, A.; Hoeke, V.; Schneider, L.; Kuepper, K.; Glaser, T.; Heinzmann, U. Crystallographic order and decomposition of [MnIII6CrIII]3+ single-molecule magnets deposited in submonolayers and monolayers on HOPG studied by means of molecular resolved atomic force microscopy (AFM) and kelvin probe force microscopy in UHV. Nanoscale Res. Lett. 2014, 9, 60. [Google Scholar] [CrossRef] [PubMed]
- Wende, H.; Antoniak, C. X-ray magnetic dichroism. In Magnetism and Synchrotron Radiation; Beaurepaire, E., Bulou, H., Scheurer, F., Kappler, J.P., Eds.; Springer Proceedings in Physics; Springer Verlag: Berlin, Germany; Heidelberg, Germany, 2010; p. 145. [Google Scholar]
- Sessoli, R.; Mannini, M.; Pineider, F.; Cornia, A.; Sainctavit, P. XAS and XMCD of single molecule magnets. In Magnetism and Synchrotron Radiation; Beaurepaire, E., Bulou, H., Scheurer, F., Kappler, J.P., Eds.; Springer Proceedings in Physics; Springer Verlag: Berlin, Germany; Heidelberg, Germany, 2010; p. 279. [Google Scholar]
- Nakamura, T.; Suzuki, M. Recent Progress of the X-ray Magnetic Circular Dichroism Technique for Element-Specific Magnetic Analysis. J. Phys. Soc. Jpn. 2013, 82, 021006. [Google Scholar] [CrossRef]
- Thole, B.T.; Carra, P.; Sette, F.; van der Laan, G. X-ray circular dichroism as a probe of orbital magnetization. Phys. Rev. Lett. 1992, 68, 1943. [Google Scholar] [CrossRef] [PubMed]
- Carra, P.; Thole, B.T.; Altarelli, M.; Wang, X. X-ray circular dichroism and local magnetic fields. Phys. Rev. Lett. 1993, 70, 694. [Google Scholar] [CrossRef] [PubMed]
- Scherz, A. Spin-Dependent X-ray Absorption Spectroscopy of 3D Transition Metals: Systematics and Applications. PhD Thesis, Freien Universität Berlin, Berlin, Germany, 2003. [Google Scholar]
- Chen, C.T.; Idzerda, Y.U.; Lin, H.J.; Smith, N.V.; Meigs, G.; Chaban, E.; Ho, G.H.; Pellegrin, E.; Sette, F. Experimental Confirmation of the X-ray Magnetic Circular Dichroism Sum Rules for Iron and Cobalt. Phys. Rev. Lett. 1995, 75, 152. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.; Wang, D.; Freeman, A.J. First principles investigation of the validity and range of applicability of the x-ray magnetic circular dichroism, sum rule. Phys. Rev. Lett. 1993, 71, 3581. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.; Freeman, A.J. Limitation of the Magnetic-Circular-Dichroism Spin Sum Rule for Transition Metals and Importance of the Magnetic Dipole Term. Phys. Rev. Lett. 1994, 73, 1994. [Google Scholar] [CrossRef] [PubMed]
- Schütz, G.; Fischer, P.; Attenkofer, K.; Knülle, M.; Ahlers, D.; Stähler, S.; Detlefs, C.; Ebert, H.; de Groot, F.M.F. X-ray magnetic circular dichroism in the near and extended absorption edge structure. J. Appl. Phys. 1994, 76, 6453. [Google Scholar] [CrossRef]
- Van der Laan, G. Sum rule practice. J. Synchrotron Rad. 1999, 6, 694–695. [Google Scholar] [CrossRef] [PubMed]
- Nesvizhskii, A.I.; Ankudinov, A.L.; Rehr, J.J. Normalization and convergence of X-ray absorption sum rules. Phys. Rev. B 2001, 63, 094412. [Google Scholar] [CrossRef]
- Teramura, Y.; Tanaka, A.; Jo, T. Effect of Coulomb Interaction on the X-ray Magnetic Circular Dichroism Spin Sum Rule in 3d Transition Elements. J. Phys. Soc. Jap. 1996, 65, 1053–1055. [Google Scholar] [CrossRef]
- Piamonteze, C.; Miedema, P.; de Groot, F.M.F. Accuracy of the spin sum rule in XMCD for the transition-metal L edges from manganese to copper. Phys. Rev. B 2009, 80, 184410. [Google Scholar] [CrossRef]
