Abstract
In this article, for the first time the chemical and mechanical properties of novel adsorbents based on the coating of activated carbons with silicon carbide are reported. The adsorbents are prepared by chemical vapor infiltration (CVI) of activated carbons with tetramethylsilane (TMS) as a precursor. A comparison of two different modified types of activated carbon, C40/4 Extra and A35/4 Extra, each infiltrated with 25%-mass at infiltration temperatures of 973.15 and 1098.15 K, respectively, is presented. Adsorption properties were characterized by measuring nitrogen isotherms and volatile organic compounds (VOC) isotherms in gas phase and excess isotherms in liquid phase. In addition, the physico-chemical properties including the bulk density, ash content, particle hardness, abrasion, conductivity, water-soluble components, and pH value were determined. Furthermore, the first experiments in a fluidized bed adsorber are presented. The results show that the adsorption properties of the modified adsorbents are mainly maintained. The particle hardness and the abrasion resistance increases with increasing infiltration temperature, which leads to an overall increasing of mechanical stability. A modification of the chemical stability as a result of the infiltration experiments is not observed.
1. Introduction
Activated carbon is one of the most important adsorbents due to its high porosity, large specific surface area, and chemical properties []. It is used in many technical applications, as gas purification [,], gas separation [], decolourization [,,], and purification of different liquids and solvents [,], including water [,,]. However, activated carbon has also some disadvantages. The low mechanical and chemical stability makes it unsuitable for applications e.g. in fluidized bed adsorbers and in certain liquid adsorption processes. Furthermore, it is known that carbon reacts to form CO and/or CO2 under oxidizing conditions, which can lead to adsorber fires. Therefore, it is a promising option to utilize the properties of ceramics by coating activated carbon with oxidation resistant materials as SiC or SiO2 without significantly changing the pore structure, so that the adsorption capacity is grossly maintained. SiC is characterized by its hardness and chemical inertness []. A common method to produce SiC functionalized materials, which cover a broad range of applications from high voltage and electronic devices to high temperature diffusion barriers, is chemical vapor deposition (CVD) [,,,,,,,,,]. This process may also take place in porous materials, then called chemical vapor infiltration (CVI). It was previously used to modify activated carbons [,]. Moene et al. investigated high surface area silicon carbide catalysts [,]. The functionality of activated carbon by metal oxides prepared by CVD was also shown by Busch et al. []. SiC-adsorbents were derived from the vapor infiltration of activated carbon with silicon chloride [,,]. In all cases, the initial morphology of the template was well adapted or only briefly modified by the infiltration process. However, the usage of silicon chloride as a precursor is often technically undesirable due to the production of HCl, which is quite corrosive, mainly in humid environments. In our former studies, tetramethylsilane (TMS) turned out to be an appropriate alternative as a precursor for the CVI of SiC in activated carbons. The advantages of TMS are a simple handling, moderate costs, and high availability. It was shown that it is possible to modify the properties of activated carbon by SiC coating without significantly altering the adsorption capacity []. Furthermore, the high temperatures resistance in an oxygen atmosphere is increased when compared to uncoated activated carbon [].
The present work presents the characterization of such coated activated carbons helping to judge possible improvements for technical applications. Two different types of activated carbon were infiltrated at 973.15 and 1098.15 K, respectively. Experimental results to characterize physico-chemical and adsorption properties of these modified adsorbents are shown. Experiments in a fluidized bed adsorber demonstrate the modification of mechanical stability.
2. Results and Discussion
The infiltration of TMS leads to a coating of the outer surface and of the inner pore system of the activated carbon. At a coating temperature of 973.15 K, a coating with silicon dioxide (SiO2), while at 1098.15 K, a coating with silicon carbide (SiC) was proved [,], both after contact with air.
The surface morphology on the microscopic scale was analyzed by a high resolution scanning electron microscope (SEM). Figure 1 shows the SEM images of activated carbon C40/4 Extra (a) uncoated, (b) coated with SiO2 and (c) coated with SiC. In contrast to the relatively flat but rough surface of the uncoated C40/4 Extra, the SiO2 coating leads to a smooth and uniform surface coverage with sharp edges. Overall, the deposited film seems to be well adapted to the surface morphology of the carbon. The SiC coated materials have small accumulations of smoothed edged particles distributed over the entire surface. Presumably there is both, film growth and particle agglomeration. A more detailed discussion of surface morphology of uncoated and coated materials can be found in Pflitsch et al. []. In addition, this publication contains a detailed energy dispersive X-ray spectroscopy (EDX) study to investigate the composition of coated and uncoated materials.

