
Exploring Genetic Diversity and Phylogenetic Relationships in Camellia reticulata Cultivars Using Novel Low-Copy Nuclear Gene Markers
Laboratory of Landscape Plants, Faculty of Architecture and City Planning, Kunming University of Science and Technology, Kunming 650500, China
*
Author to whom correspondence should be addressed.
Horticulturae 2024, 10(3), 303; https://doi.org/10.3390/horticulturae10030303
Submission received: 24 January 2024
/
Revised: 12 March 2024
/
Accepted: 20 March 2024
/
Published: 21 March 2024
(This article belongs to the Special Issue Breeding of Ornamental Plants—Genetic Resources, New Challenges and Prospects)
Abstract
:This study delves into the exploration of genetic diversity and phylogenetic relationships within Camellia reticulata cultivars, providing a vital reference for horticultural research on this hetero-hexaploid species. Utilizing available transcriptome data from C. reticulata, novel low-copy nuclear gene sequences were successfully identified. With tailored primer design, these genes were amplified and sequenced from 100 C. reticulata cultivars originating from Kunming, Chuxiong, Dali, and Tengchong in China. Five distinct low-copy nuclear gene sequences were found to collectively span 3481 bp, showcasing 71 polymorphic mutation sites (Pi = 0.0077) and 91 haplotypes (Hd = 0.9974). The genetic diversity among cultivars from the four provenances ranked Tengchong > Dali > Kunming > Chuxiong. AMOVA analysis revealed that 96.50% of the genetic variation exists within the provenances. Low genetic distance and differentiation (Fst = 0.0199) were observed among cultivars from the four provenances. Ninety-nine cultivars were clustered into four clades, corresponding to the three ancestors of C. reticulata (diploid C. reticulata, C. pitardii, and C. saluenensis). The novel low-copy nuclear gene sequence markers developed in this study provide an effective tool for analyzing genetic diversity, phylogenetic relationships, and origination of C. reticulata cultivars.
1. Introduction
Camellia reticulata Lindl. (known as Yunnan camellia), an exclusive Theaceae species native to China, is a national key second-level protected plant and proudly serves as the official city flower of Kunming [1]. As the tallest ornamental Camellia species worldwide, C. reticulata can ascend to an impressive height of 28.1 m. Renowned for its sizable flowers reaching up to 22 cm in diameter, vibrant colors, and a diverse array of cultivars (with 823 registered on the World Camellia Plant Variety Registry Database) [2], these camellias are extensively used as potted and landscaping plants throughout Yunnan, China. With a rich history spanning over 1300 years, they have transcended national borders and become cherished ornamentals in numerous countries [3,4].
Despite their widespread cultivation, it is noteworthy that C. reticulata has predominantly been grown in controlled environments, such as greenhouses, deviating from their native outdoor habitats. This is attributed to their origin in the mild regions of southwest China (Figure 1), specifically at altitudes ranging from 1000 to 3200 m [5]. Although more than 100 cultivars have been successfully cultivated, they are susceptible to freezing damage below –5 °C, and their growth is impeded beyond 32 °C [1], restricting the outdoor planting range for C. reticulata. To overcome these limitations and expand the outdoor cultivation footprint, there is a compelling need to develop new cultivars endowed with enhanced heat and cold resistance.
Understanding the genetic diversity of C. reticulata is important for the development of new cultivars. Recent investigations into the genetic diversity of C. reticulata cultivars have encompassed both morphological markers [2] and molecular studies. While AFLP (Amplified Fragment Length Polymorphism) markers have been employed to explore genetic diversity and structure [6], challenges arise from the hexaploid nature of C. reticulata, which also includes octaploid and decaploid cultivars [7,8]. Traditional molecular markers, such as AFLP, ITS (Internal Transcribed Spacer), and SSR (Simple Sequence Repeat), may present complications, such as double bands during amplification and multi-peaks during sequencing in polyploid groups [9,10,11]. It has been reported that the phylogenetic relationships of C. reticulata cultivars, based on ITS copies from clones, are confounded by the presence of numerous ITS pseudogenes [12]. Prior attempts utilizing ITS sequences for genetic diversity and phylogenetic relationships faced challenges due to incomplete consistency evolution and the necessity for high-quality DNA and specific PCR conditions [4,12,13,14].
Recognizing the constraints of traditional markers, this study advocates for the utilization of low-copy nuclear genes. Characterized by ease of acquisition and parental inheritance traits, low-copy nuclear genes offer an optimal choice for studying hybrid and polyploidy populations, particularly suited for phylogenetic analysis [9,15,16,17]. Notable examples include the use of the single-copy nuclear gene waxy for molecular phylogeny analysis of Camellia [13], and the utilization of the low-copy nuclear gene RPB2 (RNA polymerase II) in the authentication of C. chekiangolosa and its closely related species [18]. Moreover, the phylogeography of C. taliensis [19] and the population genetic structure and phylogeography of C. flavida [20] have been successfully explored using the low-copy nuclear gene PAL (Phenylalanine Ammonia Lyase).
In light of the existing abundance of C. reticulata cultivars and the scarcity of available low-copy nuclear gene markers, this study aims to meticulously select low-copy nuclear genes utilizing available transcriptome data. The subsequent screening process identified stable primers with high-quality amplification, facilitating a comprehensive exploration of the genetic diversity and phylogenetic relationships among C. reticulata cultivars. The insights gained from this endeavor are anticipated to serve as a valuable reference for the collection, preservation, identification, and innovative breeding of C. reticulata cultivars.
