
Neonatal Calf Diarrhea Is Associated with Decreased Bacterial Diversity and Altered Gut Microbiome Profiles
 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection and Processing
2.2. Microbial DNA Extraction and Sequencing
2.3. Bioinformatics and Statistical Analyses
3. Results
3.1. Analysis of 16S rRNA Gene Sequencing
3.2. Diarrhea Is Associated with Decreased Alpha-Diversity of Gut Bacterial Microbiota
3.3. Relationship between Diarrhea and the Fecal Microbiome Beta Diversity
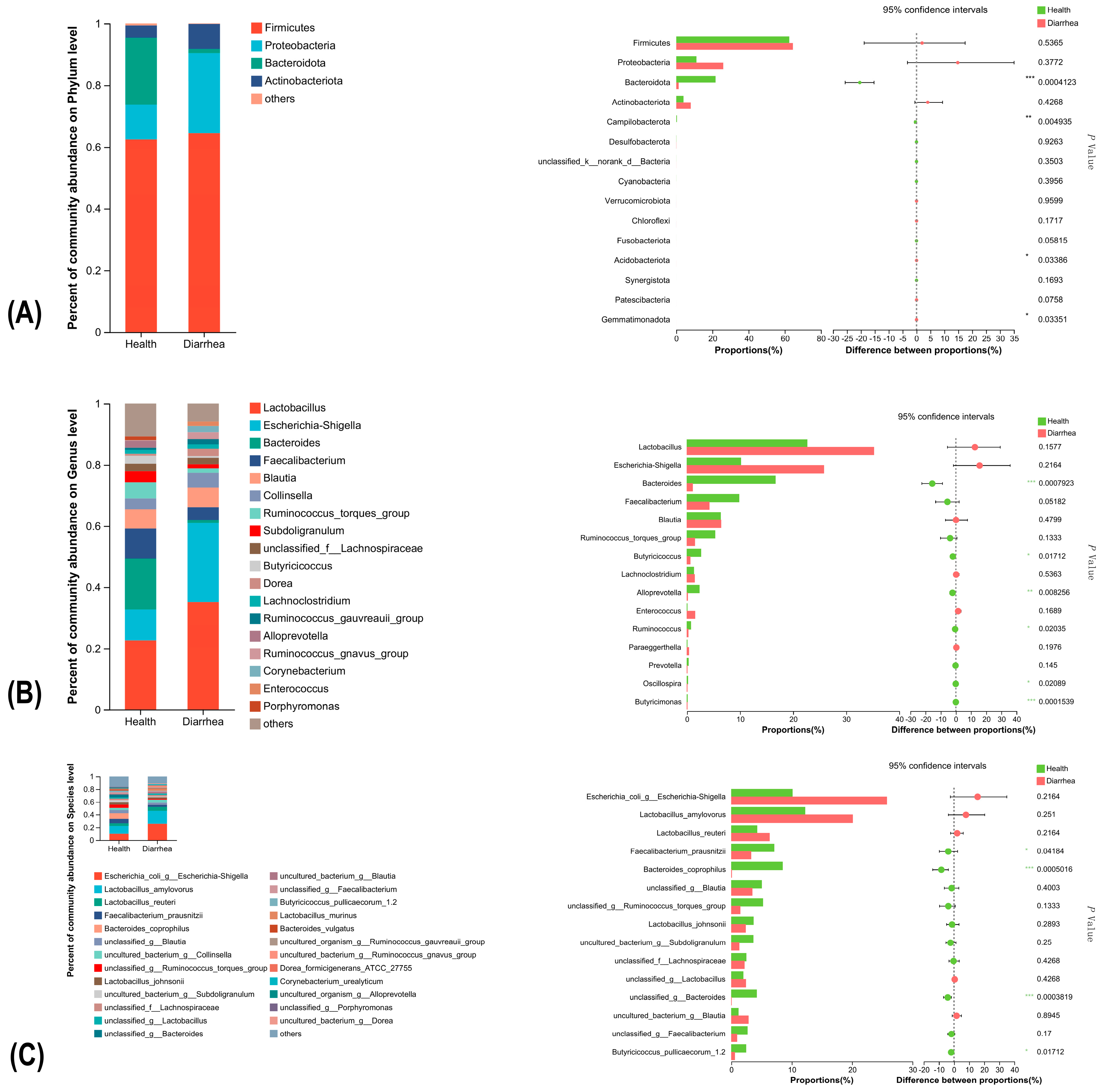
3.4. Significant Alterations of Bacterial Taxonomic Composition in Diarrheic Calves
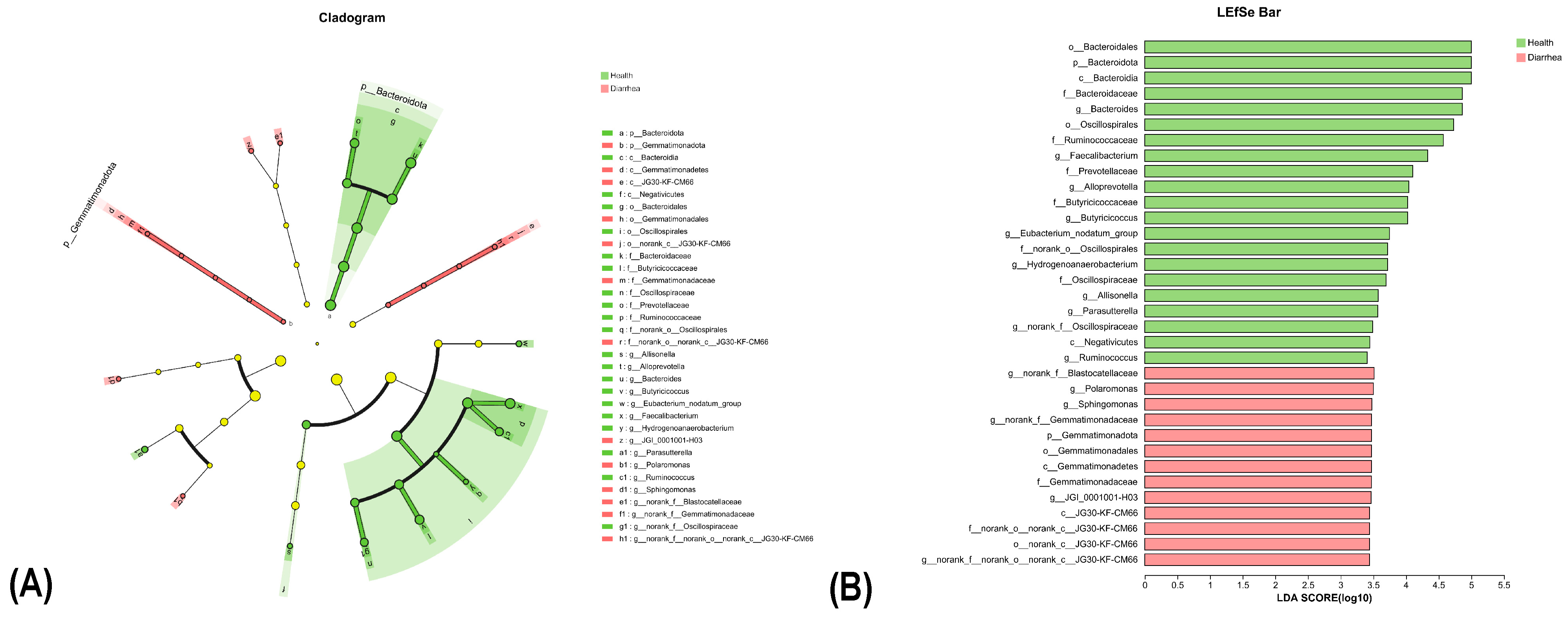
3.5. LEfSe Analysis Reveals Distinct Microbial Profiles between Healthy and Diarrheic Calves from Phylum to Species Level
3.6. Prokaryotic Taxa Identified across the Study Sample
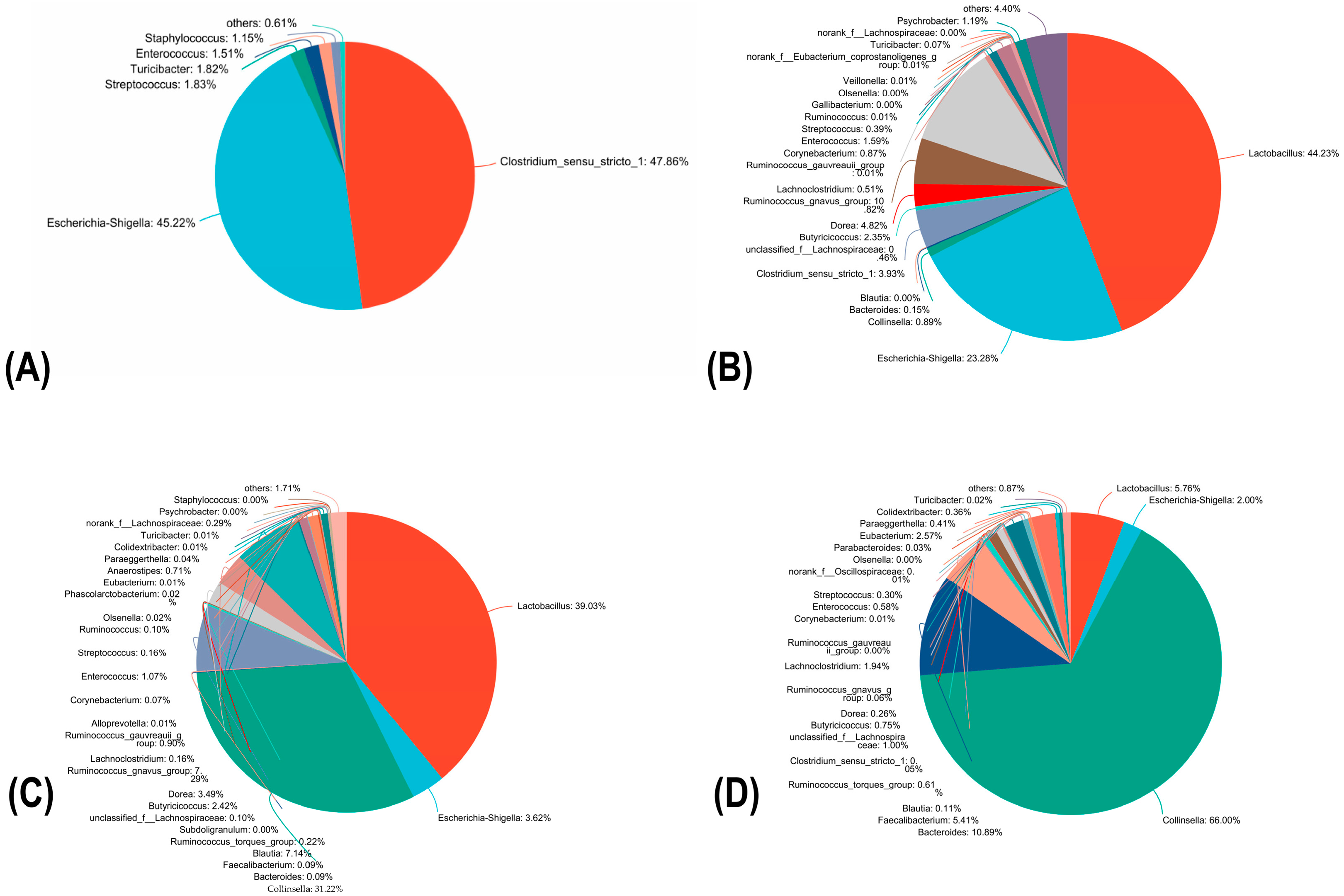
3.7. Fecal Microbiota Changes of Diarrhea Calves during Prodrome, Obvious Symptom, Conversion Period and Death
3.8. 16S rRNA Functional Prediction
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sharp, C.P.; Gregory, W.F.; Mason, C.; Bronsvoort, B.M.d.C.; Beard, P.M. High Prevalence and Diversity of Bovine Astroviruses in the Faeces of Healthy and Diarrhoeic Calves in South West Scotland. Vet. Microbiol. 2015, 178, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Fritzen, J.T.; Oliveira, M.V.; Lorenzetti, E.; Miyabe, F.M.; Viziack, M.P.; Rodrigues, C.A.; Ayres, H.; Alfieri, A.F.; Alfieri, A.A. Longitudinal Surveillance of Rotavirus A Genotypes Circulating in a High Milk Yield Dairy Cattle Herd after the Introduction of a Rotavirus Vaccine. Vet. Microbiol. 2019, 230, 260–264. [Google Scholar] [CrossRef] [PubMed]
- Blanchard, P.C. Diagnostics of Dairy and Beef Cattle Diarrhea. Vet. Clin. N. Am. Food Anim. Pract. 2012, 28, 443–464. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.R. Field Disease Diagnostic Investigation of Neonatal Calf Diarrhea. Vet. Clin. Food Anim. Pract. 2012, 28, 465–481. [Google Scholar] [CrossRef] [PubMed]