Figure 1.
Scanning electron microscopy images of activated carbon C40/4 Extra (a) uncoated, (b) coated with silicon dioxide (SiO2) and (c) coated with silicon carbide (SiC).
Table 1 shows the BET surface area, as well as the pore volume of the activated carbons used. Both carbons C40/4 and A35/4 showed a 35% (973.15 K) and 25% (1073.15 K) decrease in the BET surface area as a result of the coating, respectively. While the pore volume decreased by about 40% in the materials coated at 973.15 K, a decrease of about 30% occurred in the materials coated at 1098.15 K. Since a mass increase of 25% was set for all coating processes, the lower decrease in the pore volume at 1098.15 K leads to the conclusion that at this temperature a larger proportion of the mass increase takes place on the outer surface. This is supported by the fact that the Thiele modulus is larger at 1098.15 K due to the higher temperature. In addition, these coated activated carbons have a silver discoloration, which indicates coating with SiC.

Table 1.
Basic properties of materials.
The pore volume decreased as a result of the coating (Figure 2). However, the decreased pore volume did not result in a shift in the pore sizes. This suggests that the coating of the inner surface mainly results in a partial pore blocking, which closes the part of the pore system behind the blocking C40/4 showed a uniform decrease in the pore volume over the entire pore range, whereas for A35/4, the decrease occurred primarily in pores larger than 1 nm.
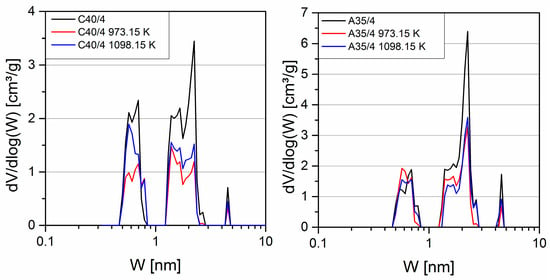
Figure 2.
Pore size distribution by non-localized density functional theory (NLDFT) method.
In addition to the structural properties, adsorption properties were investigated by the adsorption of acetone, ethanol, and toluene. In the representation of the isotherms, the load was normalized to the surface and plotted against relative pressure. Normalization eliminates the influence of differences in surface area on the load so that differences between the isotherms can be attributed to differences in surface chemistry.
Figure 3 shows the adsorption isotherms of the polar solvent acetone at 298.15 K. The load was significantly higher on uncoated A35/4 compared to C40/4. Both carbons were manufactured from the same starting material, but A35/4 was activated in a water vapor atmosphere over a longer period of time. As a result of the longer activation time under oxidizing conditions in the original production process, A35/4 had a higher number of polar surface groups, which favored the adsorption of polar acetone. The loads of the samples coated at 973.15 K are of the same order of magnitude for both types of activated carbons, since the differences in surface properties are reduced as a consequence of the coating of the inner pore system, and the associated modification of surface chemistry. While the load on the coated C40/4 increased in comparison to the uncoated activated carbon due to stronger interactions between the polar SiO2 coating and the polar acetone, load on the polar A35/4 only changed slightly. The samples coated at 1098.15 K also had comparable loads. Because of the non-polar SiC coating and the resulting weaker interactions, the load is below the load at the samples coated at 973.15 K.
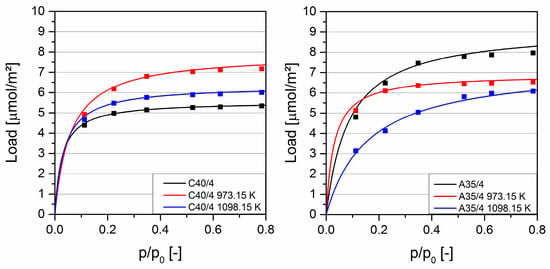
Figure 4 shows the adsorption isotherms of ethanol at 298.15 K. Both course and sequence of the isotherms are comparable to the adsorption measurements of acetone. This can be attributed to the fact that both ethanol and acetone are polar solvents, and thus form comparable interactions with the respective activated carbon. The slightly higher load of ethanol can be attributed to the smaller molecular size and the resulting smaller space demand on the surface.
