
Optimized Linear DNA Recombineering for CRISPR-Cpf1 System in Corynebacterium glutamicum

Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Strains and Culture
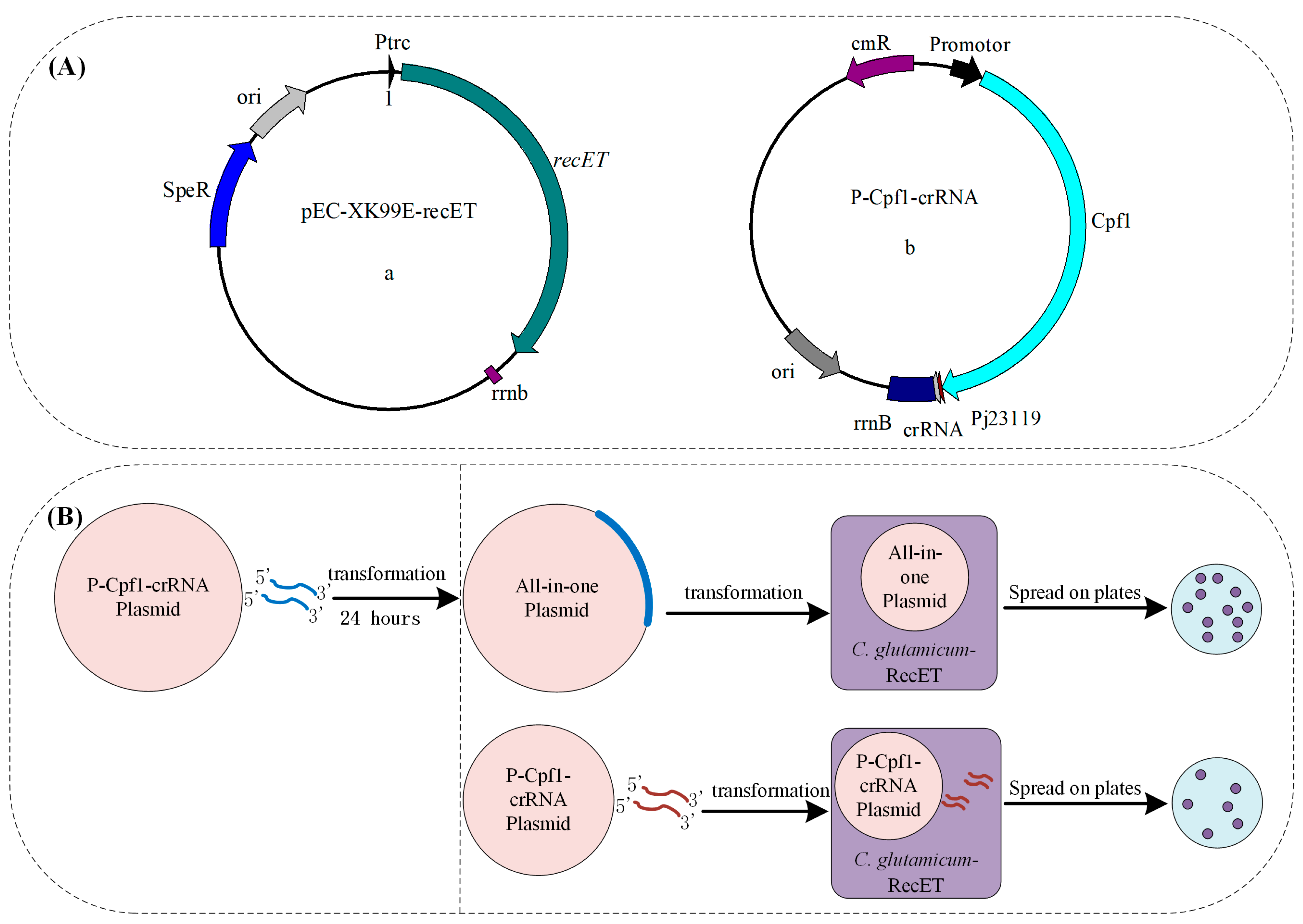
2.3. Construction of Plasmids
2.4. Preparation of Linear DNA Fragments
2.5. Preparation and Transformation of Chemotransformed Competent Cells
2.6. Competent Cells Preparation and Electrotransformation
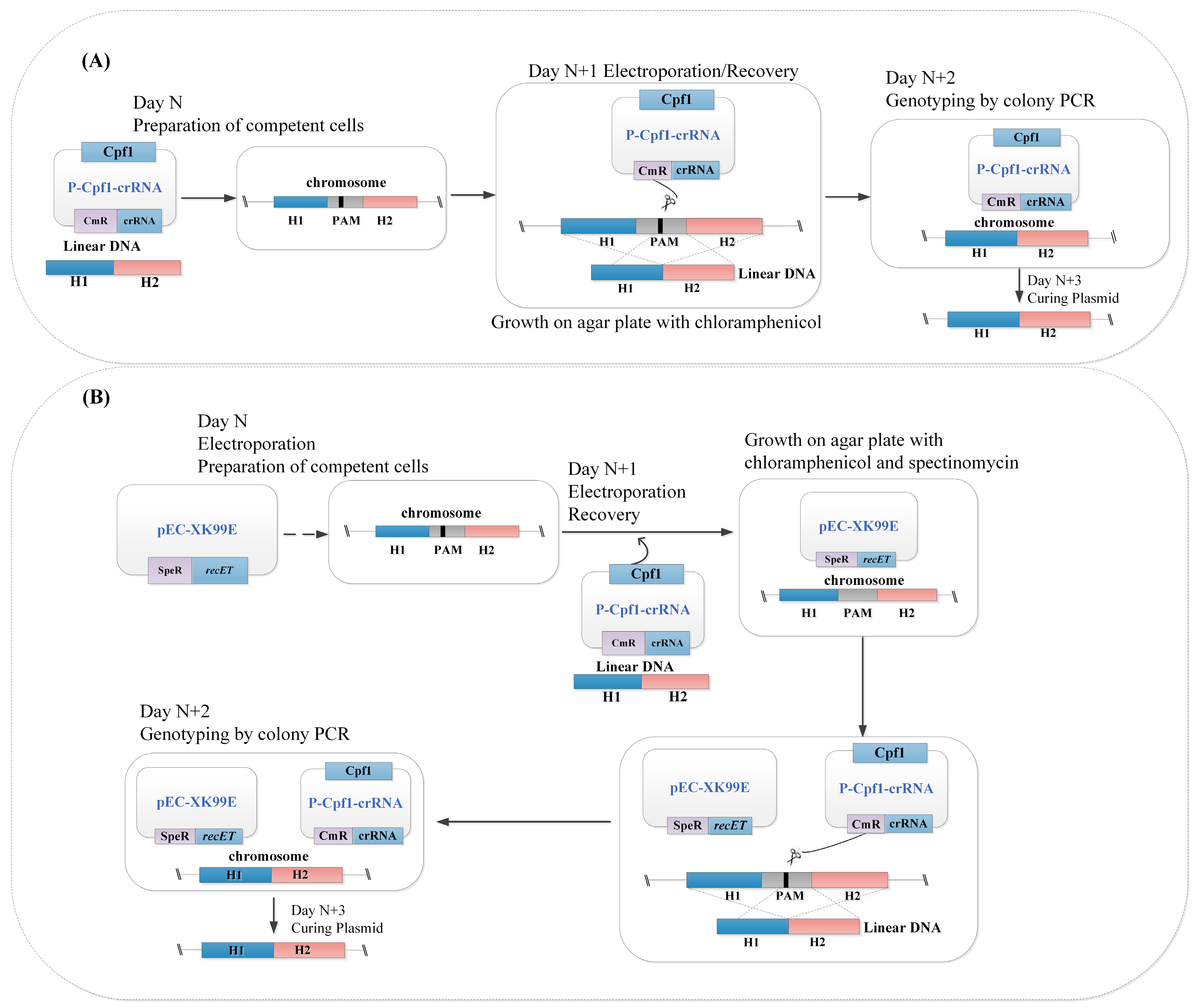
2.7. RecET-Assisted CRISPR-Cpf1 Genome Editing Procedure Using Linear DNA
2.8. Plasmid Loss
2.9. Determination of Genome Editing Efficiency
2.10. Fermentor (5 L) Culture
2.11. Substrate and Product Analysis
3. Results
3.1. Construction of the Preliminary CRISPR-Cpf1 Gene Editing System
3.2. Optimization of the CRISPR-Cpf1 Gene Editing System
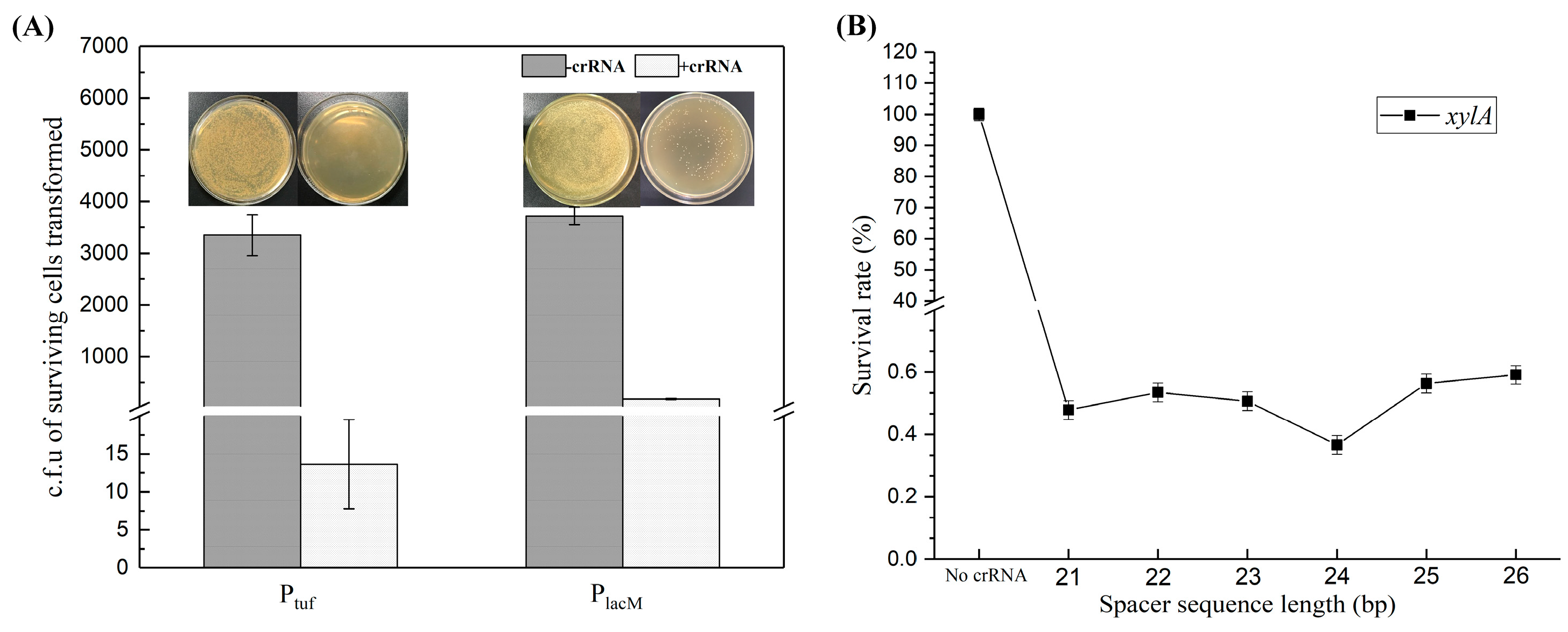
3.2.1. Optimization of the Cleavage System
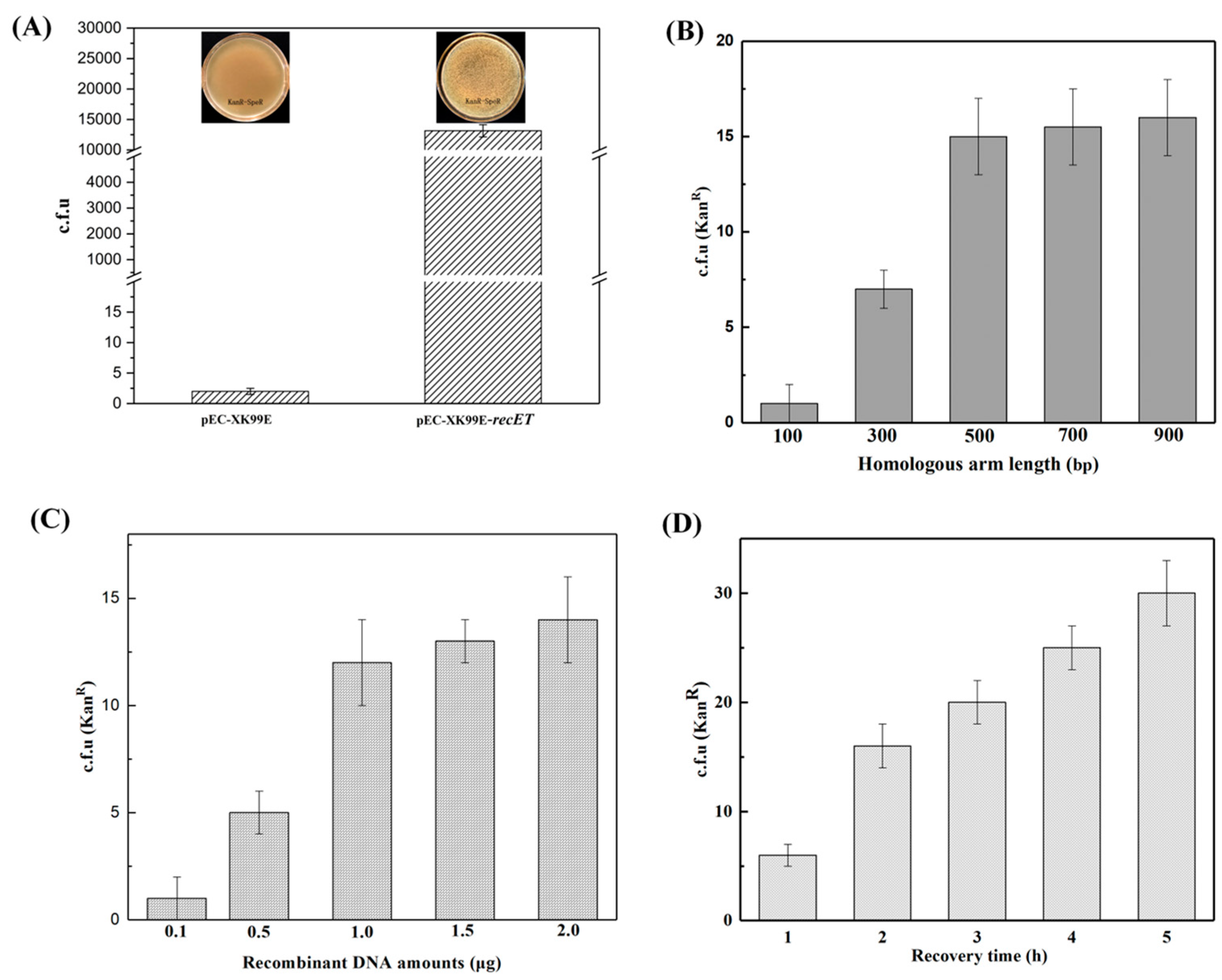
3.2.2. Optimization of the Recombination System
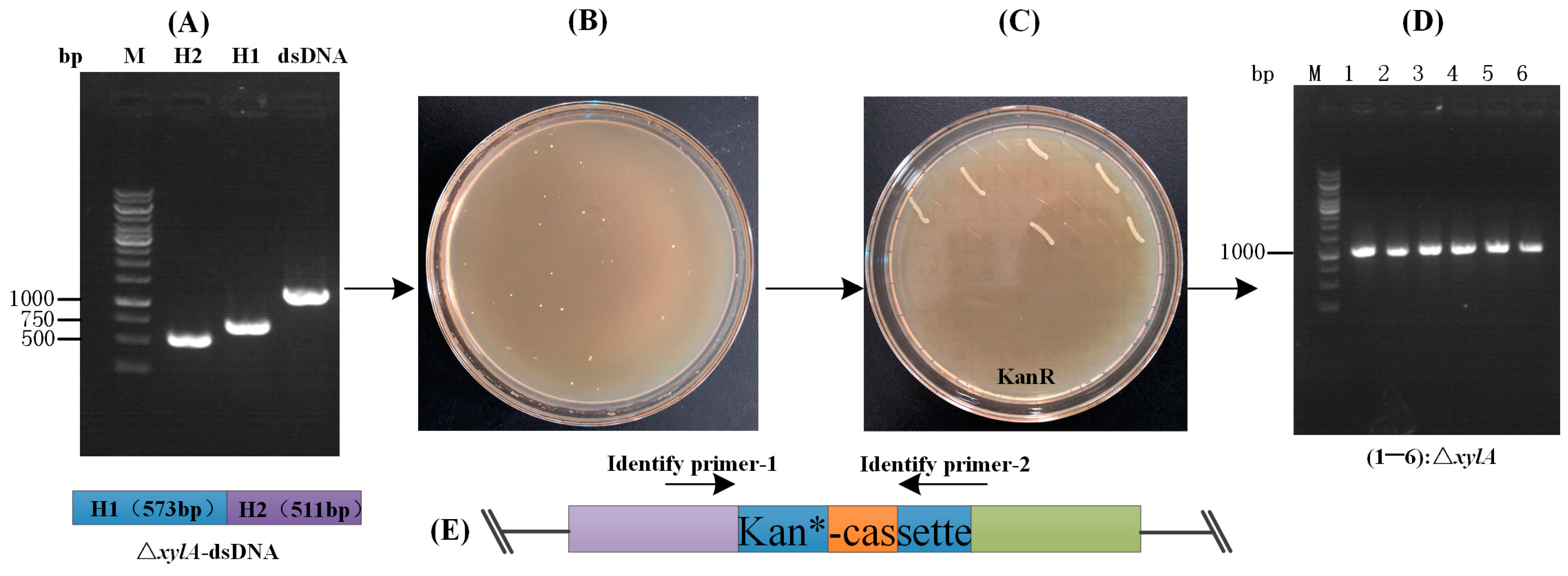
3.3. Statistics of the Linear DNA-Mediated CRISPR-Cpf1 Gene Editing System
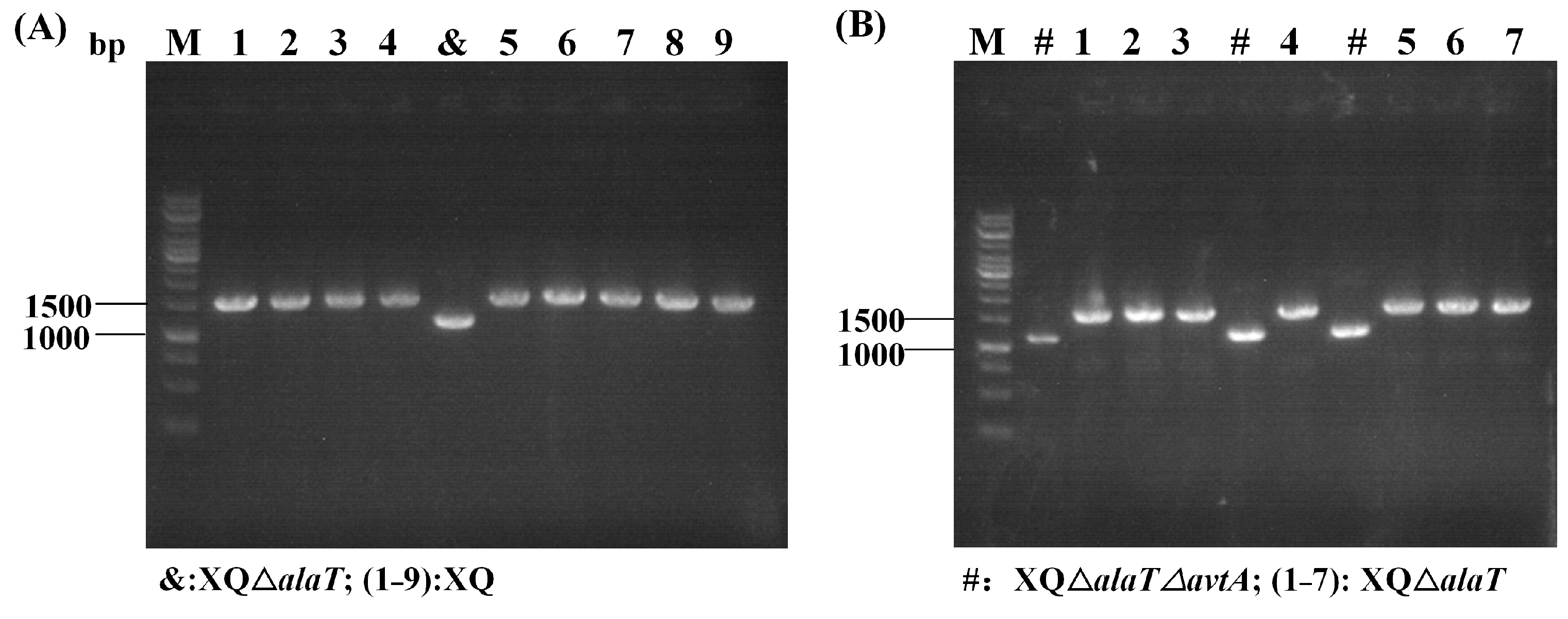
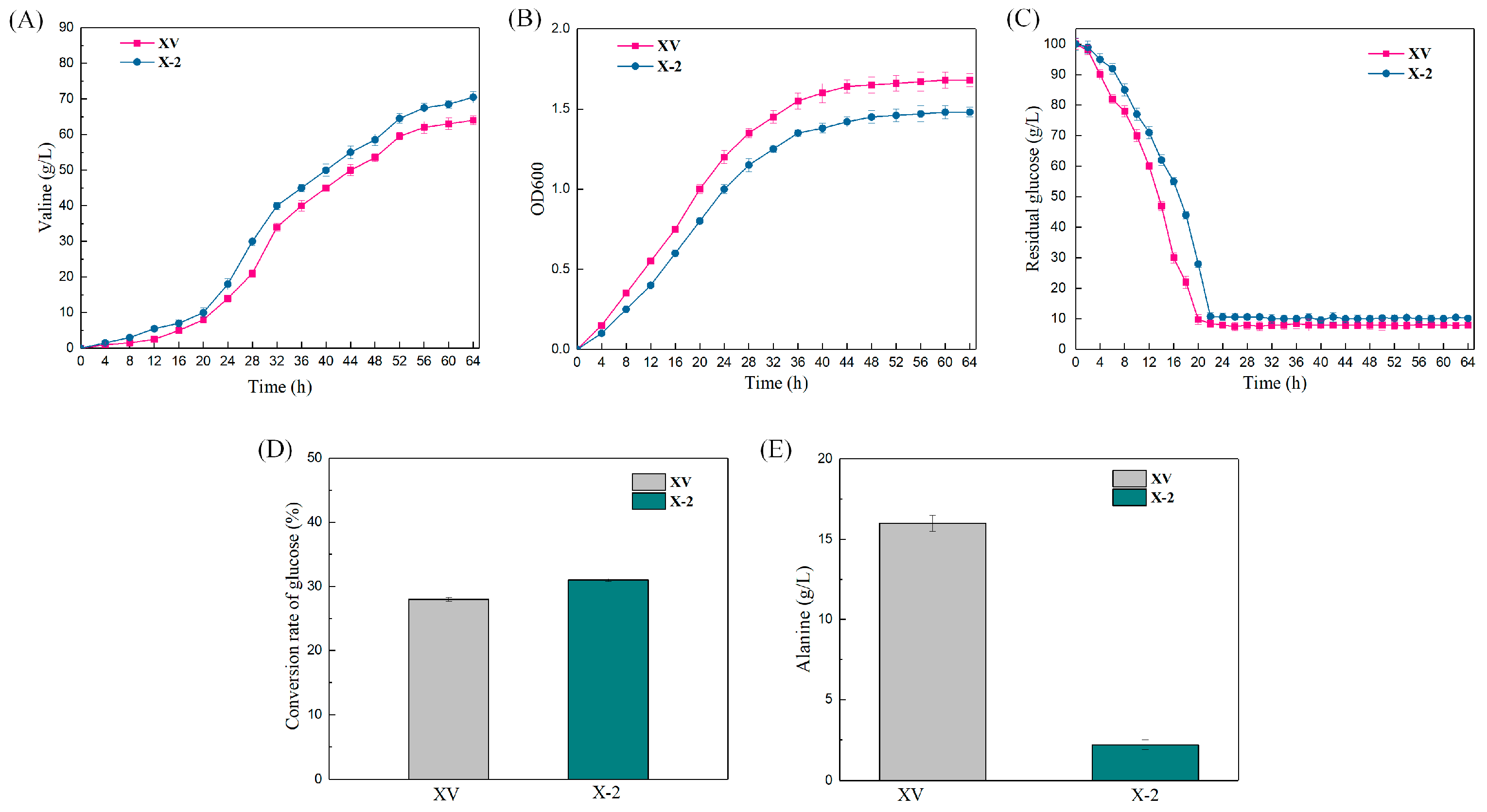
3.4. Construction of an L-Valine-Producing Strain
4. Discussion
Supplementary Materials
Author Contributions
Funding
InstitutionaL Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bao, Z.; Cartinhour, S.; Swingle, B. Substrate and target sequence length influence RecTE(Psy) recombineering efficiency in Pseudomonas syringae. PLoS ONE 2012, 7, e50617. [Google Scholar] [CrossRef] [PubMed]
