Effect of Cellulose Nanocrystals on the Coating of Chitosan Nanocomposite Film Using Plasma-Mediated Deposition of Amorphous Hydrogenated Carbon (a–C:H) Layers

 ,
,  , and
, and 
Abstract
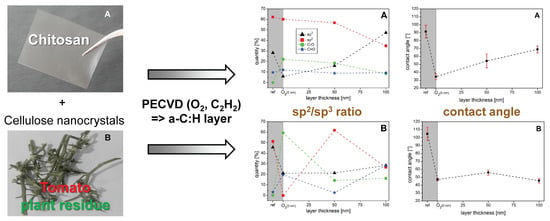
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Samples Preparation
2.2. Coating Process
2.3. Surface Structure
2.4. Chemical Composition
3. Results and Discussion
3.1. Processing of Films
3.2. FTIR Characterization of the Samples
3.3. Topography and Wettability
3.4. Chemical Composition
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Yadav, M.; Behera, K.; Chang, Y.-H.; Chiu, F.-C. Cellulose Nanocrystal Reinforced Chitosan Based UV Barrier Composite Films for Sustainable Packaging. Polymers (Basel) 2020, 12, 202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlebrowski, T.; Beucher, L.; Bazzi, H.; Hahn, B.; Wehner, S.; Fischer, C.B. Changing Contents of Carbon Hybridizations in Amorphous Hydrogenated Carbon Layers (a–C:H) on Sustainable Polyhydroxybutyrate (PHB) Exhibit a Significant Deterioration in Stability, Depending on Thickness. C J. Carbon Res. 2019, 5, 52. [Google Scholar] [CrossRef] [Green Version]
- El Achaby, M.; Kassab, Z.; Barakat, A.; Aboulkas, A. Alfa fibers as viable sustainable source for cellulose nanocrystals extraction: Application for improving the tensile properties of biopolymer nanocomposite films. Ind. Crop. Prod. 2018, 112, 499–510. [Google Scholar] [CrossRef]
- Kassab, Z.; Abdellaoui, Y.; Salim, M.H.; Bouhfid, R.; Qaiss, A.E.K.; El Achaby, M. Micro- and nano-celluloses derived from hemp stalks and their effect as polymer reinforcing materials. Carbohydr. Polym. 2020, 245, 116506. [Google Scholar] [CrossRef] [PubMed]
- Schlebrowski, T.; Acharchi, H.; Hahn, B.; Wehner, S.; Fischer, C.B. Refinement of Sustainable Polybutylene Adipate Terephthalate (PBAT) with Amorphous Hydrogenated Carbon Films (a-C:H) Revealing Film Instabilities Influenced by a Thickness-Dependent Change of sp2/sp3 Ratio. Materials 2020, 13, 1077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlebrowski, T.; Rouabeh, W.; Wehner, S.; Fischer, C.B. Specifying the interlayer turning point and dehydrogenation in a-C:H layers plasma deposited on high-density polyethylene with X-ray synchrotron techniques. Thin Solid Films 2019, 691, 137617. [Google Scholar] [CrossRef]
- Grill, A. Plasma-deposited diamondlike carbon and related materials. IBM J. Res. Dev. 1999, 43, 147–162. [Google Scholar] [CrossRef]
- Mohagheghpour, E.; Rajabi, M.; Gholamipour, R.; Larijani, M.M.; Sheibani, S. Ion beam energy dependence of surface and structural properties of amorphous carbon films deposited by IBSD method on Ni–Cu alloy. J. Mater. Res. 2017, 32, 1258–1266. [Google Scholar] [CrossRef]
- Robertson, J. Diamond-like amorphous carbon. Mater. Sci. Eng. R. Rep. 2002, 37, 129–281. [Google Scholar] [CrossRef] [Green Version]
- Robertson, J. Plasma Deposition of Diamond-Like Carbon. Jpn. J. Appl. Phys. 2011, 50, 01AF01. [Google Scholar] [CrossRef]
- Schlebrowski, T.; Beucher, L.; Bazzi, H.; Hahn, B.; Wehner, S.; Fischer, C.B. Prediction of a-C:H layer failure on industrial relevant biopolymer polylactide acide (PLA) foils based on the sp2/sp3 ratio. Surf. Coat. Technol. 2019, 368, 79–87. [Google Scholar] [CrossRef]