- House, J.A. Economic Impact of Rotavirus and Other Neonatal Disease Agents of Animals. J. Am. Vet. Med. Assoc. 1978, 173, 573–576. [Google Scholar]
- Van Der Waaij, D.; Berghuis-de Vries, J.M.; Lekkerkerk-van Der Wees, J.E.C. Colonization Resistance of the Digestive Tract in Conventional and Antibiotic-Treated Mice. J. Hyg. 1971, 69, 405–411. [Google Scholar] [CrossRef]
- Li, H.; Li, B.; Liang, Q.; Jin, X.; Tang, L.; Ding, Q.; Wang, Z.; Wei, Z. Porcine Deltacoronavirus Infection Alters Bacterial Communities in the Colon and Feces of Neonatal Piglets. Microbiologyopen 2020, 9, e1036. [Google Scholar] [CrossRef]
- Burrello, C.; Garavaglia, F.; Cribiù, F.M.; Ercoli, G.; Lopez, G.; Troisi, J.; Colucci, A.; Guglietta, S.; Carloni, S.; Guglielmetti, S.; et al. Therapeutic Faecal Microbiota Transplantation Controls Intestinal Inflammation through IL10 Secretion by Immune Cells. Nat. Commun. 2018, 9, 5184. [Google Scholar] [CrossRef]
- Li, X.-Y.; He, C.; Zhu, Y.; Lu, N.-H. Role of Gut Microbiota on Intestinal Barrier Function in Acute Pancreatitis. World J. Gastroenterol. 2020, 26, 2187–2193. [Google Scholar] [CrossRef]
- Gomez, D.E.; Arroyo, L.G.; Costa, M.C.; Viel, L.; Weese, J.S. Characterization of the Fecal Bacterial Microbiota of Healthy and Diarrheic Dairy Calves. J. Vet. Intern. Med. 2017, 31, 928–939. [Google Scholar] [CrossRef]
- Jang, J.-Y.; Kim, S.; Kwon, M.-S.; Lee, J.; Yu, D.-H.; Song, R.-H.; Choi, H.-J.; Park, J. Rotavirus-Mediated Alteration of Gut Microbiota and Its Correlation with Physiological Characteristics in Neonatal Calves. J. Microbiol. 2019, 57, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wang, X.; Zhang, W.; Kulyar, M.F.-A.; Ullah, K.; Han, Z.; Qin, J.; Bi, C.; Wang, Y.; Li, K. Comparative Analysis of Gut Microbiota in Healthy and Diarrheic Yaks. Microb. Cell Factories 2022, 21, 111. [Google Scholar] [CrossRef]
- Yang, Q.; Huang, X.; Zhao, S.; Sun, W.; Yan, Z.; Wang, P.; Li, S.; Huang, W.; Zhang, S.; Liu, L. Structure and Function of the Fecal Microbiota in Diarrheic Neonatal Piglets. Front. Microbiol. 2017, 8, 502. [Google Scholar] [CrossRef] [PubMed]
