
Magnetic Behaviour of Transition Metal Complexes with Functionalized Chiral and C60-Filled Nanotubes as Bridging Ligands: A Theoretical Study


Abstract
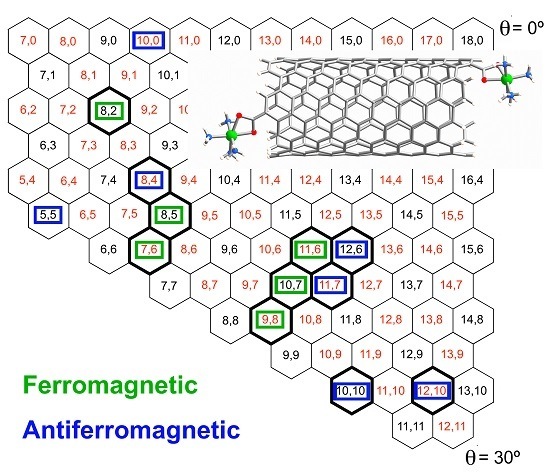
:
1. Introduction
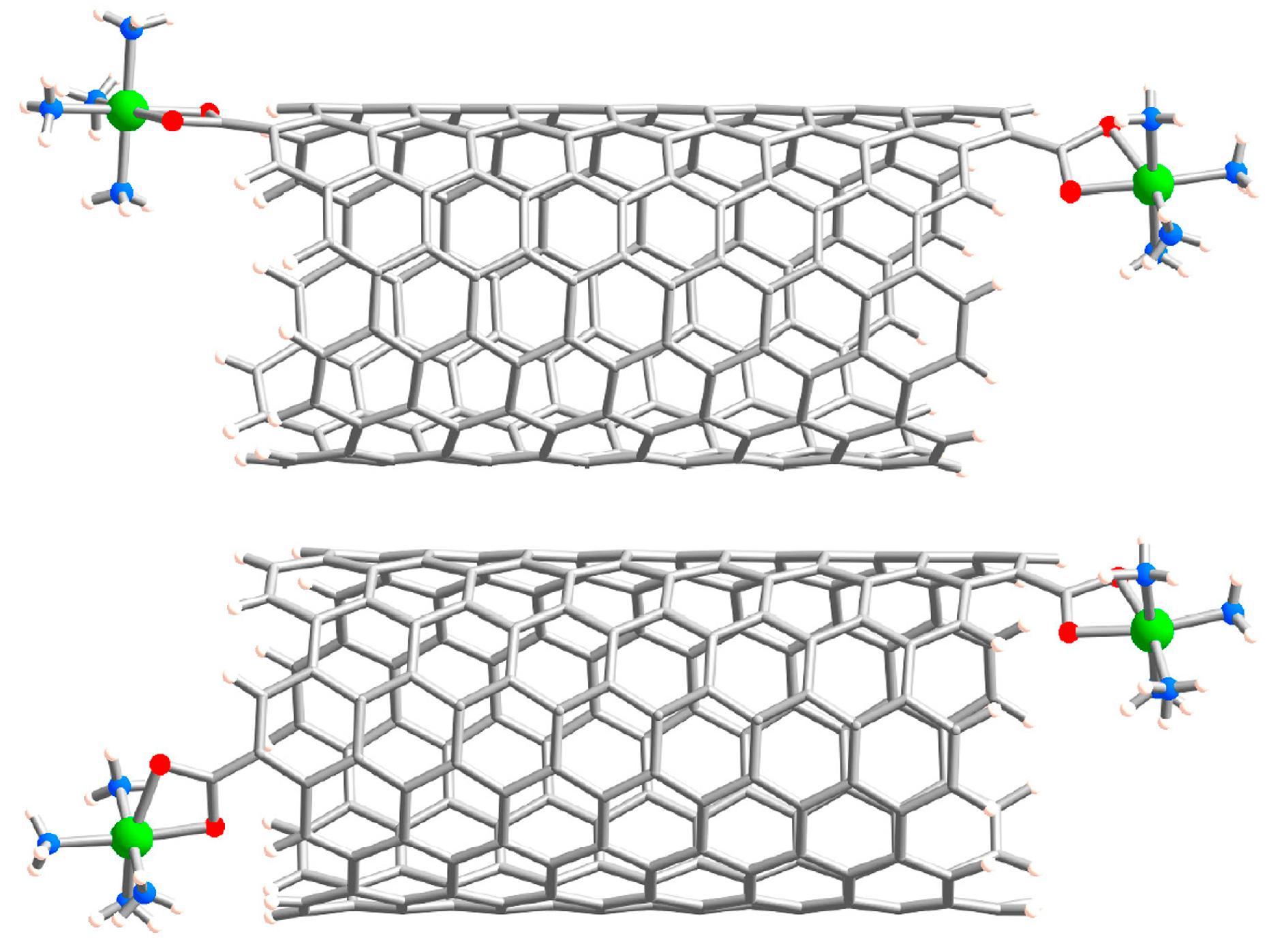
2. Results and Discussion
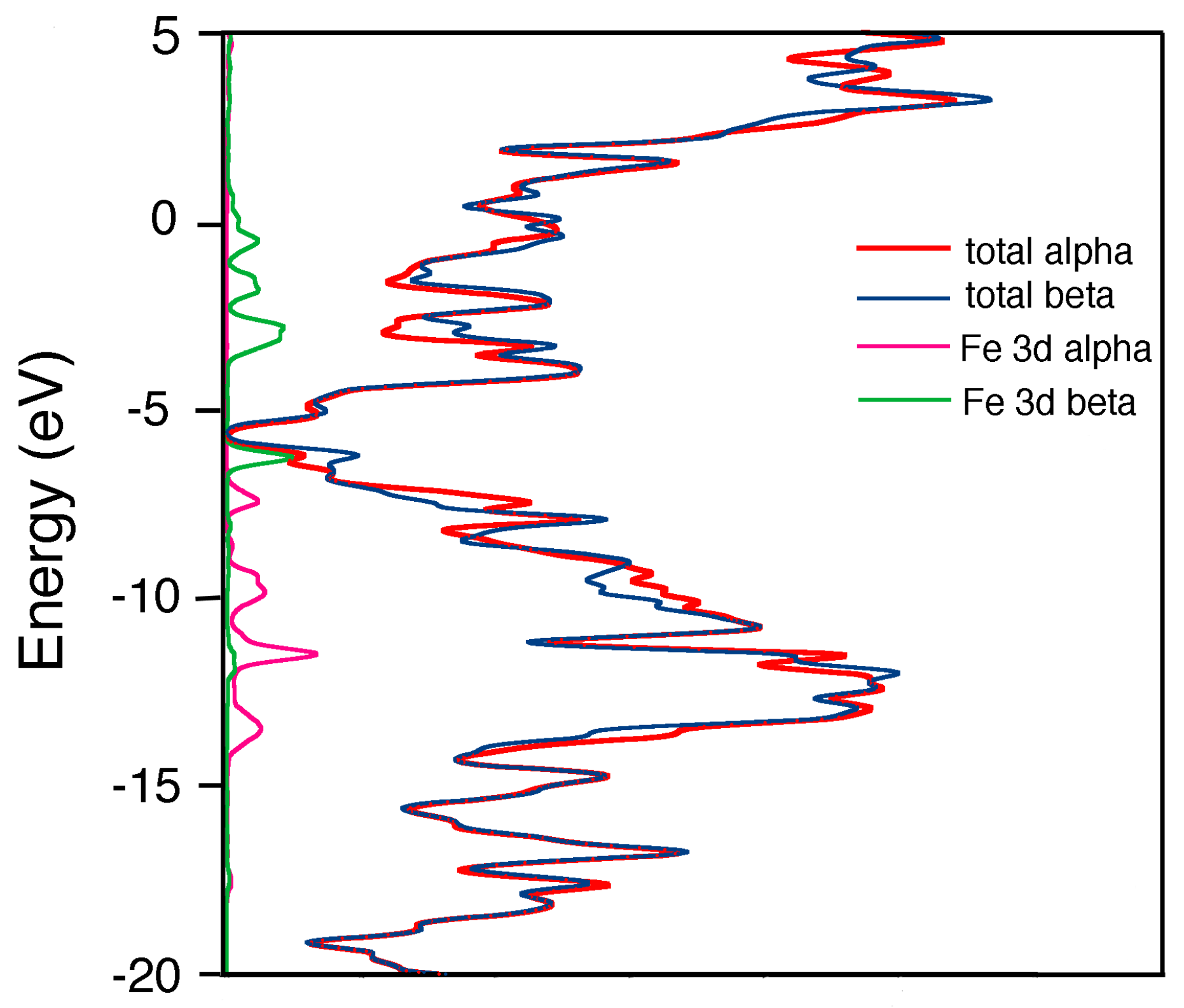
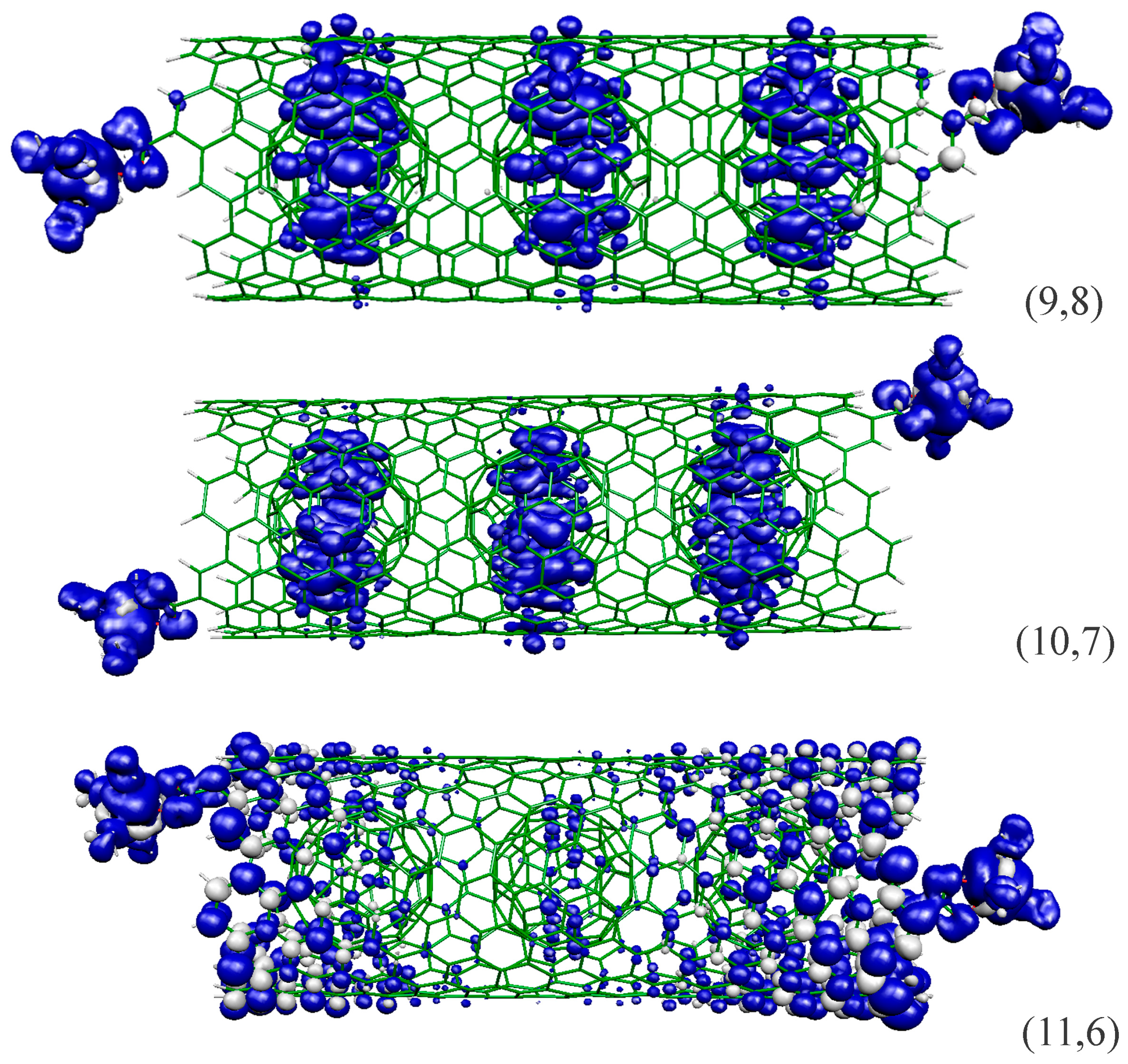
2.1. Metal Systems with Chiral Nanotubes

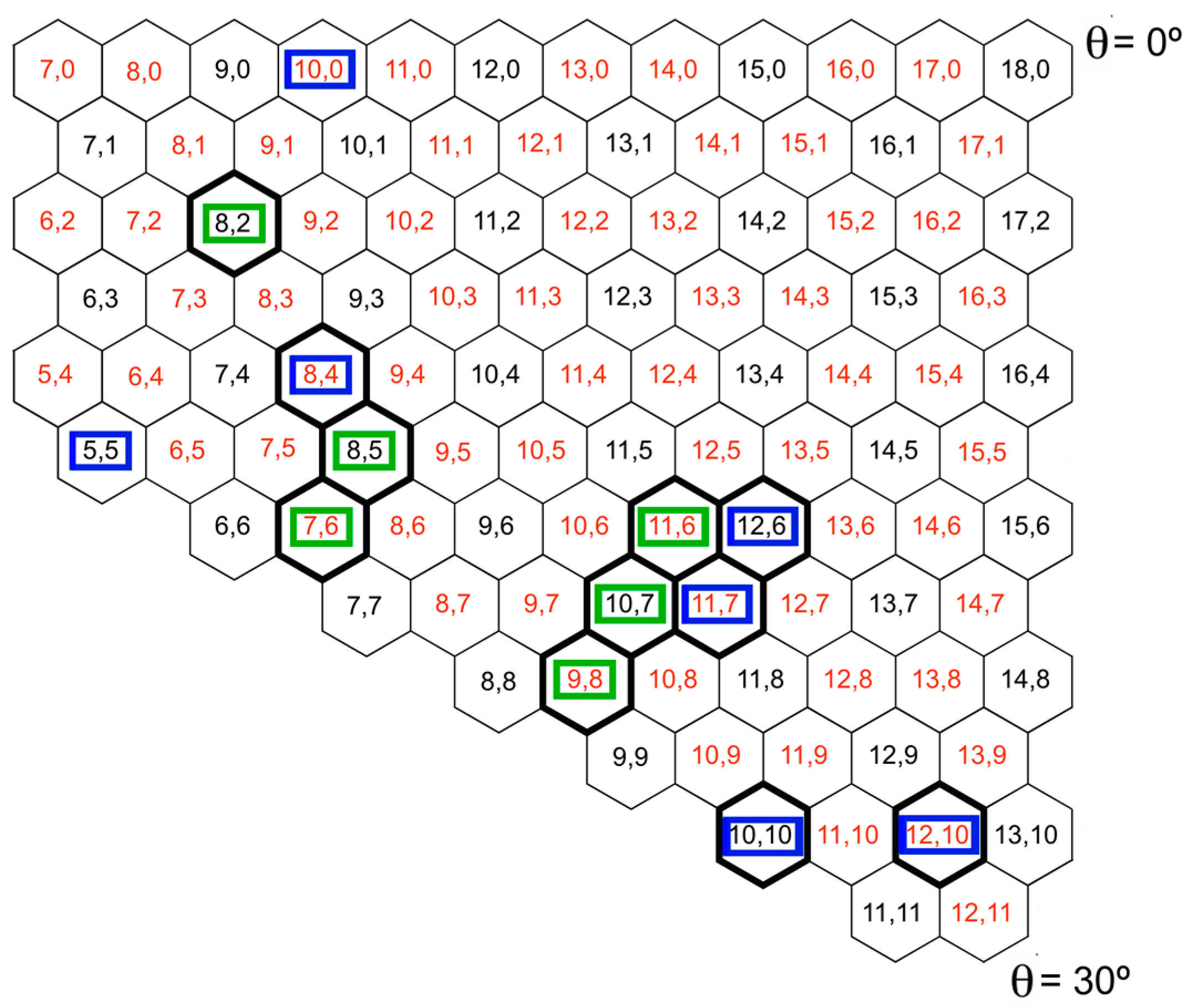

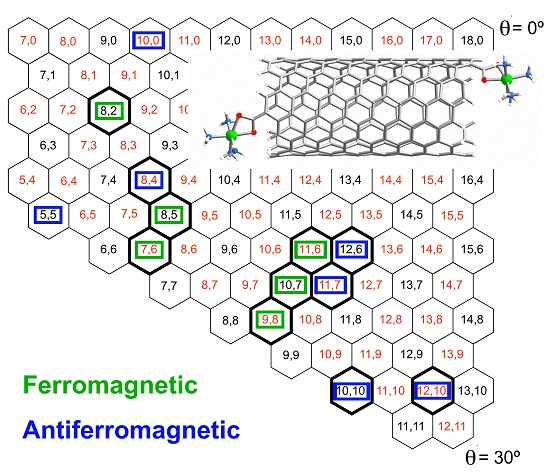{kind=link}
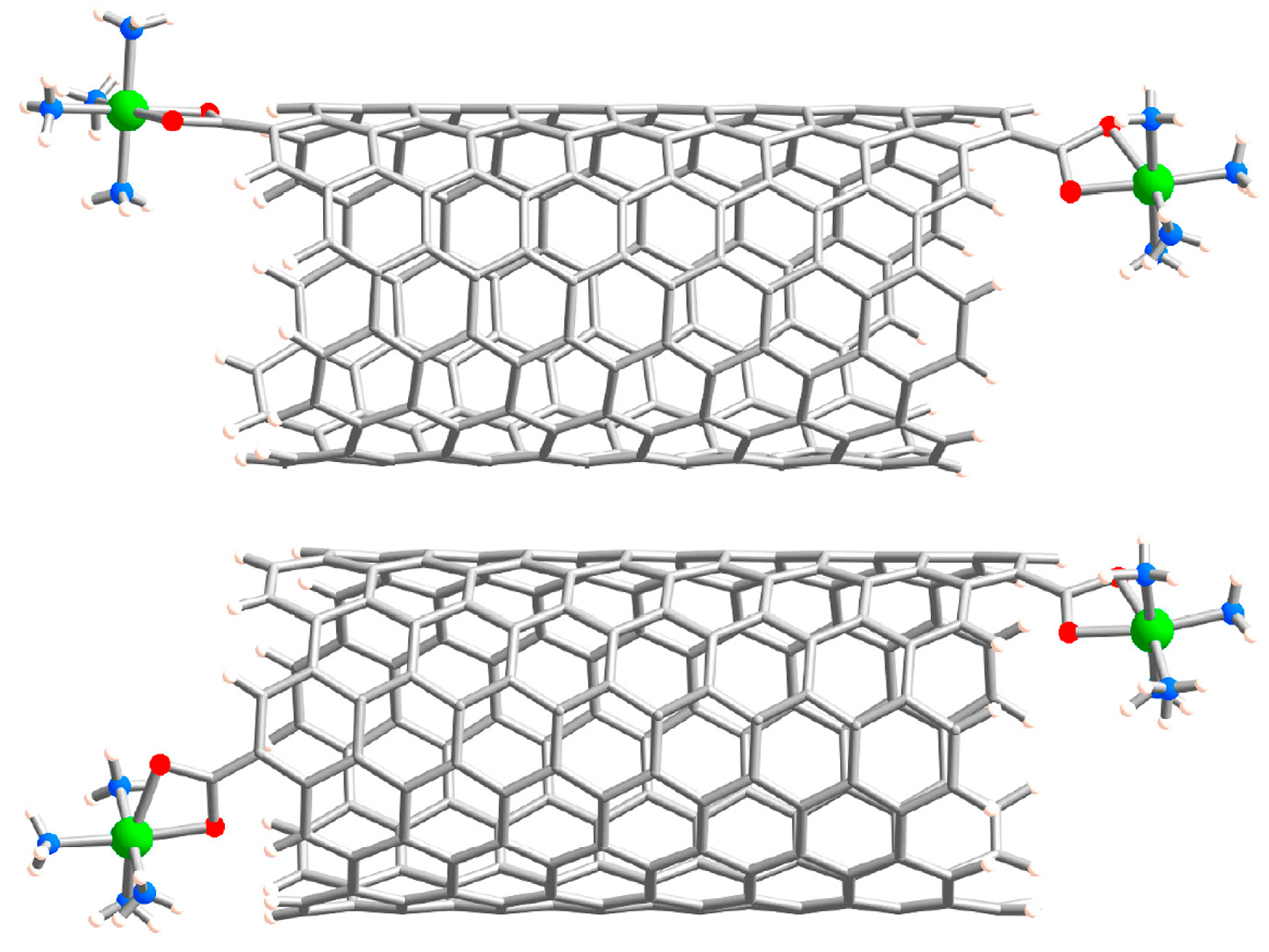{kind=link}
{kind=link}
{kind=link}
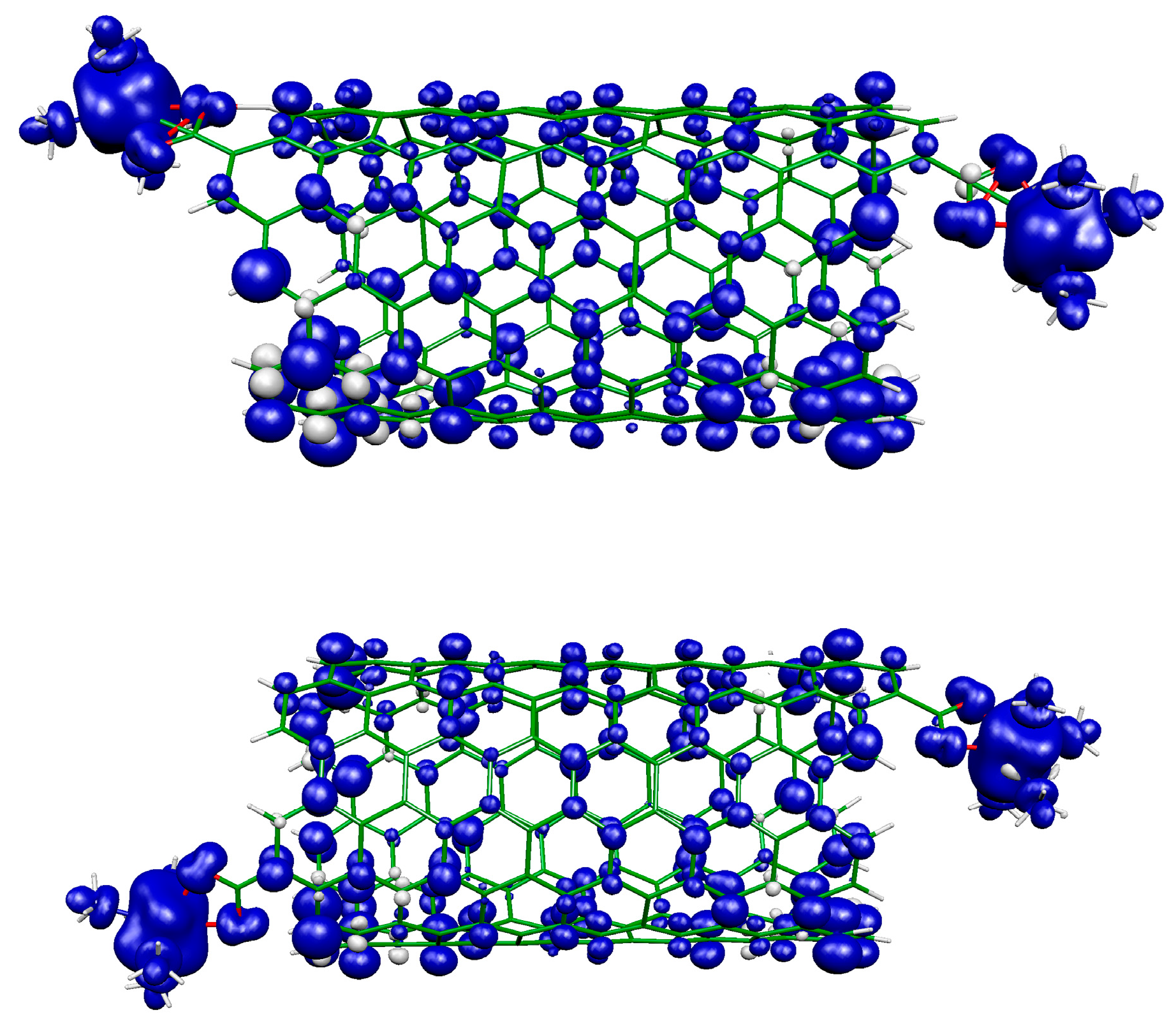{kind=link}
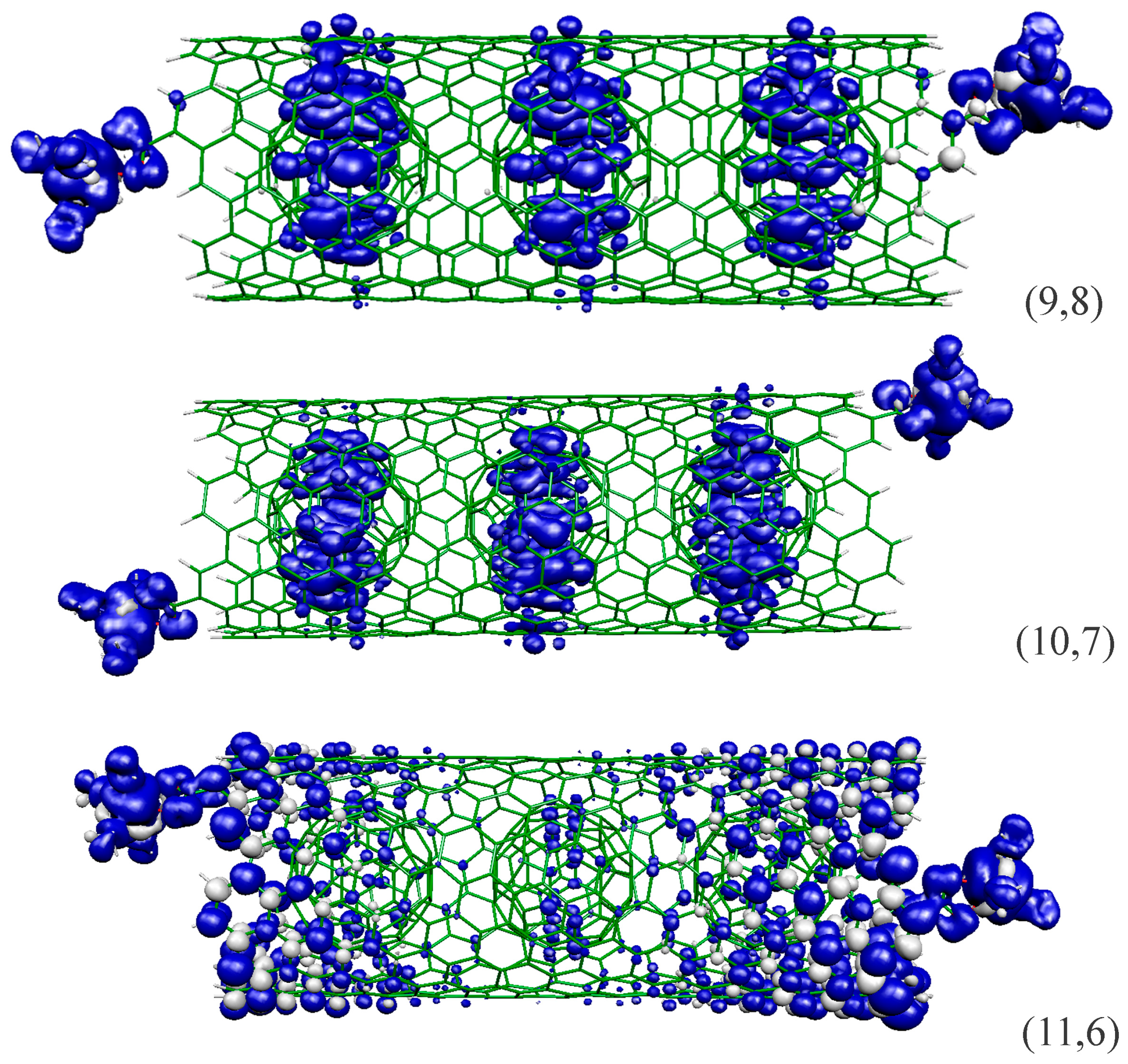{kind=link}
{kind=link}
| Chirality | Transport | Diameter | d(Fe···Fe) | Jcalc |