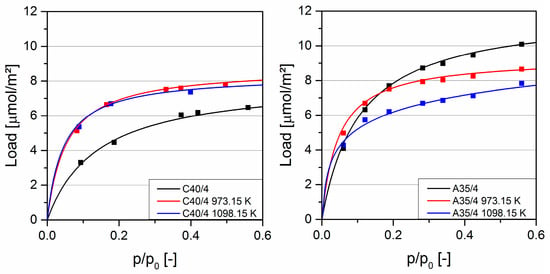
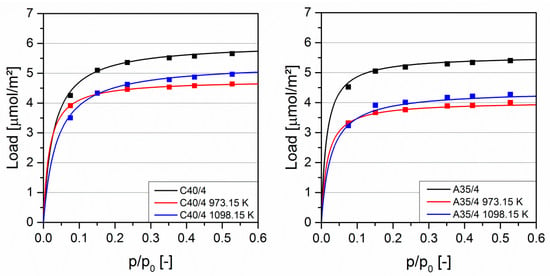
Figure 5 shows the adsorption isotherm of the aromatic toluene at 298.15 K. For both C40/4 and A35/4, the uncoated samples show the highest load, while the samples coated at 973.15 and 1098.15 K have nearly identical loads in both types of activated carbon. In contrast to the adsorption measurements with polar solvents, the comparison of C40/4 and A35/4 shows a very good agreement. The lower load on the coated materials occurs because toluene preferentially adsorbs on aromatic groups which decrease by the coating with polar SiO2 at 973.15 K and non-polar SiC at 1098.15 K. The comparable loads on the coated materials suggest similarly strong interactions between toluene and polar SiO2, as well as toluene and non-polar SiC. Repeated adsorption-desorption showed no change in adsorption capacity for N2 nor for the solvents.
Physico-chemical properties of the coated as well as the uncoated activated carbons are shown in Table 2. The experimental error (standard deviation) of the particle hardness determined from the standard deviation ranged between 20–30%. This large error can be attributed to the marked differences in length and diameter of the pellets, as well as the heterogeneity in porosity, which strongly influenced particle hardness. Other methods have errors in the range of 2–3%, which mainly represents instrumental errors.
Table 2.
Physico-chemical properties of activated carbons.
In the materials coated at 973.15 K, mainly the inner pore volume was coated, so that the mass but not the size of the activated carbon pellets increased. Thus, the bulk density of the coated activated carbons increased by about 25%. As a result of the additional outer coating of the materials coated at 1098.15 K, the size of the pellets increased slightly, whereby bulk density increased less prominently as compared to the materials coated at 973.15 K. The infiltrated silicon increased ash content from approximately 8% for uncoated activated carbons to 20–25% for all coated materials.
The particle hardness of the uncoated carbon C40/4 was 50% higher than the hardness of carbon A35/4. This was attributed to the much stronger porosity of A35/4 and the associated lower mechanical stability. Coating of the inner pore volume at 973.15 K with SiO2 increased particle hardness by about 40 N for both activated carbons. Since the activated carbons were coated at 1098.15 K with much harder SiC, the increase (70 N) was larger than the coating formed at 973.15 K. Another important property is abrasion stability. While particle hardness describes the resistance to an external force on the stationary particle, abrasion represents a measure for the mass loss as a result of friction between moving particles. For uncoated materials, the abrasion of A35/4 (about 9%) was about three times higher than of C40/4 (about 3%). While abrasion of activated carbons coated at 973.15 K exhibited only a slight increase, abrasion of samples coated at 1098.15 K decreased significantly below 2% for A35/4 and below 1% for C40/4. The significant reduction in abrasion was attributed to the coating of the outer pellet surface, which led to an encapsulation of the activated carbon pellets.
Figure 6 shows time-dependent abrasion in fluidized bed tests of uncoated C40/4 and C40/4 coated at 1098.15 K. For each measurement, 100 g of activated carbon was weighed. Comparable flow conditions with a Reynolds number of Re ≈ 900 (250–270 liters per minute, see Table 2) were used in the experiments. For the uncoated activated carbon, abrasion increased sharply within the first 30 min and then continued to increase constantly with time. Presumably, the strong initial abrasion was due to the dust particles being removed from the pellet surface. While abrasion of the uncoated activated carbon was 40 g after 6 h, abrasion of the coated activated carbon was as below 3 g; an abrasion reduction of more than 90%. This result strongly confirms the presumption of an encapsulation of the surface by forming an outer SiC layer.