2. Materials and Methods
To assess the genetic diversity within the C. reticulata cultivars, a total of 100 cultivars were collected from Kunming Botanical Garden, Chuxiong Elu Park, and Tengchong Laifengshan Camellia Garden. This diverse collection encompassed 34 cultivars from Kunming, 19 from Chuxiong, 12 from Dali, and 35 from Tengchong (Table 1, Figure 1), as documented by the Kunming Association for Science and Technology [1] and the World Camellia Plant Variety Registry Database [2]. To enrich the analysis, we also included a set of closely related diploid species, including C. reticulata, C. saluenensis, C. yunnanensis, C. pitardii, C. tuberculate, and C. japonica obtained from Kunming Botanical Garden. Fresh and healthy adult leaves were carefully collected from all species, thoroughly dried with discolored silicone, sealed in plastic zip lock bags, and stored at −20 °C for DNA extraction.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Sample information of Camellia reticulata cultivars.
| Provenance | Cultivars |
|---|---|
| Kunming (34 cultivars) | ‘Shizitou’ (8x), ‘Juban’(Figure 2b, 8x), ‘Songzilin’ (8x), ‘Liuye Yinhong’, ‘Damanao’ (6x), ‘Maye Taohong’ (8x), ‘Yujie’, ‘Yipinhong’, ‘Xiaoguiye’ (6x), ‘Zuijiaohong’, ‘Xiaotaohong’, ‘Houye Diechi’, ‘Baoyuhong’, ‘Caiyun’, ‘Taohongpao’ (6x), ‘Dianchi Xiuqiu’, ‘Mudancha’ (8x), ‘Dianchi Mingzhu’ (8x), ‘Zipao’ (8x), ‘Zaomudan’ (8x), ‘Saitaohong’ (8x), ‘Guiye Yanghong’ (6x), ‘Dayinhong’ (8x), ‘Maye Yinhong’, ‘Dataohong’, ‘Baiyi Zaotaohong’ (8x), ‘Lianrui’, ‘Fenhudie’, ‘Dayulan’ (6x), ‘Yanhe’, ‘Jinrui Furong’, ‘Yulancha’, ‘Hongwancha’, ‘Jingancha’ (8x) |
| Chuxiong (19 cultivars) | ‘Dandinghe’ (8x), ‘Zibao’ (8x), ‘Miyilu’ (8x), ‘Chuxiongcha’, ‘Baize’ (8x), ‘Fozuolian’ (6x), ‘Zehe’ (6x), ‘Ziyu’, ‘Lifang’, ‘Weixihong’, ‘Zifen’, ‘Luchengchun’ (6x), ‘Ziyan’, ‘Liuye Meihong’, ‘Yundie’, ‘Jinrui Dahong’, ‘Jianye Diechi’, ‘Zilian’, ‘Weichu’ |
| Dali (12 cultivars) | ‘Daguiye’ (6x), ‘Saijuban’, ‘Yanhong Songzike’, ‘Meihong Guiye’ (6x), ‘Tongzimian’ (8x), ‘Baozhucha’ (6x), ‘Dalicha’, ‘Duxin Dalicha’, ‘Hentiangao’ (10x), ‘Pumencha’ (6x), ‘Songzike’ (6x), ‘Xiguiye’ |
| Tengchong (35 cultivars) | ‘Fentongcao’ (6x), ‘Mudankui’ (Figure 2a), ‘Fengshancha’, ‘Yunzhen’, ‘Wujiao Xiuqiu’ (6x), ‘Hehua Xianzi’ (8x), ‘Tuanye Diechi’ (6x), ‘Shuimeiren’ (6x), ‘Biyu’, ‘Yunfeng’ (8x), ‘Honghua Youcha’ (8x), ‘Fenzhen Mudan’, ‘Huahun’, ‘Jinrui Diechi’, ‘Xiaojiaojiao’, ‘Fenzhaoyun’, ‘Yushizi’, ‘Xianyecha’, ‘Yumeiren’, ‘Hongmei’, ‘Xiyingchun’, ‘Taohong Mudan’, ‘Yunhuacha’, ‘Fentianjiao’, ‘Saierqiao’, ‘Taihe Mudan’, ‘Manao Dahongcha’, ‘Jiaohe’, ‘Naochun Xiuqiu’, ‘Jiaoxiaohong’, ‘Fenhong Xiuqiu’, ‘Yanghong Songzike’, ‘Yulin’, ‘Nansongzi’, ‘Heidahong’ |
Note: the cell ploidy of cultivars was labeled according to the literature published by Xu et al. [7].
To generate low-copy nuclear gene markers, the genome coding sequences (CDS) from transcriptome data (SRX1343024) [21] were downloaded and filtered to select single isoform mRNA strands for ease of processing. Genes within the range of 500–1100 bp were chosen to ensure they could be easily amplified using traditional PCR methods. Next, the transcriptome reads were cleaned to remove adapter sequences, low-complexity sequences, contamination, and PCR duplicates [16,17]. Sequences that match plastid, chloroplast, ribosomal, transposon, or mitochondrial loci in the National Center for Biotechnology Information (NCBI) database (http://www.ncbi.nlm.nih.gov/, accessed on 9 December 2023) were then filtered out. Finally, sequences that show hits to RepeatMasker (http://repeartmasker.org/, accessed on 11 December 2023) were removed to ensure the selected gene markers were specific to the low-copy nuclear genes of interest.
Primer pairs were designed using Primer 4.0 [22] with a focus on conservative coding sequences. The primer selection phase involved the utilization of eight samples, including three randomly selected cultivars and the aforementioned five congeneric species.
Total DNA from C. reticulata leaf samples weas extracted using an improved CTAB method [23]. The PCR reaction mixture consisted of 25 µL Taq premixture, 1 µL DNA, 22 µL ddH2O, and 1 µL primers. The PCR reaction program followed a sequence of steps: initial denaturation at 98 °C for 2 min, 35 cycles including denaturation at 98 °C for 10 s, annealing at 60 °C for 10 s, and extension at 72 °C for 20 s, and final extension at 72 °C for 5 min. PCR products were sequenced using the ABI 3730 sequencer (Applied Biosystems, Beijing). Subsequently, mRNAs associated with amplified gene partial sequences were identified through an online Blast search (https://blast.ncbi.nlm.nih.gov/, accessed on 19 December 2023).