|---|---|---|---|---|
| (7,6) | SC | 8.82 | 25.4, 25.9 | +17.2, +19.3 |
| (8,2) | M | 7.18 | 31.1, 32.4 | +9.0, +11.0 |
| (8,4) | SC | 8.29 | 25.9, 25.6 | −230, −260 |
| (8,5) | M | 8.89 | 25.7, 26.2 | +21.3, +28.6 |
| (9,8) | SC | 11.53 | 38.8 | +6.1 |
| (10,7) | M | 11.59 | 40.2, 26.7 | +94.8, +13.6 |
| (10,10) | M | 13.56 | 40.91 | −17.6 |
| (11,6) | SC | 11.69 | 40.2 | +11.7 |
| (11,7) | SC | 12.30 | 40.1 | −150 |
| (12,6) | M | 12.43 | 38.8 | −22.0 |
| (12,10) | SC | 14.94 | 47.51 | −199 |

2.2. Systems Based on Peapods
| Chirality | Transport | Diameter | d(Fe···Fe) | Jnano | Jpeapod | Einter |
|---|---|---|---|---|---|---|
| (9,8) | SC | 11.53 | 38.8 | +6.1 | +18.2 | 58.9 |
| (10,7) | SC | 11.59 | 40.2 | +94.8 | +15.4 | 52.9 |