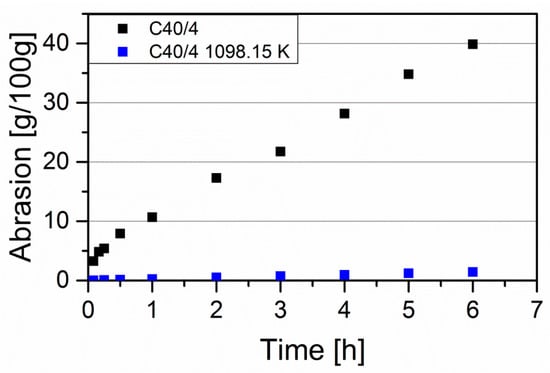
Figure 6.
Fluidized bed abrasion tests of uncoated C40/4 and C40/4 coated at 1098.15 K (Table S5).
The differences in the results with the abrasion tester PTF10ER and in the fluidized bed are clearly due to the different experimental conditions. Using the test device, abrasion occurred by rubbing activated carbon pellets on the drum surface. In contrast, the fluidized bed abrasion was caused by a collision of activated carbon pellets with each other and with the tubes. Furthermore, due to high volume flows, a higher velocity in the fluidized bed resulted, which in turn led to stronger mechanical impacts.
Encapsulation of the outer carbon surface of the material coated at 1098.15 K was also evident from the amounts of water-soluble components and from conductivity (Table 2). Compared to the uncoated carbons, the amount of water-soluble components and the conductivity were significantly lower.
At a coating temperature of 973.15 K, no encapsulation of the surface occurred. When compared to the uncoated materials, conductivity as well as the amount of water-soluble components increased, possibly due to partial hydrolysis of the infiltrated SiO2 yielding silicic acids.
Figure 7 shows the excess isotherms of the system acetone-toluene at 298.15 K. Toluene excess is plotted against the mole fraction of toluene. The more mesoporous carbon A35/4 was chosen because it should have faster kinetics in liquid phase adsorption experiments.
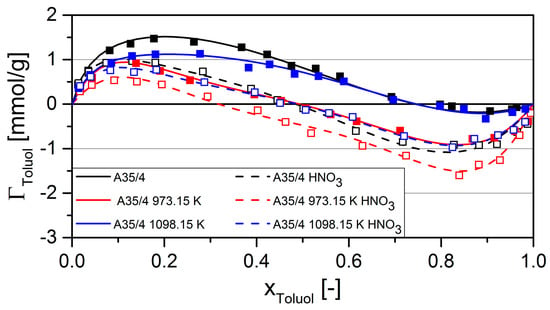
The activated carbon A35/4 exhibited preferred adsorption of the aromatic toluene over a wide range, with an azeotropic point at a toluene mole fraction of about 0.75. This simply means that the ratio of aromatic to polar functional groups was about 3:1. As a result of the coating with polar SiO2, the azeotropic point of the activated carbon coated at 973.15 K shifted to 0.5. This implied a decrease of ratio of aromatic to polar functional groups and a lower selectivity for toluene. This can also be seen from the solvent isotherms (see Figure 3 and Figure 5), where—compared to the uncoated material—the capacity of the activated carbon coated at 973.15 K decreased by 25% for acetone, and by 33% for toluene. Coating obviously decreased the number of aromatic groups more than the number of polar groups.
The excess isotherm of the activated carbon coated at 1098.15 K showed only a very slight shift to smaller mole fractions when compared to the uncoated activated carbon. This may be explained by coating with non-polar SiC, which results in a similar reduction in aromatic and polar groups, so that the ratio remains nearly constant. As less aromatic and polar groups are available, and thus weaker interactions occur, the adsorption excess decreases.
For all activated carbons, the azeotropic point in the curves of materials treated with HNO3 shifted to the left, which indicated a more polar surface. There was no different shift of uncoated and coated activated carbons. Thus, the surface chemistry seems to change by oxidation with HNO3 in a similar way. Under the given experimental conditions, an improved chemical resistance of the coated activated carbons could not be detected. Since a very aggressive treatment with HNO3 was carried out, more experiments are required in the future, which cover a broader range of temperature, exposure time, or concentration of HNO3 in order to check the generality of this finding.