- Hennessy, M.; Indugu, N.; Vecchiarelli, B.; Redding, L.; Bender, J.; Pappalardo, C.; Leibstein, M.; Toth, J.; Stefanovski, D.; Katepalli, A. Comparison of the Fecal Bacterial Communities in Diarrheic and Nondiarrheic Dairy Calves from Multiple Farms in Southeastern Pennsylvania. J. Dairy Sci. 2021, 104, 7225–7232. [Google Scholar] [CrossRef]
- Larson, L.L.; Owen, F.G.; Albright, J.L.; Appleman, R.D.; Lamb, R.C.; Muller, L.D. Guidelines Toward More Uniformity in Measuring and Reporting Calf Experimental Data. J. Dairy Sci. 1977, 60, 989–991. [Google Scholar] [CrossRef]
- Constable, P.D.; Hinchcliff, K.W.; Done, S.H.; Grünberg, W. Veterinary Medicine: A Textbook of the Diseases of Cattle, Horses, Sheep, Pigs and Goats; Elsevier Health Sciences: Amsterdam, The Netherlands, 2016. [Google Scholar]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An Ultra-Fast All-in-One FASTQ Preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Magoč, T.; Salzberg, S.L. FLASH: Fast Length Adjustment of Short Reads to Improve Genome Assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef]
- Edgar, R.C. UPARSE: Highly Accurate OTU Sequences from Microbial Amplicon Reads. Nat. Methods 2013, 10, 996–998. [Google Scholar] [CrossRef]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naive Bayesian Classifier for Rapid Assignment of RRNA Sequences into the New Bacterial Taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef]
- Hall, M.; Beiko, R.G. 16S RRNA Gene Analysis with QIIME2. In Microbiome Analysis: Methods and Protocols; Springer: Berlin/Heidelberg, Germany, 2018; pp. 113–129. [Google Scholar] [CrossRef]
- Uetake, K. Newborn Calf Welfare: A Review Focusing on Mortality Rates: Newborn Calf Welfare. Anim. Sci. J. 2013, 84, 101–105. [Google Scholar] [CrossRef]
- Youmans, B.P.; Ajami, N.J.; Jiang, Z.-D.; Campbell, F.; Wadsworth, W.D.; Petrosino, J.F.; DuPont, H.L.; Highlander, S.K. Characterization of the Human Gut Microbiome during Travelers’ Diarrhea. Gut Microbes 2015, 6, 110–119. [Google Scholar] [CrossRef]