Upon sequencing with selected primers, low-copy gene sequences were compared through the MAFFT method [24] executed by the GeneiousR10 software. After alignment, crucial genetic parameters, including the number of haplotypes (h), haplotype diversity (Hd), and nucleotide diversity (Pi), were calculated using the DnaSP5.1 software [25]. The distribution of genetic variation within and between provenances was analyzed with the software Arlequin3.0 [26]. Analysis of Molecular Variance (AMOVA) was performed, and the genetic differentiation coefficient Fst was calculated. Gene flow (Nm) was estimated using the formula Nm = (1 − Fst)/4Fst [27]. The model-based cluster analysis was performed using the program STRUCTURE v2.3.1 [28]. The optimum number of clusters (K) was processed and identified by STRUCTURE HARVESTER through comparing log probabilities of data for each value of K [29]. The output of structure analyses was visualized using the software CLUMPP v1.1.2 [30] and DISTRUCT v1.1 [31].
The Kimura 2-parameter model was employed to calculate the genetic distance that was used to construct a Neighbor-Joining (NJ) phylogenetic tree, wherein the bootstrap method was used for testing through 1000 replicates. The Median-joining method [32], facilitated by Network5.0 (http://www.fluxus-engineering.com/sharenet.htm, accessed on 29 December 2023), was used for a comprehensive analysis of haplotype data, including the determination of phylogenetic relationships.
3. Results
3.1. Identification of Low-Copy Nuclear Genes of C. reticulata
A total of 966 partial sequences of low-copy genes were obtained through an integrated analysis of the C. reticulata transcriptome. Following the selection process, 30 low-copy nuclear gene sequences were identified, ranging from 538 to 1038 bp in length. Among the corresponding 30 primer pairs, 18 failed to amplify targets or exhibited faint bands, and seven displayed a problematic bimodal pattern during sequencing. Consequently, five primer pairs emerged with clear amplification bands and single-peaked sequencing outcomes (Table 2). These five distinct low-copy nuclear gene sequences, registered in GenBank (MH257911-48), have been identified as partial coding sequences associated with unique mRNAs (Table 2).
3.2. Nucleotide and Haploid Diversity of C. reticulata Cultivars
Among five low-copy genes, C1179 showed the most single nucleotide polymorphism (SNP, 20), while C10214 showed the least number of polymorphic markers. The cumulative length of five amplified sequences was 3481 bp. A total of 71 mutation sites were identified (Figure A1), accounting for 2.04% of the overall sites. The overall nucleotide diversity (Pi) within C. reticulata cultivars was determined to be 0.0077, accompanied by an average nucleotide difference (k) of 26.65. Among four provenances, Tengchong displayed the highest nucleotide genetic diversity (Pi = 0.0084), followed by Dali (Pi = 0.0081), Kunming (Pi = 0.0068), and Chuxiong (Pi = 0.0066).
Locus variation analysis revealed a total of 91 haplotypes (Figure A1), with 35 from Kunming, 14 from Chuxiong, 10 from Dali, and 36 from Tengchong (Table 3). The collective haplotype diversity (Hd) attained an impressive 0.9974. Within four provenances, Dali and Kunming exhibited the highest haplotype diversity (Hd = 1.0000), closely followed by Tengchong (Hd = 0.9958) and Chuxiong (Hd = 0.9825).
AMOVA further revealed that a substantial 96.50% of the genetic variation within C. reticulata cultivars is distributed within the provenances, with only 3.50% accounting for the genetic variation between provenances (Table 4).
3.3. Genetic Distance, Genetic Structure and Differentiation of C. reticulata Cultivars
Within four provenances, the genetic distance between provenance pairs is consistently comparable, ranging from 0.0070 to 0.0080 (Table 5). Notably, Tengchong exhibited a higher genetic distance from the other provenances. The genetic differentiation coefficient of C. reticulata cultivars stands out between Chuxiong and Kunming, registering the highest value at 0.0911. In contrast, the genetic differentiation coefficients between Dali and the remaining are relatively low.
The optimum number of clusters (K) was processed and identified by STRUCTURE, which was K = 5 (Figure 3), indicating that the Camellia reticulata cultivars were mainly composed of 5 gene pools (represented in red, blue, yellow, peach, and green, respectively). As shown in Figure 3, on the whole, the proportions of five gene pools in the four provenances are relatively uniform, indicating that the C. reticulata cultivars from four provenances have similar genetic structures. Among four provenances, Kunming showed relatively high proportion of peach gene pool (33%), while Tengchong showed relatively high proportion of red gene pool (34%).
The total genetic differentiation coefficient Fst for C. reticulata cultivars stood at 0.0351, with a corresponding Nst of 0.0199 (p < 0.05), falling below the threshold of 0.02. This suggests a relatively low genetic differentiation among diverse provenances of C. reticulata cultivars. The gene flow index Nm was 6.8827, significantly exceeding 1.0, pointing to a frequent exchange of genes between distinct provenances.
3.4. Phylogenetic Relationships of C. reticulata Cultivars
Cluster analysis divided 100 C. reticulata cultivars into four groups (Figure 4). Notably, three of these groups displayed a close relationship with the diploid ancestors of the allogeneic hexaploid C. reticulata. These three groups were specifically linked to the diploid C. reticulata, encompassing a total of 47 cultivars (19 from Kunming, 9 from Chuxiong, 5 from Dali, and 14 from Tengchong), the diploid C. pitardii, comprising 38 varieties (13 from Kunming, 9 from Chuxiong, 5 from Dali, and 11 from Tengchong), and the diploid C. saluenensis, featuring a collective count of 14 varieties (4 from Kunming, 1 from Chuxiong, and 9 from Tengchong). The fourth and final branch only has one cultivar, namely ‘Taihe Mudan’, originating from Tengchong.
Haploid network analysis showed that haplotypes H12 and H80 were located at the center of the network, while haplotype H53, distinguished by its extensive distribution and highest frequency, played a prominent role in shaping the network’s structure (Figure 5). As shown in Figure 5, there were no obvious diffusion trends among C. reticulata cultivars in any specific direction. Instead, a mixed and monogenic distribution pattern was observed in the chart, strongly indicating a close genetic kinship among cultivars from different provenances.