- Becker, J.; Giesselmann, G.; Hoffmann, S.L.; Wittmann, C. Corynebacterium glutamicum for Sustainable Bioproduction: From Metabolic Physiology to Systems Metabolic Engineering. Adv. Biochem. Eng. Biotechnol. 2018, 162, 217–263. [Google Scholar] [CrossRef]
- Wang, Q.; Zhang, J.; Al Makishah, N.H.; Sun, X.; Wen, Z.; Jiang, Y.; Yang, S. Advances and Perspectives for Genome Editing Tools of Corynebacterium glutamicum. Front. Microbiol. 2021, 12, 654058. [Google Scholar] [CrossRef] [PubMed]
- Wendisch, V.F.; Bott, M.; Eikmanns, B.J. Metabolic engineering of Escherichia coli and Corynebacterium glutamicum for biotechnological production of organic acids and amino acids. Curr. Opin. Microbiol. 2006, 9, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Liang, B.; Sun, G.; Zhang, X.; Nie, Q.; Zhao, Y.; Yang, J. Recent advances, challenges and metabolic engineering strategies in the biosynthesis of 3-hydroxypropionic acid. Biotechnol. Bioeng. 2022, 119, 2639–2668. [Google Scholar] [CrossRef] [PubMed]
- Kallscheuer, N.; Vogt, M.; Stenzel, A.; Gatgens, J.; Bott, M.; Marienhagen, J. Construction of a Corynebacterium glutamicum platform strain for the production of stilbenes and (2S)-flavanones. Metab. Eng. 2016, 38, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Gordillo Sierra, A.R.; Alper, H.S. Progress in the metabolic engineering of bio-based lactams and their omega-amino acids precursors. Biotechnol. Adv. 2020, 43, 107587. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y.; Kim, H.U. Systems strategies for developing industrial microbial strains. Nat. Biotechnol. 2015, 33, 1061–1072. [Google Scholar] [CrossRef]
- Srivastava, P.; Deb, J.K. Gene expression systems in corynebacteria. Protein Expr. Purif. 2005, 40, 221–229. [Google Scholar] [CrossRef]
- Vertes, A.A.; Inui, M.; Yukawa, H. Manipulating corynebacteria, from individual genes to chromosomes. Appl. Environ. Microbiol. 2005, 71, 7633–7642. [Google Scholar] [CrossRef]
- Yang, J.; Ma, X.; Wang, X.; Zhang, Z.; Wang, S.; Qin, H.; Mao, S.; Lu, F. Advances in gene editing of Corynebacterium glutamate. Sheng Wu Gong Cheng Xue Bao 2020, 36, 820–828. [Google Scholar] [CrossRef] [PubMed]
- Schafer, A.; Tauch, A.; Jager, W.; Kalinowski, J.; Thierbach, G.; Puhler, A. Small mobilizable multi-purpose cloning vectors derived from the Escherichia coli plasmids pK18 and pK19: Selection of defined deletions in the chromosome of Corynebacterium glutamicum. Gene 1994, 145, 69–73. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Wang, X.; Mao, Y.; Wang, Z.; Chen, T.; Zhao, X. Development of a markerless gene replacement system in Corynebacterium glutamicum using upp as a counter-selection marker. Biotechnol. Lett. 