- Deng, L.; Lee, J.W.; Tan, K.S. Infection with Pathogenic Blastocystis ST7 Is Associated with Decreased Bacterial Diversity and Altered Gut Microbiome Profiles in Diarrheal Patients. Parasites Vectors 2022, 15, 312. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Wu, H.; Pan, H. Comparative Analysis of the Fecal Microbiome and Metabolomics of Healthy versus Captive South China Tigers with Mild Diarrhea. Can. J. Microbiol. 2022, 68, 758–768. [Google Scholar] [CrossRef] [PubMed]
- Schippa, S.; Conte, M. Dysbiotic Events in Gut Microbiota: Impact on Human Health. Nutrients 2014, 6, 5786–5805. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.-Z.; Wang, S.-Y.; Wang, H.; Cui, D.-A.; Yang, Y.-J.; Liu, X.-W.; Kong, X.-J.; Li, J.-Y. Differences in the Intestinal Microbiota between Uninfected Piglets and Piglets Infected with Porcine Epidemic Diarrhea Virus. PLoS ONE 2018, 13, e0192992. [Google Scholar] [CrossRef] [PubMed]
- Suchodolski, J.S.; Foster, M.L.; Sohail, M.U.; Leutenegger, C.; Queen, E.V.; Steiner, J.M.; Marks, S.L. The Fecal Microbiome in Cats with Diarrhea. PLoS ONE 2015, 10, e0127378. [Google Scholar] [CrossRef]
- Costa, M.C.; Arroyo, L.G.; Allen-Vercoe, E.; Stämpfli, H.R.; Kim, P.T.; Sturgeon, A.; Weese, J.S. Comparison of the Fecal Microbiota of Healthy Horses and Horses with Colitis by High Throughput Sequencing of the V3-V5 Region of the 16S RRNA Gene. PLoS ONE 2012, 7, e41484. [Google Scholar] [CrossRef] [PubMed]
- Ling, Z.; Liu, X.; Jia, X.; Cheng, Y.; Luo, Y.; Yuan, L.; Wang, Y.; Zhao, C.; Guo, S.; Li, L.; et al. Impacts of Infection with Different Toxigenic Clostridium Difficile Strains on Faecal Microbiota in Children. Sci. Rep. 2014, 4, 7485. [Google Scholar] [CrossRef] [PubMed]
- Stojanov, S.; Berlec, A.; Štrukelj, B. The Influence of Probiotics on the Firmicutes/Bacteroidetes Ratio in the Treatment of Obesity and Inflammatory Bowel Disease. Microorganisms 2020, 8, 1715. [Google Scholar] [CrossRef] [PubMed]
- Duvallet, C.; Gibbons, S.; Gurry, T.; Irizarry, R.; Alm, E. Meta Analysis of Microbiome Studies Identifies Shared and Disease-Specific Patterns. bioRxiv 2017, 134031. [Google Scholar] [CrossRef]