3. Materials and Methods
The activated carbon C40/4 Extra and A35/4 Extra from CarboTech AC GmbH were chosen as base substrates for the infiltration experiments in the present study. They are commercially available materials for air purification and solvent recovery. In our experiments we used cylindrical pellets with a diameter of 4 mm with a BET surface area of 1227 m2/g (C40/4) and 1443 m2/g (A35/4) and a large pore volume 0.659 cm3/g (C40/4) respectively 0.867 cm3/g (A35/4).
3.1. Coating
The precursor TMS was purchased from Merck KGaA with a purity of 99.7%. The activated carbons were infiltrated at 973.15 and 1098.15 K with TMS in a horizontal hot wall CVD reactor consisting of a cylindrical ceramic tube with an inner diameter of 60 mm and a heated length of 950 mm placed in a single zone furnace. The reactor pressure was 2 hPa. For degassing the activated carbons, they were preheated at 150 °C for 3 h in a nitrogen atmosphere to remove the initial load of water of about 5% (mass). The carbon pellets were moved during infiltration by rotating the sample holder. The sample holder had 36 holes of 1 mm diameter in order to allow for gas flow towards the pellets. It was positioned in the center of the tube furnace and rotated by an electrical motor. The infiltration procedure was as follows: the reactor was heated up to the infiltration temperature while a nitrogen flow of 100 sccm (standard cubic centimeters per minute at 1.013 bar and 273.15 K) provided an inert atmosphere inside of the ceramic tube. A stream of 50-sccm nitrogen (purity: 99.99%) was saturated with TMS vapor in a bubbler at a temperature of 293.15 K, and the mixture fed to the reactor. An additional nitrogen flow of 50 sccm was added in order to provide the desired precursor gas phase concentration. After the infiltration experiments, the samples were taken out of the furnace and cooled down in an inert gas atmosphere. They were subsequently analyzed after getting into contact with the laboratory atmosphere, as detailed described elsewhere [,,].
3.2. Characterization
The aim of the infiltration experiments was to produce activated carbons with an improved mechanical and chemical resistance while maintaining the adsorption properties. Accordingly, mechanical and chemical properties were investigated in addition to the structural and adsorption properties. To characterize the uncoated and coated activated carbons, nitrogen isotherms were measured at 77 K using the BELSORB-max (BEL JAPAN, Inc., Tokyo, Japan). The specific surface area was determined using the BET method according to DIN ISO 9277 and the pore volume by the Gurvich method at a relative pressure of p/p0 = 0.98 []. The activated carbon pore size distribution (PSD) was calculated using the non-localized density functional theory (NLDFT) []. The influence of different interactions with the surface and molecules of different sizes on adsorption was investigated by gravimetric adsorption measurements of acetone, ethanol, and toluene with the thermogravimetric balance (TGA) STA 449 F3 Jupiter® of NETZSCH-Gerätebau GmbH (Selb, Germany) at 298.15 K.
Bulk density, ash content, particle hardness, abrasion, conductivity, water-soluble components and pH-value were investigated using standardized measuring methods. Bulk density was determined according to ASTM D 854-96 (American Society for Testing and Materials, West Conshohocken, PA, USA), and ash content according to ASTM D 2866-94 (American Society for Testing and Materials).
Particle hardness, defined as the force required to break a test specimen, was measured with a tablet hardness tester (TBH 225; ERWEKA GmbH, Heusenstamm, Germany). In contrast to tablets, the activated carbon pellets used in this study have substantial fluctuations in diameter and length. This leads to higher uncertainties in the determination of the particle hardness. In order to reduce these variations analyses were repeated 30 times.
Abrasion was determined using the friability tester (PTF10ER; Pharma Testapparatebau AG, Hainburg, Germany). Specifically, 100 mL of the activated carbon was weighed and filled into the drum of the apparatus. The drum was rotated at 50 revolutions per minute. After two and a half hours, the activated carbon was reweighed. Abrasion represents the ratio of the mass difference between the initial and the back weighed mass, referred to the initial mass. Water-soluble components were measured according to ASTM D 5029-3 (American Society for Testing and Materials), conductivity according to ASTM D 1125-14 (American Society for Testing and Materials), and pH value according to ASTM D 3838-80 (American Society for Testing and Materials).