2015, 37, 609–617. [Google Scholar] [CrossRef] [PubMed]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Cleto, S.; Jensen, J.V.; Wendisch, V.F.; Lu, T.K. Corynebacterium glutamicum Metabolic Engineering with CRISPR Interference (CRISPRi). ACS Synth. Biol. 2016, 5, 375–385. [Google Scholar] [CrossRef]
- Jeong, S.H.; Lee, H.J.; Lee, S.J. Recent Advances in CRISPR-Cas Technologies for Synthetic Biology. J. Microbiol. 2023, 61, 13–36. [Google Scholar] [CrossRef] [PubMed]
- Okano, K.; Sato, Y.; Hizume, T.; Honda, K. Genome editing by miniature CRISPR/Cas12f1 enzyme in Escherichia coli. J. Biosci. Bioeng. 2021, 132, 120–124. [Google Scholar] [CrossRef]
- Yan, M.Y.; Yan, H.Q.; Ren, G.X.; Zhao, J.P.; Guo, X.P.; Sun, Y.C. CRISPR-Cas12a-Assisted Recombineering in Bacteria. Appl. Environ. Microbiol. 2017, 83, e00947-17. [Google Scholar] [CrossRef]
- Feng, X.; Zhao, D.; Zhang, X.; Ding, X.; Bi, C. CRISPR/Cas9 Assisted Multiplex Genome Editing Technique in Escherichia coli. Biotechnol. J. 2018, 13, e1700604. [Google Scholar] [CrossRef]
- Zetsche, B.; Gootenberg, J.S.; Abudayyeh, O.O.; Slaymaker, I.M.; Makarova, K.S.; Essletzbichler, P.; Volz, S.E.; Joung, J.; van der Oost, J.; Regev, A.; et al. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell 2015, 163, 759–771. [Google Scholar] [CrossRef]