- Bäumler, A.J.; Sperandio, V. Interactions between the Microbiota and Pathogenic Bacteria in the Gut. Nature 2016, 535, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Abbracchio, M.P.; Burnstock, G.; Boeynaems, J.-M.; Barnard, E.A.; Boyer, J.L.; Kennedy, C.; Knight, G.E.; Fumagalli, M.; Gachet, C.; Jacobson, K.A.; et al. International Union of Pharmacology LVIII: Update on the P2Y G Protein-Coupled Nucleotide Receptors: From Molecular Mechanisms and Pathophysiology to Therapy. Pharmacol. Rev. 2006, 58, 281–341. [Google Scholar] [CrossRef] [PubMed]
- Van Nhieu, G.T.; Clair, C.; Bruzzone, R.; Mesnil, M.; Sansonetti, P.; Combettes, L. Connexin-Dependent Inter-Cellular Communication Increases Invasion and Dissemination of Shigella in Epithelial Cells. Nat. Cell Biol. 2003, 5, 720–726. [Google Scholar] [CrossRef] [PubMed]
- Crane, J.K.; Olson, R.A.; Jones, H.M.; Duffey, M.E. Release of ATP during Host Cell Killing by Enteropathogenic E. coli and Its Role as a Secretory Mediator. Am. J. Physiol. Gastrointest. Liver Physiol. 2002, 283, G74–G86. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Liu, B.; Li, A.; Iqbal, M.; Mehmood, K.; Jamil, T.; Chang, Y.-F.; Zhang, H.; Wu, Q. Microbiome Analysis Reveals the Attenuation Effect of Lactobacillus from Yaks on Diarrhea via Modulation of Gut Microbiota. Front. Cell. Infect. Microbiol. 2021, 10, 610781. [Google Scholar] [CrossRef] [PubMed]
- Dempsey, E.; Corr, S.C. Lactobacillus spp. for Gastrointestinal Health: Current and Future Perspectives. Front. Immunol. 2022, 13, 840245. [Google Scholar] [CrossRef] [PubMed]
- Nishida, S.; Ishii, M.; Nishiyama, Y.; Abe, S.; Ono, Y.; Sekimizu, K. Lactobacillus paraplantarum 11-1 Isolated from Rice Bran Pickles Activated Innate Immunity and Improved Survival in a Silkworm Bacterial Infection Model. Front. Microbiol. 2017, 8, 436. [Google Scholar] [CrossRef]
- Wolin, M.J. Fermentation in the Rumen and Human Large Intestine. Science 1981, 213, 1463–1468. [Google Scholar] [CrossRef]