- Catena, A.; Agnello, S.; Rösken, L.M.; Bergen, H.; Recktenwald, E.; Bernsmann, F.; Busch, H.; Cannas, M.; Gelardi, F.M.; Hahn, B.; et al. Characteristics of industrially manufactured amorphous hydrogenated carbon (a-C:H) depositions on high-density polyethylene. Carbon 2016, 96, 661–671. [Google Scholar] [CrossRef]
- Catena, A.; Guo, Q.; Kunze, M.R.; Agnello, S.; Gelardi, F.M.; Wehner, S.; Fischer, C.B. Morphological and Chemical Evolution of Gradually Deposited Diamond-Like Carbon Films on Polyethylene Terephthalate: From Subplantation Processes to Structural Reorganization by Intrinsic Stress Release Phenomena. ACS Appl. Mater. Interfaces 2016, 8, 10636–10646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rohrbeck, M.; Körsten, S.; Fischer, C.B.; Wehner, S.; Kessler, B. Diamond-like carbon coating of a pure bioplastic foil. Thin Solid Films 2013, 545, 558–563. [Google Scholar] [CrossRef]
- Fischer, C.B.; Rohrbeck, M.; Wehner, S.; Richter, M.; Schmeißer, D. Interlayer formation of diamond-like carbon coatings on industrial polyethylene: Thickness dependent surface characterization by SEM, AFM and NEXAFS. Appl. Surf. Sci. 2013, 271, 381–389. [Google Scholar] [CrossRef]
- Kassab, Z.; Kassem, I.; Hannache, H.; Bouhfid, R.; Qaiss, A.E.K.; El Achaby, M. Tomato plant residue as new renewable source for cellulose production: Extraction of cellulose nanocrystals with different surface functionalities. Cellulose 2020, 27, 4287–4303. [Google Scholar] [CrossRef]
- Catena, A.; McJunkin, T.; Agnello, S.; Gelardi, F.M.; Wehner, S.; Fischer, C.B. Surface morphology and grain analysis of successively industrially grown amorphous hydrogenated carbon films (a-C:H) on silicon. Appl. Surf. Sci. 2015, 347, 657–667. [Google Scholar] [CrossRef]
- Catena, A.; Kunze, M.R.; Agnello, S.; Gelardi, F.M.; Wehner, S.; Fischer, C.B. Amorphous hydrogenated carbon (a-C:H) depositions on polyoxymethylene: Substrate influence on the characteristics of the developing coatings. Surf. Coat. Technol. 2016, 307, 658–665. [Google Scholar] [CrossRef]
- Kleinen, L.; Böde, U.; Laube, N. Ex-vivo investigations on the friction behavior of amorphous carbon coated ureteral stents. Diam. Relat. Mater. 2008, 17, 1746–1750. [Google Scholar] [CrossRef]
- Kleinen, L.; Böde, U.; Schenk, K.; Busch, H.; Bradenahl, J.; Müller, S.C.; Hillebrands, B.; Laube, N. Amorphous Carbon Coatings Inhibit Crystalline Biofilm Formation on Urological Implants. Plasma Process. Polym. 2007, 4, S386–S391. [Google Scholar] [CrossRef]
- Aguiar, P.H.L.; Oliveira, É.C.; Cruz, S.A. Modification of clarified polypropylene by oxygen plasma to improve the adhesion of thin amorphous hydrogenated carbon films deposited by plasma enhanced chemical vapor deposition. Polym. Eng. Sci. 2013, 53, 1065–1072. [Google Scholar] [CrossRef]
- Mandolfino, C. Polypropylene surface modification by low pressure plasma to increase adhesive bonding: Effect of process parameters. Surf. Coat. Technol. 2019, 366, 331–337. [Google Scholar] [CrossRef]
- Ozeki, K.; Kobayashi, S.; Hirakuri, K.K.; Aoki, H.; Fukui, Y. Oxygen plasma pre-treatment improves the wear properties of a diamond-like carbon film coated on UHMWPE and PMMA for biomaterials. Biomed. Mater. Eng. 2007, 17, 175–182. [Google Scholar] [PubMed]
- Vishnuvarthanan, M.; Rajeswari, N. Effect of mechanical, barrier and adhesion properties on oxygen plasma surface modified PP. Innov. Food Sci. Emerg. Technol. 2015, 30, 119–126. [Google Scholar] [CrossRef]