- Zetsche, B.; Heidenreich, M.; Mohanraju, P.; Fedorova, I.; Kneppers, J.; DeGennaro, E.M.; Winblad, N.; Choudhury, S.R.; Abudayyeh, O.O.; Gootenberg, J.S.; et al. Multiplex gene editing by CRISPR-Cpf1 using a single crRNA array. Nat. Biotechnol. 2017, 35, 31–34. [Google Scholar] [CrossRef] [PubMed]
- Dong, D.; Ren, K.; Qiu, X.; Zheng, J.; Guo, M.; Guan, X.; Liu, H.; Li, N.; Zhang, B.; Yang, D.; et al. The crystal structure of Cpf1 in complex with CRISPR RNA. Nature 2016, 532, 522–526. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Liu, Q.; Shi, H.; Xie, J.; Zhang, Q.; Ouyang, Z.; Li, N.; Yang, Y.; Liu, Z.; Zhao, Y.; et al. Engineering CRISPR/Cpf1 with tRNA promotes genome editing capability in mammalian systems. Cell Mol. Life Sci. 2018, 75, 3593–3607. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Qin, R.; Li, H.; Li, D.; Li, L.; Wei, P.; Yang, J. Generation of targeted mutant rice using a CRISPR-Cpf1 system. Plant Biotechnol. J. 2017, 15, 713–717. [Google Scholar] [CrossRef] [PubMed]
- Ellis, H.M.; Yu, D.; DiTizio, T.; Court, D.L. High efficiency mutagenesis, repair, and engineering of chromosomal DNA using single-stranded oligonucleotides. Proc. Natl. Acad. Sci. USA 2001, 98, 6742–6746. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, Y.; Liu, J.; Guo, Y.; Fan, L.; Ni, X.; Zheng, X.; Wang, M.; Zheng, P.; Sun, J.; et al. MACBETH: Multiplex automated Corynebacterium glutamicum base editing method. Metab. Eng. 2018, 47, 200–210. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Li, L.; Xie, S.; Zhao, N.; Han, S.; Lin, Y.; Zheng, S. Recombineering using RecET in Corynebacterium glutamicum ATCC14067 via a self-excisable cassette. Sci. Rep. 2017, 7, 7916. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Hu, Q.; Zhang, Y.; Shi, R.; Chai, X.; Liu, Z.; Shang, X.; Zhang, Y.; Wen, T. A RecET-assisted CRISPR-Cas9 genome editing in Corynebacterium glutamicum. Microb. Cell Fact. 2018, 17, 63. [Google Scholar] [CrossRef]
- Luo, G.; Zhao, N.; Jiang, S.; Zheng, S. Application of RecET-Cre/loxP system in Corynebacterium glutamicum ATCC14067 for L-leucine production. Biotechnol. Lett. 2021, 43, 297–306. [Google Scholar] [CrossRef]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef]
- Zhao, N.; Li, L.; Luo, G.; Xie, S.; Lin, Y.; Han, S.; Huang, Y.; Zheng, S. Multiplex gene editing and large DNA fragment deletion by the CRISPR/Cpf1-RecE/T system in Corynebacterium glutamicum. J. Ind. Microbiol. Biotechnol. 2020, 47, 599–608. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Qian, F.; Yang, J.; Liu, Y.; Dong, F.; Xu, C.; Sun, B.; Chen, B.; Xu, X.; Li, Y.; et al. CRISPR-Cpf1 assisted genome editing of Corynebacterium glutamicum. Nat. Commun. 2017, 8, 15179. [Google Scholar] [CrossRef] [PubMed]
- Su, R.; Wang, T.; Bo, T.; Cai, N.; Yuan, M.; Wu, C.; Jiang, H.; Peng, H.; Chen, N.; Li, Y. Enhanced production of D-pantothenic acid in Corynebacterium glutamicum using an efficient CRISPR-Cpf1 genome editing method. Microb. Cell Fact. 2023, 22, 3. [Google Scholar] [CrossRef] [PubMed]