- Kim, E.T.; Lee, S.J.; Kim, T.Y.; Lee, H.G.; Atikur, R.M.; Gu, B.H.; Kim, D.H.; Park, B.Y.; Son, J.K.; Kim, M. Dynamic Changes in Fecal Microbial Communities of Neonatal Dairy Calves by Aging and Diarrhea. Animals 2021, 11, 1113. [Google Scholar] [CrossRef]
- Ju, T.; Kong, J.Y.; Stothard, P.; Willing, B.P. Defining the Role of Parasutterella, a Previously Uncharacterized Member of the Core Gut Microbiota. ISME J. 2019, 13, 1520–1534. [Google Scholar] [CrossRef]
- Zhang, X.; Deeke, S.A.; Ning, Z.; Starr, A.E.; Butcher, J.; Li, J.; Mayne, J.; Cheng, K.; Liao, B.; Li, L. Metaproteomics Reveals Associations between Microbiome and Intestinal Extracellular Vesicle Proteins in Pediatric Inflammatory Bowel Disease. Nat. Commun. 2018, 9, 2873. [Google Scholar] [CrossRef] [PubMed]
- Cornuault, J.K.; Petit, M.-A.; Mariadassou, M.; Benevides, L.; Moncaut, E.; Langella, P.; Sokol, H.; De Paepe, M. Phages Infecting Faecalibacterium prausnitzii Belong to Novel Viral Genera that Help to Decipher Intestinal Viromes. Microbiome 2018, 6, 65. [Google Scholar] [CrossRef] [PubMed]
- Eeckhaut, V.; Machiels, K.; Perrier, C.; Romero, C.; Maes, S.; Flahou, B.; Steppe, M.; Haesebrouck, F.; Sas, B.; Ducatelle, R.; et al. Butyricicoccus pullicaecorum in Inflammatory Bowel Disease. Gut 2013, 62, 1745–1752. [Google Scholar] [CrossRef] [PubMed]
- Facchin, S.; Vitulo, N.; Calgaro, M.; Buda, A.; Romualdi, C.; Pohl, D.; Perini, B.; Lorenzon, G.; Marinelli, C.; D’Incà, R.; et al. Microbiota Changes Induced by Microencapsulated Sodium Butyrate in Patients with Inflammatory Bowel Disease. Neurogastroenterol. Motil. 2020, 32, 13914. [Google Scholar] [CrossRef]
- Wang, W.; Chen, L.; Zhou, R.; Wang, X.; Song, L.; Huang, S.; Wang, G.; Xia, B. Increased Proportions of Bifidobacterium and the Lactobacillus Group and Loss of Butyrate-Producing Bacteria in Inflammatory Bowel Disease. J. Clin. Microbiol. 2014, 52, 398–406. [Google Scholar] [CrossRef]
- Guthrie, E.; Creed, F.; Dawson, D.; Tomenson, B. A Controlled Trial of Psychological Treatment for the Irritable Bowel Syndrome. Gastroenterology 1991, 100, 450–457. [Google Scholar] [CrossRef]