| (11,6) | SC | 11.69 | 40.2 | +11.7 | +7.0 | 44.0 |
| (11,7) | SC | 12.30 | 40.1 | −150 | −143 | 13.8 |
| (12,6) | M | 12.43 | 38.8 | −22.0 | −33.9 | 10.3 |
| (10,10) | M | 13.56 | 40.91 | −17.6 | +43 | −1.9 |
| (12,10) | SC | 14.94 | 47.51 | −199 | −182 | −2.0 |

3. Computational Details
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Dresselhaus, M.S.; Dresselhaus, D.; Avouris, G. Carbon Nanotubes: Synthesis, Structure, Properties and Applications; Springer-Verlag: Berlin, Germany, 2001. [Google Scholar]
- Schnorr, J.M.; Swager, T.M. Emerging applications of carbon nanotubes. Chem. Mater. 2011, 23, 646–657. [Google Scholar] [CrossRef]
- Anantram, M.P.; Leonard, F. Physics of carbon nanotube electronic devices. Rep. Prog. Phys. 2006, 69, 507–561. [Google Scholar] [CrossRef]
- Ruiz, E.; Nunzi, F.; Alvarez, S. Magnetic communication through functionalized nanotubes: A theoretical study. Nano Lett. 2006, 6, 380–384. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Rinzler, A.G.; Dai, H.J.; Hafner, J.H.; Bradley, R.K.; Boul, P.J.; Lu, A.; Iverson, T.; Shelimov, K.; Huffman, C.B.; et al. Fullerene pipes. Science 1998, 280, 1253–1256. [Google Scholar] [CrossRef] [PubMed]
- Pederson, M.R.; Broughton, J.Q. Nanocapillarity in fullerene tubules. Phys. Rev. Lett. 1992, 69, 2689–2692. [Google Scholar] [CrossRef] [PubMed]
- Okada, S.; Saito, S.; Oshiyama, A. Energetics and electronic structures of encapsulated C60 in a carbon nanotube. Phys. Rev. Lett. 2001, 86, 