- Nefedov, A.; Wöll, C. Advanced Applications of NEXAFS Spectroscopy for Functionalized Surfaces. In Surface Science Techniques; Bracco, G., Holst, B., Eds.; Springer Berlin Heidelberg: Berlin, Heidelberg, Germany, 2013; pp. 277–303. ISBN 978-3-642-34242-4. [Google Scholar]
- Watts, B.; Thomsen, L.; Dastoor, P.C. Methods in carbon K-edge NEXAFS: Experiment and analysis. J. Electron. Spectros. Relat. Phenomena. 2006, 151, 105–120. [Google Scholar] [CrossRef]
- D’Souza, L.; Devi, P.; Kamat, T.; Naik, C.G. Diffuse reflectance infrared fourier transform spectroscopic (DRIFTS) investigation of E. coli, Staphylococcus aureus and Candida albicans. Indian J. Mar. Sci. 2009, 38, 45–51. [Google Scholar]
- Armaroli, T.; Bécue, T.; Gautier, S. Diffuse Reflection Infrared Spectroscopy (Drifts): Application to the in Situ Analysis of Catalysts. Oil Gas Sci. Technol. 2004, 59, 215–237. [Google Scholar] [CrossRef]
- Günzler, H.; Gremlich, H.-U. IR spectroscopy. An Introduction; Wiley-VCH: Weinheim, Germany, 2002; ISBN 3-527-28896-1. [Google Scholar]
- Koidl, P.; Wild, C.; Dischler, B.; Wagner, J.; Ramsteiner, M. Plasma Deposition, Properties and Structure of Amorphous Hydrogenated Carbon Films. Mater. Sci. Forum 1991, 52–53, 41–70. [Google Scholar] [CrossRef]
- El Miri, N.; Abdelouahdi, K.; Zahouily, M.; Fihri, A.; Barakat, A.; Solhy, A.; El Achaby, M. Bio-nanocomposite films based on cellulose nanocrystals filled polyvinyl alcohol/chitosan polymer blend. J. Appl. Polym. Sci. 2015, 132, 1–13. [Google Scholar] [CrossRef]
- Banerjee, D.; Mukherjee, S.; Chattopadhyay, K.K. Controlling the surface topology and hence the hydrophobicity of amorphous carbon thin films. Carbon 2010, 48, 1025–1031. [Google Scholar] [CrossRef]
- Barthlott, W.; Neinhuis, C. Purity of the sacred lotus, or escape from contamination in biological surfaces. Planta 1997, 202, 1–8. [Google Scholar] [CrossRef]
- Feng, L.; Li, S.; Li, Y.; Li, H.; Zhang, L.; Zhai, J.; Song, Y.; Liu, B.; Jiang, L.; Zhu, D. Super-Hydrophobic Surfaces: From Natural to Artificial. Adv. Mater. 2002, 14, 1857–1860. [Google Scholar] [CrossRef]
- Ostrovskaya, L.; Perevertailo, V.; Ralchenko, V.; Dementjev, A.; Loginova, O. Wettability and surface energy of oxidized and hydrogen plasma-treated diamond films. Diam. Relat. Mater. 2002, 11, 845–850. [Google Scholar] [CrossRef]
- Ostrovskaya, L.Y. Studies of diamond and diamond-like film surfaces using XAES, AFM and wetting. Vacuum 2002, 68, 219–238. [Google Scholar] [CrossRef]
- Piazza, F.; Morell, G. Wettability of hydrogenated tetrahedral amorphous carbon. Diam. Relat. Mater. 2009, 18, 43–50. [Google Scholar] [CrossRef]
- Tay, B.K.; Sheeja, D.; Lau, S.P.; Guo, J.X. Study of surface energy of tetrahedral amorphous carbon films modified in various gas plasma. Diam. Relat. Mater. 2003, 12, 2072–2076. [Google Scholar] [CrossRef]
- Paul, R.; Das, S.N.; Dalui, S.; Gayen, R.N.; Roy, R.K.; Bhar, R.; Pal, A.K. Synthesis of DLC films with different sp2/sp3 ratios and their hydrophobic behaviour. J. Phys. D Appl. Phys. 2008, 41, 55309. [Google Scholar] [CrossRef]
- Werder, T.; Walther, J.H.; Jaffe, R.L.; Halicioglu, T.; Koumoutsakos, P. On the Water−Carbon Interaction for Use in Molecular Dynamics Simulations of Graphite and Carbon Nanotubes. J. Phys. Chem. B 2003, 107, 1345–1352. [Google Scholar] [CrossRef]