4. Discussion
4.1. Genetic Diversity of C. reticulata Cultivars
In comparing our AMOVA analysis results (96.50%) with those from previous studies utilizing AFLP markers [6], a remarkable consistency emerges, with the earlier study reporting 96.31% genetic variation within populations. This phenomenon can be explained from the following two aspects. Firstly, the genetic diversity within the wild populations of C. reticulata are relatively high [33], while cultivars are mainly selected from different wild variations. Secondly, the long-term breeding work may enhance genetic diversity within the provenances. Furthermore, the genetic distance between distinct provenances in our study (0.0070–0.0080) aligns closely with AFLP marker results (0.0077–0.0300) [6]. This robust alignment underscores the efficacy of low-copy nuclear gene markers developed in our study for a comprehensive analysis of C. reticulata cultivar genetic diversity.
Our study reveals an order in the genetic diversity of C. reticulata cultivars among different provenances: Tengchong > Dali > Kunming > Chuxiong. Contrastingly, a previous AFLP markers-based study suggested Kunming > Dali > Tengchong > Chuxiong [6]. This discrepancy likely stems from variations in sample composition. The AFLP study incorporated 190 samples, including 56 from Tengchong, 37 from Chuxiong, 28 from Dali, and 66 from Kunming [6], but only 85 of these were distinct C. reticulata cultivars, some of which were collected across different provenances. In the present study, we focused on a selected cohort of 100 cultivars, with a more balanced representation from Tengchong (35), Chuxiong (19), Dali (12), and Kunming (34).
Typically, Fst < 0.05 signifies a low level of genetic differentiation and suggests close relationships [6,34]. This pattern may be attributed to historical cultivation practices spanning 1300 years, during which cultivars were frequently introduced from their native provenances to new cultivation regions. These introduced cultivars then served as ancestors for breeding programs, associating the new cultivation regions as their provenances. This observation also likely explains the high gene flow (Nm = 6.8827), the high genetic variation (96.50%), and the similar genetic structures within populations.
4.2. Genetic Relationships among C. reticulata Cultivars
In this study, 99 C. reticulata cultivars were clustered in three clades (Figure 4), with each clade aligning with one of three diploid ancestors of C. reticulata, namely diploid C. reticulata, C. pitardii, and C. saluenensis. This clustering based on genetic relationships rather than provenances reaffirms the accuracy of recognizing diploid C. reticulata, C. pitardii, and C. saluenensis as ancestors of the hetero-hexaploid C. reticulata. Notably, the cultivar ‘Taihe Peony’ stands alone on a single branch, hinting at the potential involvement of other yet unknown parents in the genetic makeup of C. reticulata cultivars.
In terms of leaf morphology, ‘Xiaoguiye’ and ‘Xiguiye’ exhibit a striking resemblance to the diploid ancestor C. saluenensis. However, these two cultivars did not cluster with C. saluenensis but rather aligned with 2x C. reticulata and C. pitardii, respectively. This observation suggests that low-copy nuclear genes of allopolyploid C. reticulata stem from diverse ancestral species and, in this study, only a random copy from either source was amplified. Consequently, the five amplified low-copy nuclear gene copies of ‘Xiaoguiye’ and ‘Xiguiye’ predominantly correlated with two other ancestral species, diverging from the expected alignment with C. saluenensis.
Analyzing phylogenetic relationships in polyploids demands an extensive amplification of low-copy nuclear genes. The clone technology can be used to amplify numerous copies of a single sequence [12], while genome resequencing provides access to a large number of different low-copy nuclear genes [21]. In this study, the utilization of five low-copy nuclear gene markers across 100 cultivars resulted in a substantial dataset of 500 low-copy nuclear sequences. This abundance not only facilitates a more accurate study of phylogenetic relationships but also enables the tracing of the diverse ancestors of C. reticulata cultivars.
4.3. Evolutionary Dynamics of C. reticulata Cultivars
In addition to the well-known 6x cultivars, C. reticulata also encompasses 8x cultivars [7], such as ‘Honghua Youcha’ and ‘Zipao’ [35]. However, the origins of 8x cultivars remain a mystery.
Despite being recognized as an allopolyploid species involving 2x, 4x, and 6x variations, C. reticulata has recently been regarded as a part of polyploid complexes [5,36,37]. Genomic in situ hybridization (GISH) studies have revealed a fascinating evolutionary process. The diploid C. reticulata and C. pitardii gave rise to the allotetraploid C. reticulata. Subsequently, the allotetraploid C. reticulata and the diploid C. saluenensis led to the formation of the hexaploid C. reticulata in the Jinshajiang valley of south Sichuan [37]. The 6x C. reticulata then migrated from its origins to the Yunnan Province (Figure 1).
Interestingly, while 2x and 4x C. reticulata variants are confined to the Jinshajiang valley, no instances of 8x C. reticulata have been discovered in this region [38]. This raises the possibility that 8x C. reticulata may not originate through the duplication of the 4x variant. Furthermore, the widespread cultivation of 6x and 8x C. reticulata cultivars in Kunming, Chuxiong, Dali, and Tengchong, where 2x or 4x C. reticulata have not been documented, suggests that the 8x C. reticulata may have developed through additional hybrid polyploidization of the 6x variant during its southward diffusion. Notably, the unique clade ‘Taihe Mudan’ may be linked to unknown parents in this process.
5. Conclusions
This study successfully identified five pairs of primers suitable for amplifying low-copy nuclear gene sequences in Camellia. Our analysis encompassed 100 cultivars, revealing significant genetic diversity and low genetic differentiation of C. reticulata among four provenances. The majority of the cultivars (99) were categorized into three distinct clades, aligning with the diploid ancestors of the hetero-hexaploid C. reticulata. Newly developed low-copy nuclear gene markers in this study offer valuable tools for the genetic enhancement of C. reticulata and the advancement of related research endeavors involving Camellia species.
Author Contributions
W.Z. and X.X.: conceived and designed the study and collected plant material; Y.G.: performed field and laboratory experiments; W.Z., X.X. and Y.G.: performed the analysis of data; X.X. and Y.G.: completed the original draft; W.Z.: reviewed the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This study was supported by the National Natural Science Foundation of China (Grant Numbers 32060691, 32060093, and 32260093).