3835–3838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dion, M.; Rydberg, H.; Schröder, E.; Langreth, D.C.; Lundqvist, B.I. Van der Waals Density Functional for General Geometries. Phys. Rev. Lett. 2004, 92, 246401. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Portal, D.; Ordejón, P.; Artacho, E.; Soler, J.M. Density-functional method for very large systems with LCAO basis sets. Int. J. Quantum Chem. 1997, 65, 453–461. [Google Scholar] [CrossRef]
- Artacho, E.; Sánchez-Portal, D.; Ordejón, P.; García, A.; Soler, J.M. Linear-scaling ab-initio calculations for large and complex systems. Phys. Status Solidi A 1999, 215, 809. [Google Scholar] [CrossRef]
- Artacho, E.; Anglada, E.; Diéguez, O.; Gale, J.D.; García, A.; Junquera, J.; Martin, R.M.; Ordejón, P.; Pruneda, J.M.; Sánchez-Portal, D. The siesta method; developments and applicability. J. Phys.: Condens. Matter 2008, 20, 064208. [Google Scholar] [CrossRef] [PubMed]
- Soler, J.M.; Artacho, E.; Gale, J.D.; García, A.; Junquera, J.; Ordejón, P.; Sánchez-Portal, D. The siesta method for ab initio order-N materials simulation. J. Phys.: Condens. Matter 2002, 14, 2745. [Google Scholar]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Troullier, N.; Martins, J.L. Efficient pseudopotentials for plane-wave calculations. Phys. Rev. B 1991, 43, 1993. [Google Scholar] [CrossRef]
- Ruiz, E.; Rodriguez-Fortea, A.; Tercero, J.; Cauchy, T.; Massobrio, C. Exchange coupling in transition-metal complexes via density-functional theory: Comparison and reliability of different basis set approaches. J. Chem. Phys. 2005, 123, 074102. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, E.; Alvarez, S.; Cano, J.; Polo, V. About the calculation of exchange coupling constants using density-functional theory: The role of the self-interaction error. J. Chem. Phys. 2005, 123, 164110. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, E.; Rodriguez-Fortea, A.; Cano, J.; Alvarez, S.; Alemany, P. About the calculation of exchange coupling constants in polynuclear transition metal complexes. J. Comput. Chem. 2003, 24, 982–989. [Google Scholar] [CrossRef] [PubMed]
- Mintmire, J.W.; Dunlap, B.I.; White, C.T. Are fullerene tubules metallic? Phys. Rev. Lett. 1992, 68, 631–634. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gómez-Coca, S.; Ruiz, E. Magnetic Behaviour of Transition Metal Complexes with Functionalized Chiral and C60-Filled Nanotubes as Bridging Ligands: A Theoretical Study. Magnetochemistry 2015, 1, 62-71. https://doi.org/10.3390/magnetochemistry1010062
Gómez-Coca S, Ruiz E. Magnetic Behaviour of Transition Metal Complexes with Functionalized Chiral and C60-Filled Nanotubes as Bridging Ligands: A Theoretical Study. Magnetochemistry. 2015; 1(1):62-71. https://doi.org/10.3390/magnetochemistry1010062
Chicago/Turabian StyleGómez-Coca, Silvia, and Eliseo Ruiz. 2015. "Magnetic Behaviour of Transition Metal Complexes with Functionalized Chiral and C60-Filled Nanotubes as Bridging Ligands: A Theoretical Study" Magnetochemistry 1, no. 1: 62-71. https://doi.org/10.3390/magnetochemistry1010062