- Garst, A.D.; Bassalo, M.C.; Pines, G.; Lynch, S.A.; Halweg-Edwards, A.L.; Liu, R.; Liang, L.; Wang, Z.; Zeitoun, R.; Alexander, W.G.; et al. Genome-wide mapping of mutations at single-nucleotide resolution for protein, metabolic and genome engineering. Nat. Biotechnol. 2017, 35, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Peng, F.; Wang, X.; Sun, Y.; Dong, G.; Yang, Y.; Liu, X.; Bai, Z. Efficient gene editing in Corynebacterium glutamicum using the CRISPR/Cas9 system. Microb. Cell Fact. 2017, 16, 201. [Google Scholar] [CrossRef] [PubMed]
- Marienhagen, J.; Eggeling, L. Metabolic function of Corynebacterium glutamicum aminotransferases AlaT and AvtA and impact on L-valine production. Appl. Environ. Microbiol. 2008, 74, 7457–7462. [Google Scholar] [CrossRef] [PubMed]
- Altenbuchner, J. Editing of the Bacillus subtilis Genome by the CRISPR-Cas9 System. Appl. Environ. Microbiol. 2016, 82, 5421–5427. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Lin, Z.; Huang, C.; Zhang, Y.; Wang, Z.; Tang, Y.J.; Chen, T.; Zhao, X. Metabolic engineering of Escherichia coli using CRISPR-Cas9 meditated genome editing. Metab. Eng. 2015, 31, 13–21. [Google Scholar] [CrossRef]
- Liu, J.; Wang, Y.; Lu, Y.; Zheng, P.; Sun, J.; Ma, Y. Development of a CRISPR/Cas9 genome editing toolbox for Corynebacterium glutamicum. Microb. Cell Fact. 2017, 16, 205. [Google Scholar] [CrossRef]