Data Availability Statement
Data are contained within the article.
Acknowledgments
The authors are grateful to Zhonglang Wang, Weizhu Shao, Wei Yi and Suo Li for assistance with sample collections.
Conflicts of Interest
The authors of the manuscript state that there are no competing interests regarding financial or individual conflicts.
Appendix A
Figure A1.
Variable sites of the aligned sequences in the 91 haplotypes in Camellia reticulata. All sequences are compared to the reference haplotype H1. The number at the top indicates polymorphic sites. Dots represent nucleotide variants identical to the first sequence.
Figure A1.
Variable sites of the aligned sequences in the 91 haplotypes in Camellia reticulata. All sequences are compared to the reference haplotype H1. The number at the top indicates polymorphic sites. Dots represent nucleotide variants identical to the first sequence.

References
- Kunming Association for Science and Technology. Wonderful Flower of Yunnan—Camellia Reticulata; Yunnan Science and Technology Press: Kunming, China, 2010. [Google Scholar]
- Wu, G.; Chen, L.; Wei, Q.; Hu, Y.; Xu, J.; Li, D.; Chen, S.; Wang, Z.; Geng, F. Statistics and phenotypic traits analysis of Camellia reticulata registered cultivars. Acta Hortic. Sin. 2023, 50, 2157–2170. [Google Scholar]
- Zhang, W.; Ming, T. A cytogeographical study of Camellia, Sect Camellia. Acta Bot. Yun. 1998, 20, 321–328. [Google Scholar]
- Vijayan, K.; Zhang, W.; Tsou, C.H. Molecular taxonomy of Camellia (Theaceae) inferred from nrITS sequences. Am. J. Bot. 2009, 96, 1348–1360. [Google Scholar] [CrossRef] [PubMed]
- Min, T.; Bartholomew, B. Theaceae. In Flora of China; Wu, Z.Y., Raven, P.H., Hong, D.Y., Eds.; Science Press: Beijing, China; Missouri Botanical Garden Press: St. Louis, MI, USA, 2007; Volume 12, pp. 368–372. [Google Scholar]
- Xin, T.; Huang, W.; Riek, J.D.; Zhang, S.; Ahmed, S.; Huylenbroeck, J.V.; Long, C. Genetic diversity, population structure, and traditional culture of Camellia reticulata. Ecol. Evol. 2017, 7, 8915–8926. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Shao, W.; Zheng, W. Ploidy study on polyploidy cultivars of Camellia reticulata. Sci. Silvae Sin. 2018, 54, 44–48. [Google Scholar]
- Xu, X.; Zheng, W.; Harris, A.J.; Wang, W.; Shao, W.; Wen, J. Assessing the maternal origin in the polyploid complex of Camellia reticulata based on the chloroplast rpl16 intron sequences: Implications for camellia cross breeding. Mol. Breed. 2018, 38, 123. [Google Scholar] [CrossRef]
- Elizabeth, A.Z.; Wen, J. Using nuclear gene data for plant phylogenetics: Progress and prospects II. Next-gen approaches. J. Syst. Evol. 2015, 53, 371–379. [Google Scholar]
- Guo, S.; Ji, P.; Wang, J.; He, Y.; Zhang, Y.; Zhang, F.; Yun, Y.; Zhang, G. Estimation of genetic diversity between and within biparental clones and full-sib families of the Chinese pine using SSR markers. Horticulturae 2023, 9, 1205. [Google Scholar] [CrossRef]
- Chalbi, A.; Chikh-Rouhou, H.; Mezghani, N.; Slim, A.; Fayos, O.; Bel-Kadhi, M.S.; Garcés-Claver, A. Genetic diversity analysis of onion (Allium cepa L.) from the arid region of Tunisia using phenotypic traits and SSR markers. Horticulturae 2023, 9, 1098. [Google Scholar] [CrossRef]
- Zhou, A.; Yue, L.; Li, M.; Liu, D.; Ding, Y. Intra-genomic polymorphism in the nrDNA ITS sequence of Camellia reticulata. Plant Sci. J. 2013, 31, 1–10. [Google Scholar] [CrossRef]
- Yang, J.; Li, H.; Yang, S.; Li, D. The application of four DNA sequences to studying molecular phylogeny of Camellia (Theaceae). Acta Bot. Yun. 2006, 28, 108–114. [Google Scholar]
- Vijayan, K.; Tsou, C.H. Technical report on the molecular phylogeny of Camellia with nrITS: The need for high quality DNA and PCR amplification with Pfu-DNA polymerase. Bot. Stud. 2008, 49, 177–188. [Google Scholar]
- Meseguer, A.S.; Sanmartín, I.; Marcussen, T.; Pfeil, B.E. Utility of low-copy nuclear markers in phylogenetic reconstruction of Hypericum L. (Hypericaceae). Plant Syst. Evol. 2014, 300, 1503–1514. [Google Scholar] [CrossRef]
- Chery, J.G.; Sass, C.; Specht, C.D. Development of single-copy nuclear intron markers for species-level phylogenetics: Case study with Paullinieae (Sapindaceae). Appl. Plant Sci. 2017, 5, 1700051. [Google Scholar] [CrossRef] [PubMed]
- Du, S.H.; Wang, Z.S.; Zhang, J.G. A novel set of single-copy nuclear DNA markers for the genetic study of Salicaceae. Genet. Mol. Res. 2014, 13, 4911–4917. [Google Scholar] [CrossRef]