- Kuepper, K.; Raekers, M.; Taubitz, C.; Uhlarz, M.; Piamonteze, C.; de Groot, F.M.F.; Arenholz, E.; Galakhov, V.R.; Mukovskii, Y.M.; Neumann, M. The X-ray magnetic circular dichroism spin sum rule for 3d4 systems: Mn3+ ions in colossal magnetoresistance manganites. J. Phys. Condens. Matter 2012, 24, 435602. [Google Scholar] [CrossRef] [PubMed]
- Müller, N.; Lischke, T.; Weiss, M.R.; Heinzmann, U. Spin resolved photoelectron spectroscopy from paramagnetic Gd at the 4d -> 4f resonance using circularly-polarized radiation, a cross-comparison with MCD. J. Electron Spectrosc. Relat. Phenom. 2001, 114–116, 777–782. [Google Scholar] [CrossRef]
- Heinzmann, U. Photoemission and absorption spectroscopy of solids and interfaces with synchrotron radiation. In Proceedings of the International School of Physics “Enrico Fermi”; Campagna, M., Rosei, R., Eds.; North-Holland Publishing Company: Amsterdam, The Netherland, 1990; p. 469. [Google Scholar]
- Müller, N.; David, R.; Snell, G.; Kuntze, R.; Drescher, M.; Böwering, N.; Stoppmanns, P.; Yu, S.W.; Heinzmann, U.; Viefhaus, J.; et al. Spin-resolved Auger electron spectroscopy after photoexcitation with circularly polarized radiation from the BESSY crossed undulator. J. Electron Spectrosc. Relat. Phenom. 1995, 72, 187–193. [Google Scholar] [CrossRef]
- Cherepkov, N.A.; Kuznetsov, V.V. Optical activity of polarized atoms. J. Phys. B: At. Mol. Opt. Phys. 1989, 22, L405–L409. [Google Scholar] [CrossRef]
- Heinzmann, U.; Dil, J.H. Spin orbit induced photoelectron spin polarization in angle resolved photoemission from both atomic and condensed matter targets. J. Phys. Condens. Matter 2012, 24, 173001. [Google Scholar] [CrossRef] [PubMed]
- Van der Laan, G.; Thole, B.T. Spin polarization and magnetic dichroism in photoemission from core and valence states in localized magnetic systems. IV. Core-hole polarization in resonant photoemission. Phys. Rev. B 1995, 52, 15355. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. 2008, A64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Sluis, P.V.D.; Spek, A.L. BYPASS: An effective method for the refinement of crystal structures containing disordered solvent regions. Acta Crystallogr. 1990, A46, 194–201. [Google Scholar] [CrossRef]
- The Cambridge Crystallographic Data Centre. Available online: http://www.ccdc.cam.ac.uk/data_request/cif (accessed on 20 August 2015).
- Mukherjee, C.; Stammler, A.; Bögge, H.; Glaser, T. Do trinuclear triplesalen complexes exhibit cooperative effects?—Synthesis, characterization, and enantioselective catalytic sulfoxidation by chiral trinuclear FeIII triplesalen complexes. Chem. Eur. J. 2010, 16, 10137–10149. [Google Scholar] [CrossRef] [PubMed]
- Glaser, T. Exchange Coupling Mediated by Extended Phloroglucinol Ligands: Spin-Polarization vs. Heteroradialene-Formation. Coord. Chem. Rev. 2013, 257, 140–152. [Google Scholar] [CrossRef]
- Feldscher, B.; Stammler, A.; Bögge, H.; Glaser, T. Aromatic vs. heteroradialene character in extended phloroglucinol and thiophloroglucinol ligands and their trinuclear nickel(ii) complexes. Chem. Asian J. 2014, 9, 2205–2218. [Google Scholar] [CrossRef] [PubMed]
- Godehusen, K.; (BESSY-II, Helmholtz-Zentrum Berlin, Germany). UE52-SGM beamline technical Documentation, personal communication, 2009.