- Chan, A.O. Differing Coping Mechanisms, Stress Level and Anorectal Physiology in Patients with Functional Constipation. World J. Gastroenterol. 2005, 11, 5362. [Google Scholar] [CrossRef]
- Murray, C.D.R.; Flynn, J.; Ratcliffe, L.; Jacyna, M.R.; Kamm, M.A.; Emmanuel, A.V. Effect of Acute Physical and Psychological Stress on Gut Autonomic Innervation in Irritable Bowel Syndrome. Gastroenterology 2004, 127, 1695–1703. [Google Scholar] [CrossRef]
- Yang, T.; Richards, E.M.; Pepine, C.J.; Raizada, M.K. The Gut Microbiota and the Brain–Gut–Kidney Axis in Hypertension and Chronic Kidney Disease. Nat. Rev. Nephrol. 2018, 14, 442–456. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| The Health Protocol and Management for Calves | |
|---|---|
| Breed | Simmental cattle and Angus cattle |
| Calves per year | 100 |
| Type of housing | Group pen |
| Type of bedding | The mushroom residue |
| Whether calves were born from discotic parturition | No |
| Dam vaccination | No |
| Umbilicus care | Properly sanitize the umbilical stump and ensure that it is kept clean and dry to prevent infection |
| Colostrum quality | Eligible colostrum |
| Colostrum feeding | 4 L first 4 h |
| Diet and nutrition (8–12 weeks) | milk and/or formula milk |
| Feeding method | Group bucket |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, W.; Yi, X.; Wu, B.; Li, X.; Ye, B.; Deng, Z.; A, R.; Hu, S.; Li, D.; Wu, H.; et al. Neonatal Calf Diarrhea Is Associated with Decreased Bacterial Diversity and Altered Gut Microbiome Profiles. Fermentation 2023, 9, 827. https://doi.org/10.3390/fermentation9090827
Li W, Yi X, Wu B, Li X, Ye B, Deng Z, A R, Hu S, Li D, Wu H, et al. Neonatal Calf Diarrhea Is Associated with Decreased Bacterial Diversity and Altered Gut Microbiome Profiles. Fermentation. 2023; 9(9):827. https://doi.org/10.3390/fermentation9090827
Chicago/Turabian StyleLi, Wei, Xin Yi, Baoyun Wu, Xiang Li, Boping Ye, Ziqi Deng, Runa A, Sanlong Hu, Dongdong Li, Hao Wu, and et al. 2023. "Neonatal Calf Diarrhea Is Associated with Decreased Bacterial Diversity and Altered Gut Microbiome Profiles" Fermentation 9, no. 9: 827. https://doi.org/10.3390/fermentation9090827