- Wen, Q.; Zhu, H.; Ye, J.; Xu, L.; Xu, L.; Jiang, X. Authentication of Camellia chekiangoleosa and its close species using RPB2 gene sequences. Mol. Plant Breed. 2015, 13, 2559–2565. [Google Scholar]
- Liu, Y.; Yang, S.; Ji, P.; Gao, L. Phylogeography of Camellia taliensis (Theaceae) inferred from chloroplast and nuclear DNA: Insights into evolutionary history and conservation. BMC Evol. Biol. 2012, 12, 92. [Google Scholar] [CrossRef]
- Wei, S.; Lu, Y.; Ye, Q.; Tang, S. Population genetic structure and phylogeography of Camellia flavida (Theaceae) based on chloroplast and nuclear DNA sequences. Front. Plant Sci. 2017, 8, 00718. [Google Scholar] [CrossRef]
- Zhang, Q.; Zhao, L.; Folk, R.A.; Zhao, J.; Zamora, N.A.; Yang, S.; Soltis, D.E.; Soltis, P.S.; Gao, L.; Peng, H.; et al. Phylotranscriptomics of Theaceae: Generic-level relationships, reticulation and whole-genome duplication. Ann. Bot. 2022, 129, 457–471. [Google Scholar] [CrossRef]
- Koressaar, T.; Remm, M. Enhancements and modifications of primer design program Primer3. Bioinformatics 2007, 23, 1289–1291. [Google Scholar] [CrossRef]
- Doyle, J.J.; Doyle, J.L. Isolation of plant DNA from fresh tissue. Focus 1990, 12, 39–40. [Google Scholar]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Narnia 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef] [PubMed]
- Excoffier, L.; Laval, G.; Schneider, S. Arlequin (version 3.0): An integrated software package for population genetics data analysis. Evol. Bioinform. 2005, 1, 47–50. [Google Scholar] [CrossRef]
- Wright, S. Evolution in Mendelian populations. Genetics 1931, 16, 97–159. [Google Scholar] [CrossRef]
- Raj, A.; Stephens, M.; Pritchard, J.K. FastaSTRUCTURE: Variational inference of population structure in large SNP data sets. Genetics 2014, 197, 573–589. [Google Scholar] [CrossRef]
- Earl, D.A.; Vonholdt, B.M. Structure Harvester: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 2012, 4, 359–361. [Google Scholar] [CrossRef]
- Kopelman, N.M.; Mayzel, J.; Jakobsson, M.; Rosenberg, N.A.; Mayrose, L. Clumpak: A program for identifying clustering modes and packaging population structure inference across K. Mol. Ecol. Resour. 2015, 15, 1179–1191. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, N.A. Admixture models and the breeding systems of H. S. Jennings: A GENETICS connection. Genetics 2016, 202, 9–13. [Google Scholar] [CrossRef]
- Bandelt, H.J.; Forster, P.; Röhl, A. Median-joining networks for inferring intraspecific phylogenies. Mol. Biol. Evol. 1999, 16, 37–48. [Google Scholar] [CrossRef]
- Wang, B.; Ruan, Z. Genetic diversity and differentiation in Camellia reticulata (Theaceae) polyploid complex revealed by ISSR and ploidy. Genet. Mol. Res. 2012, 11, 503–511. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Zheng, W.; Chen, L.; Wen, J. Genetic diversity and population structure of Gerbera delavayi (Asteraceae) in southwest china: Implications for conservation. Ann. Bot. Fenn. 2017, 54, 409–422. [Google Scholar] [CrossRef]
- Huang, H.; Tong, Y.; Zhang, Q.; Gao, L. Genome size variation among and within Camellia species by using flow cytometric analysis. PLoS ONE 2013, 8, e64981. [Google Scholar] [CrossRef] [PubMed]
- Xia, L.; Gu, Z.; Wang, Z.; Xiao, D.; Wang, L.; Kondo, K. Dawn on the origin of Camellia reticulata—The new discovery of its wild diploid in Jinshajiang Valley. Acta Bot. Yun. 1994, 16, 255–262. [Google Scholar]
- Gu, Z. The discovery of tetraploid Camellia reticulata and its implication in studies on the origin this species. Acta Phytotaxon. Sin. 1997, 35, 107–116. [Google Scholar]
- Liu, L.; Gu, Z. Genomic in situ hybridization identifies genome donors of Camellia reticulata (Theaceae). Plant Sci. 2011, 180, 554–559. [Google Scholar] [CrossRef]
Figure 1.
The distribution of Camellia reticulata cultivars used in the current study. The red parts in the pies represent the proportions of different sample sizes: 34 cultivars originated from Kunming (34.00%), 19 cultivars originated from Chuxiong (19.00%), 12 cultivars originated from Dali (12.00%), and 35 cultivars originated from Tengchong (35.00%).
Figure 1.
The distribution of Camellia reticulata cultivars used in the current study. The red parts in the pies represent the proportions of different sample sizes: 34 cultivars originated from Kunming (34.00%), 19 cultivars originated from Chuxiong (19.00%), 12 cultivars originated from Dali (12.00%), and 35 cultivars originated from Tengchong (35.00%).