- Mahler, W.; (FHI der MPG, BESSY-II, Helmholtz-Zentrum Berlin, Germany). UE56/2-PGM-2 beamline technical documentation, personal communication, 2013.
- Sawhney, K.J.S.; Senf, F.; Scheer, M.; Schäfers, F.; Bahrdt, J.; Gaupp, A.; Gudat, W. A novel undulator-based PGM Beamline for circularly polarized synchrotron radiation at BESSY II. Nucl. Instrum. Meth. A 1997, 390, 395–402. [Google Scholar] [CrossRef]
- Gray, L.G.; Hart, M.W.; Dunning, F.B.; Walters, G.K. Simple, compact, medium-energy Mott polarization analyzer. Rev. Sci. Instrum. 1984, 55, 88–91. [Google Scholar] [CrossRef]
- Müller, N.; Khalil, T.; Pohl, M.; Uphues, T.; Polcik, M.; Rader, O.; Heigl, F.; Starke, K.; Fritzsche, S.; Kabachnik, N.M.; et al. Interference of spin states in resonant photoemission induced by circularly polarized light from magnetized Gd. Phys. Rev. B 2006, 74, 161401R. [Google Scholar] [CrossRef]
- David, R.; Stoppmanns, P.; Yu, S.W.; Kuntze, R.; Müller, N.; Heinzmann, U. Circularly polarized undulator radiation from the new crossed double-undulator beamline at BESSY and its first use for spin resolved Auger electron emission spectroscopy. Nucl. Instrum. Meth. A 1994, 343, 650–654. [Google Scholar] [CrossRef]
- Stepanow, S.; Mugarza, A.; Ceballos, G.; Moras, P.; Cezar, J.C.; Carbone, C.; Gambardella, P. Giant spin and orbital moment anisotropies of a Cu-phthalocyanine monolayer. Phys. Rev. B 2010, 82, 014405. [Google Scholar] [CrossRef]
- Stöhr, J.; König, H. Determination of spin- and orbital-moment anisotropies in transition metals by angle-dependent X-ray magnetic circular dichroism. Phys. Rev. Lett. 1995, 75, 3748–3751. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dohmeier, N.; Helmstedt, A.; Müller, N.; Gryzia, A.; Brechling, A.; Heinzmann, U.; Heidemeier, M.; Krickemeier, E.; Stammler, A.; Bögge, H.; et al. Heptanuclear [FeIII6CrIII]3+ Complexes Experimentally Studied by Means of Magnetometry, X-ray Diffraction, XAS, XMCD and Spin-Polarized Electron Spectroscopy in Cross-Comparison with [MnIII6CrIII]3+ Single-Molecule Magnets. Magnetochemistry 2016, 2, 5. https://doi.org/10.3390/magnetochemistry2010005
Dohmeier N, Helmstedt A, Müller N, Gryzia A, Brechling A, Heinzmann U, Heidemeier M, Krickemeier E, Stammler A, Bögge H, et al. Heptanuclear [FeIII6CrIII]3+ Complexes Experimentally Studied by Means of Magnetometry, X-ray Diffraction, XAS, XMCD and Spin-Polarized Electron Spectroscopy in Cross-Comparison with [MnIII6CrIII]3+ Single-Molecule Magnets. Magnetochemistry. 2016; 2(1):5. https://doi.org/10.3390/magnetochemistry2010005
Chicago/Turabian StyleDohmeier, Niklas, Andreas Helmstedt, Norbert Müller, Aaron Gryzia, Armin Brechling, Ulrich Heinzmann, Maik Heidemeier, Erich Krickemeier, Anja Stammler, Hartmut Bögge, and et al. 2016. "Heptanuclear [FeIII6CrIII]3+ Complexes Experimentally Studied by Means of Magnetometry, X-ray Diffraction, XAS, XMCD and Spin-Polarized Electron Spectroscopy in Cross-Comparison with [MnIII6CrIII]3+ Single-Molecule Magnets" Magnetochemistry 2, no. 1: 5. https://doi.org/10.3390/magnetochemistry2010005