- Cho, J.S.; Choi, K.R.; Prabowo, C.P.S.; Shin, J.H.; Yang, D.; Jang, J.; Lee, S.Y. CRISPR/Cas9-coupled recombineering for metabolic engineering of Corynebacterium glutamicum. Metab. Eng. 2017, 42, 157–167. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Plasmid | Replicate | Colonies | Editing Efficiency | Average |
|---|---|---|---|---|
| Ptc-crRNAΔxylA | 1 | 6/21 | 28.6% | 30.4 ± 1.6% |
| 2 | 7/22 | 31.8% | ||
| 3 | 8/26 | 30.8% |
| Strain | Colonies | Editing Efficiency |
|---|---|---|
| XQΔalaT (X−1) | 1/10 | 10% |
| XQΔalaTΔavtA (X−2) | 3/10 | 30% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, T.; Jiang, X.; Lv, S.; Hu, L.; Gao, S.; Xu, Q.; Zhang, J.; Hou, D. Optimized Linear DNA Recombineering for CRISPR-Cpf1 System in Corynebacterium glutamicum. Fermentation 2024, 10, 31. https://doi.org/10.3390/fermentation10010031
Wang T, Jiang X, Lv S, Hu L, Gao S, Xu Q, Zhang J, Hou D. Optimized Linear DNA Recombineering for CRISPR-Cpf1 System in Corynebacterium glutamicum. Fermentation. 2024; 10(1):31. https://doi.org/10.3390/fermentation10010031
Chicago/Turabian StyleWang, Ting, Xiaowan Jiang, Shufang Lv, Linfeng Hu, Shuangcheng Gao, Qingyang Xu, Junhui Zhang, and Dianyun Hou. 2024. "Optimized Linear DNA Recombineering for CRISPR-Cpf1 System in Corynebacterium glutamicum" Fermentation 10, no. 1: 31. https://doi.org/10.3390/fermentation10010031