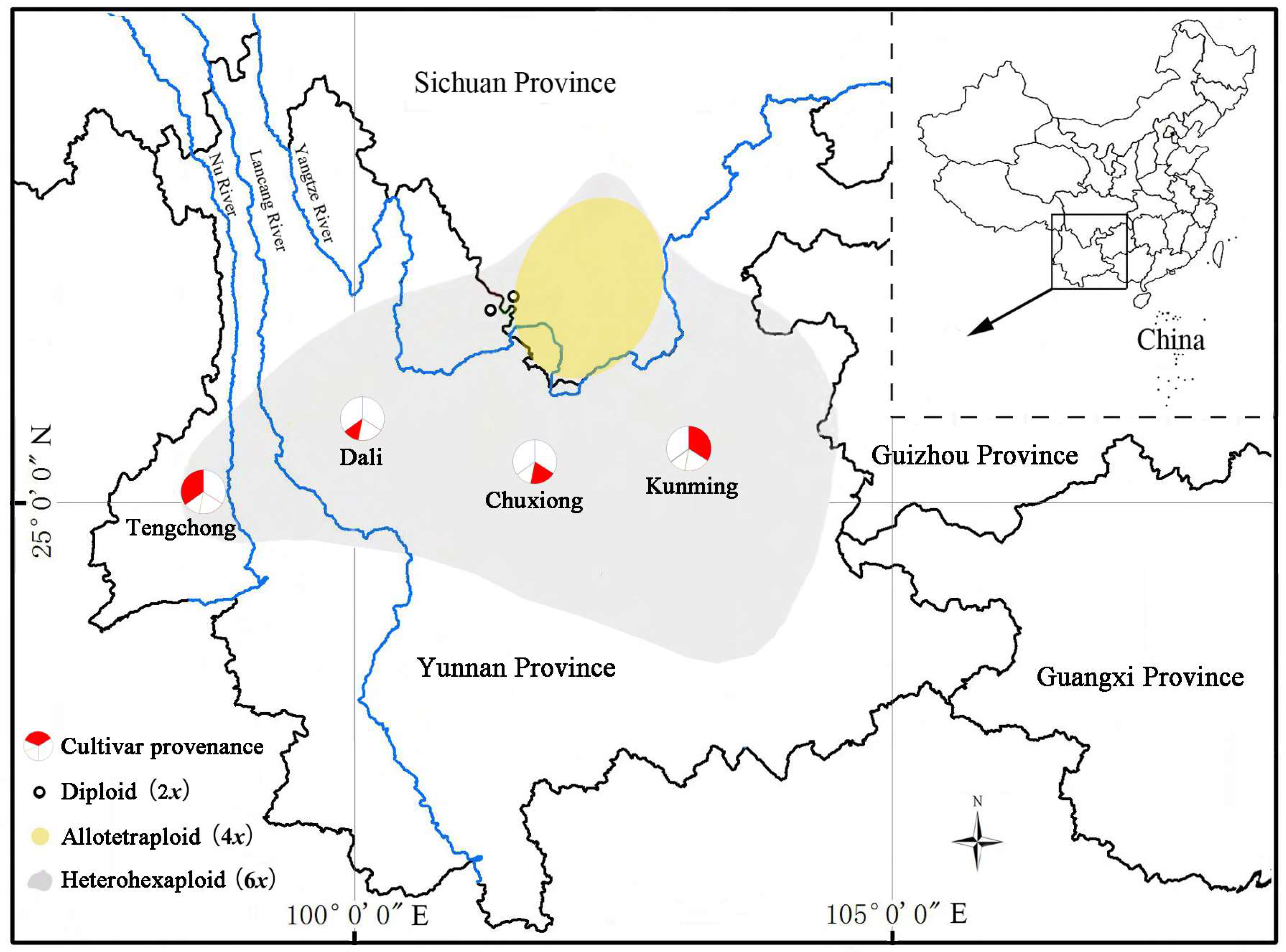
Figure 2.
The representative cultivars of Camellia reticulata: (a) ‘Mudankui’; (b) ‘Juban’.

Figure 3.
Inference of population structure based on the five novel low-copy nuclear genes. KM: Kunming; TC: Tengchong; CX: Chuxiong; DL: Dali. STRUCTURE clustering results for K = 5. (Five gene pools are represented in red, blue, yellow, peach, and green, respectively).
Figure 3.
Inference of population structure based on the five novel low-copy nuclear genes. KM: Kunming; TC: Tengchong; CX: Chuxiong; DL: Dali. STRUCTURE clustering results for K = 5. (Five gene pools are represented in red, blue, yellow, peach, and green, respectively).

Figure 4.
Neighbor-Joining (NJ) tree of Camellia reticulata cultivars based on five nuclear low-copy gene sequences. Clades in green, blue, purple, and red are closely associated with diploid C. reticulata, C. pitardii, C. saluenensis, and an unknown ancestor, respectively.
Figure 4.
Neighbor-Joining (NJ) tree of Camellia reticulata cultivars based on five nuclear low-copy gene sequences. Clades in green, blue, purple, and red are closely associated with diploid C. reticulata, C. pitardii, C. saluenensis, and an unknown ancestor, respectively.
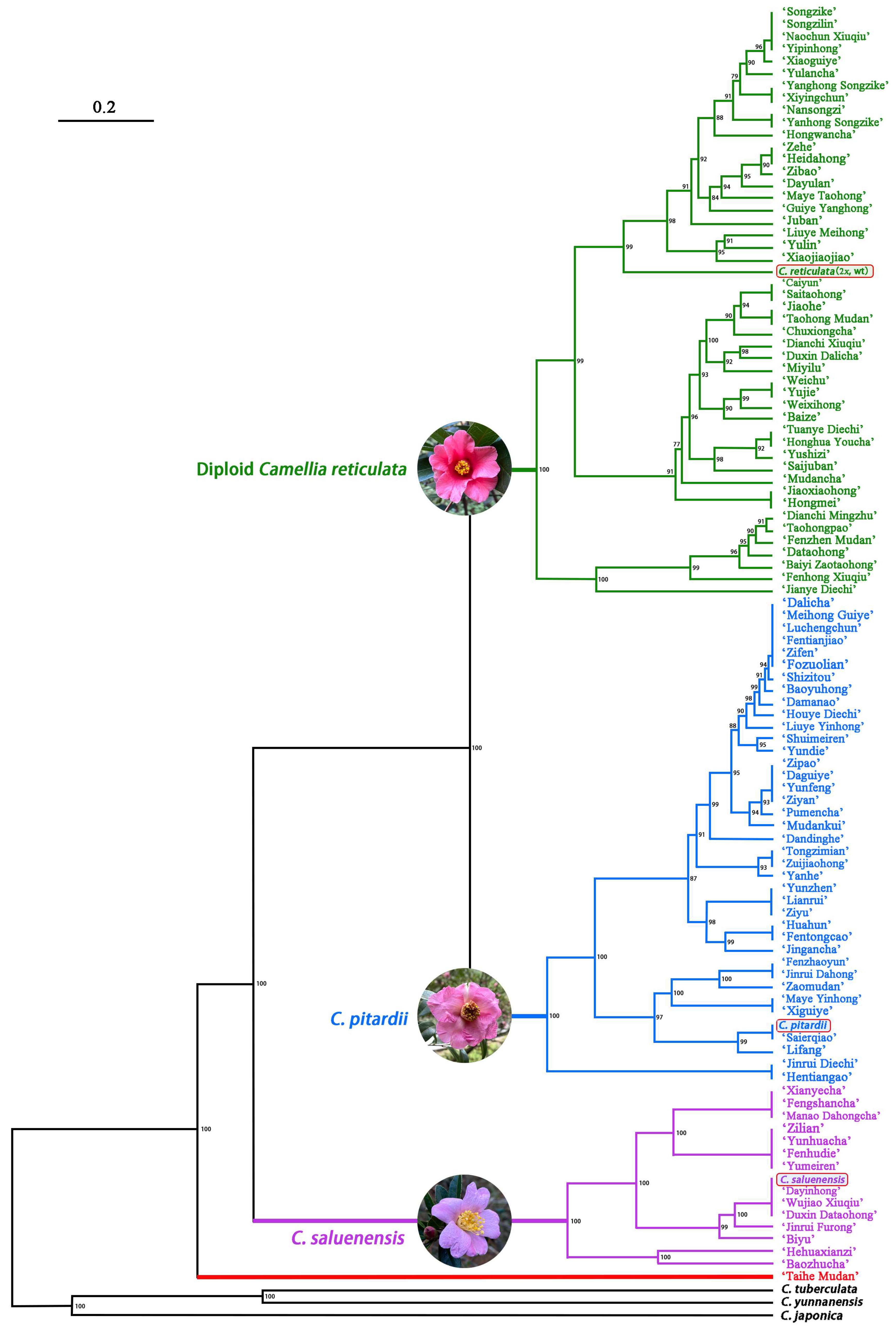
Figure 5.