- Zhou, Y.; Wang, B.; Song, X.; Li, E.; Li, G.; Zhao, S.; Yan, H. Control over the wettability of amorphous carbon films in a large range from hydrophilicity to super-hydrophobicity. Appl. Surf. Sci. 2006, 253, 2690–2694. [Google Scholar] [CrossRef]
- Dietrich, P.M.; Horlacher, T.; Girard-Lauriault, P.-L.; Gross, T.; Lippitz, A.; Min, H.; Wirth, T.; Castelli, R.; Seeberger, P.H.; Unger, W.E.S. Adlayers of dimannoside thiols on gold: Surface chemical analysis. Langmuir 2011, 27, 4808–4815. [Google Scholar] [CrossRef]
- Solomon, J.L.; Madix, R.J.; Stöhr, J. Orientation and absolute coverage of benzene, aniline, and phenol on Ag(110) determined by NEXAFS and XPS. Surf. Sci. 1991, 255, 12–30. [Google Scholar] [CrossRef]
- Tai, F.C.; Lee, S.C.; Wei, C.H.; Tyan, S.L. Correlation between ID⁄IG Ratio from Visible Raman Spectra and sp2/sp3 Ratio from XPS Spectra of Annealed Hydrogenated DLC Film. Mater. Trans. 2006, 47, 1847–1852. [Google Scholar] [CrossRef] [Green Version]
- Brüster, B.; Amozoqueño, C.; Grysan, P.; Peral, I.; Watts, B.; Raquez, J.-M.; Dubois, P.; Addiego, F. Resolving Inclusion Structure and Deformation Mechanisms in Polylactide Plasticized by Reactive Extrusion. Macromol. Mater. Eng. 2017, 302, 1700326. [Google Scholar] [CrossRef]
- Dhez, O.; Ade, H.; Urquhart, S.G. Calibrated NEXAFS spectra of some common polymers. J. Electron. Spectros. Relat. Phenomena. 2003, 128, 85–96. [Google Scholar] [CrossRef]
- Diaz, J.; Monteiro, O.R.; Hussain, Z. Structure of amorphous carbon from near-edge and extended x-ray absorption spectroscopy. Phys. Rev. B 2007, 76, 388. [Google Scholar] [CrossRef]
- Díaz, J.; Anders, S.; Zhou, X.; Moler, E.J.; Kellar, S.A.; Hussain, Z. Analysis of the π* and σ* bands of the x-ray absorption spectrum of amorphous carbon. J. Phys. D Appl. Phys. 2001, 64, 17. [Google Scholar] [CrossRef]
- Stöhr, J. NEXAFS Spectroscopy, 2. print; Springer: Berlin, Germany, 2003; ISBN 3-540-54422-4. [Google Scholar]
- Tomasella, E.; Thomas, L.; Dubois, M.; Meunier, C. Structural and mechanical properties of a-C:H thin films grown by RF-PECVD. Diam. Relat. Mater. 2004, 13, 1618–1624. [Google Scholar] [CrossRef]
- Veres, M.; Koós, M.; Pócsik, I. IR study of the formation process of polymeric hydrogenated amorphous carbon film. Diam. Relat. Mater. 2002, 11, 1110–1114. [Google Scholar] [CrossRef]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schlebrowski, T.; Kassab, Z.; El Achaby, M.; Wehner, S.; Fischer, C.B. Effect of Cellulose Nanocrystals on the Coating of Chitosan Nanocomposite Film Using Plasma-Mediated Deposition of Amorphous Hydrogenated Carbon (a–C:H) Layers. C 2020, 6, 51. https://doi.org/10.3390/c6030051
Schlebrowski T, Kassab Z, El Achaby M, Wehner S, Fischer CB. Effect of Cellulose Nanocrystals on the Coating of Chitosan Nanocomposite Film Using Plasma-Mediated Deposition of Amorphous Hydrogenated Carbon (a–C:H) Layers. C. 2020; 6(3):51. https://doi.org/10.3390/c6030051
Chicago/Turabian StyleSchlebrowski, Torben, Zineb Kassab, Mounir El Achaby, Stefan Wehner, and Christian B. Fischer. 2020. "Effect of Cellulose Nanocrystals on the Coating of Chitosan Nanocomposite Film Using Plasma-Mediated Deposition of Amorphous Hydrogenated Carbon (a–C:H) Layers" C 6, no. 3: 51. https://doi.org/10.3390/c6030051