Median-joining network of Camellia reticulata cultivars based on five nuclear low-copy gene sequences. Circumference size is proportional to the haplotype frequency. The red dots represent median vectors (mv1-50).
Figure 5.
Median-joining network of Camellia reticulata cultivars based on five nuclear low-copy gene sequences. Circumference size is proportional to the haplotype frequency. The red dots represent median vectors (mv1-50).

Table 2.
Primer pairs selected for the amplification of the low-copy nuclear genes in Camellia.
| Primer Code | Primer Sequence (5′-3′) | Amplicon Length (bp) | Associated mRNAs | GenBank No |
|---|---|---|---|---|
| c1179-F c1179-R | ATCGCCAACAGAAACAACACGC ATTCACATCTAATGAGCGAAGGTTG | 700 | Disease resistance protein RPM1 | MH257926 |
| c10316-F c10316-R | CTCCCAACCCATCGTCCTTT CCTTGCCGCTCTTGCAATC | 1038 | Endo-1,3(4)-beta-glucanase | MH257929 |
| c10214-F c10214-R | TGGAAGCTCGGCAATACCAG CCTTTGCGTTCATGGGCATT | 538 | Myosin-binding protein | MH257931 |
| c11230-F c11230-R | TCTTGTGAGGTTAAGAGGGTTT CTTGGACATTATCATTGGAGCA | 800 | S-receptor-like serine/threonine protein kinase | MH257925 |
| c11847-F c11847-R | ACATCGAAGAGCATGGCACA GCCCCAAACTAGCACTCTCT | 786 | Myb-like transcription factor | MH257932 |
Table 3.
Genetic diversity of Camellia reticulata cultivars from different provenances.
| Provenance | Number of Cultivars | Average Number of Different Nucleotides (k) | Diversity of Nucleotide (Pi) | Number of Haplotypes (h) | Diversity of Haplotype (Hd) | Variance of Haplotype Diversity (Vh) | Standard Deviation of Haplotype Diversity (Sh) |
|---|---|---|---|---|---|---|---|
| Kunming | 34 | 23.5544 | 0.0068 | 35 | 1.0000 | 0.0001 | 0.0070 |
| Chuxiong | 19 | 22.8070 | 0.0066 | 14 | 0.9825 | 0.0007 | 0.0260 |
| Dali | 12 | 28.0833 | 0.0081 | 10 | 1.0000 | 0.0027 | 0.0520 |
| Tengchong | 35 | 29.2404 | 0.0084 | 36 | 0.9958 | 0.0001 | 0.0070 |
| Total | 100 | 26.6465 | 0.0077 | 91 | 0.9974 | 0.0000 | 0.0020 |
Table 4.
Analysis of molecular variance (AMOVA) of Camellia reticulata cultivars.
| Source of Variation | df | SSD | MSD | Variance Component | Total Variance (%) | p-Value |
|---|---|---|---|---|---|---|
| Among populations | 3 | 71.8090 | 23.9630 | 0.4719 | 3.50 | <0.0010 |
| Within populations | 99 | 1247.1911 | 12.5980 | 12.9916 | 96.50 | <0.0010 |
| Total | 102 | 1319.0001 | 36.5610 | 13.4635 | 100 |
Table 5.
Genetic distance (below) and differentiation (above) between Camellia reticulata cultivars.
Table 5.
Genetic distance (below) and differentiation (above) between Camellia reticulata cultivars.
| Provenance | Kunming | Chuxiong | Dali | Tengchong |
|---|---|---|---|---|
| Kunming | – | 0.0911 | −0.0183 | 0.0331 |
| Chuxiong | 0.0070 | – | −0.0040 | 0.0471 |
| Dali | 0.0070 | 0.0070 | – | −0.0019 |
| Tengchong | 0.0080 | 0.0080 | 0.0080 | – |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Xu, X.; Gao, Y.; Zheng, W. Exploring Genetic Diversity and Phylogenetic Relationships in Camellia reticulata Cultivars Using Novel Low-Copy Nuclear Gene Markers. Horticulturae 2024, 10, 303. https://doi.org/10.3390/horticulturae10030303
AMA Style
Xu X, Gao Y, Zheng W. Exploring Genetic Diversity and Phylogenetic Relationships in Camellia reticulata Cultivars Using Novel Low-Copy Nuclear Gene Markers. Horticulturae. 2024; 10(3):303. https://doi.org/10.3390/horticulturae10030303
Chicago/Turabian StyleXu, Xiaodan, Ya Gao, and Wei Zheng. 2024. "Exploring Genetic Diversity and Phylogenetic Relationships in Camellia reticulata Cultivars Using Novel Low-Copy Nuclear Gene Markers" Horticulturae 10, no. 3: 303. https://doi.org/10.3390/horticulturae10030303
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.