In order to investigate the applicability of the modified activated carbon in moving bed systems, studies were carried out on the stability of the coated pellets in a fluidized bed. The experimental setup consisted of a cylindrical reactor with a diameter of 8 cm and a second tube with a diameter of 4 cm, which was placed inside of the reactor. To generate a fluidized bed, 100 g of activated carbon was charged into the reactor. After filling, a nitrogen stream was passed through the reactor from the bottom to the top. The activated carbon rose in the inner tube and was led down through the gap between the two tubes. The velocity of the gas stream was adjusted to allow the activated carbon pellets to circulate within the reactor without being discharged. Collisions between the activated carbon pellets among themselves and with the tubes exert mechanical stress. To test the mechanical resistance of the pellets, abrasion was measured as a function of time.
Another goal of the work was to use the modified activated carbon in processes with reactive media. Exemplarily, the chemical resistance against oxidizing nitric acid (HNO3) was investigated. The activated carbon was boiled in HNO3 at 353.15 K for three hours. After washing and drying the pellets, excess isotherms were measured with treated and untreated pellets []. Excess isotherms of a binary mixture of two adsorptives of different functionality can be used to characterize the ratio of different bonding sites in liquid phase adsorption. On activated carbon surfaces, these sites can be classified into aromatic, aliphatically polar, and aliphatic non-polar bonding sites [].
To measure an excess isotherm, the starting concentration of the two components was varied over the entire concentration range and activated carbon was added to each mixture. The samples were shaken in sealed shaking flasks until adsorption equilibrium was attained. The excess of component i Γei,exp was calculated from the concentration difference of this component due to adsorption.
Where n0 is the initial amount of component i, mAC the mass of activated carbon, x0i the initial mole fraction of component i without adsorption and xbi the equilibrium mole fraction of component i in the bulk phase. If both components adsorb in different amounts on the activated carbon surface, an excess of one component is measured. If the excess is positive, the component i is preferably adsorbed. In case of a negative excess, component i adsorbs less compared to the second component. A detailed description of the method is described elsewhere [].
Assuming that the adsorptives selectively interact with the surface groups fitting to their molecular functionalities, the adsorption behavior can be interpreted to characterize the distribution of surface groups. A characteristic feature of the excess isotherm is the azeotropic point at which the isotherm intersects the x-axis. There, the range of the preference of one component changes into the range of the preference of the other component.
If the surface of the adsorbent changes as a result of oxidation with HNO3, the concentration of polar groups will rise at the expense of the concentration of aromatic and aliphatic groups. Then, the excess of polar molecules as compared to aromatic or aliphatic molecules should increase. Consequently, a binary mixture of the polar adsorptive acetone and the aromatic adsorptive toluene was selected. We compare untreated samples with HNO3-treated samples of coated and non-coated materials to find out whether coating alters the oxidation resistance.
4. Conclusions
Using the chemical vapor infiltration of ceramic materials in commercially available activated carbons C40/4 and A35/4 from CarboTech (Essen, Germany), novel adsorbents were developed. Many properties of these materials were analyzed for the first time. The infiltration experiments were performed with TMS at 973.15 and 1098.15 K, and a constant mass increase of 25% was set. The activated carbons were coated with SiO2 at 973.15 K and SiC at 1098.15 K after air contact. The new materials were characterized with respect to their adsorption properties and their mechanical as well as chemical resistance, in order to evaluate their use in technical applications. Several improvements of the material due to the coating were shown, making them promising for technical adsorption processes.
The adsorption measurements showed a slight reduction of the BET surface and the pore system, as well as a modification of the surface chemistry of the coated activated carbons.
Particle hardness increased markedly in all coated materials since a higher stability was achieved. While abrasion of the materials coated at 973.15 K increased slightly as compared to the uncoated materials, the materials coated at 1098.15 K showed a significant decrease in abrasion. This suggests that additional to the inner pore system, the outer surface of the pellets were also coated with a stable SiC layer.
The comparison of excess isotherms showed a pronounced left shift between untreated materials and materials treated with HNO3. This indicates an oxidation of the surface for all treated materials. Thus, an improved resistance to the treatment with HNO3 could not be detected for the coated materials. It is possible that the oxidative conditions in the test were too aggressive. More experiments are required with a broader range of temperature, exposure time, or concentration of HNO3. In the future, the materials coated at 1098.15 K will be tempered at high temperatures to convert amorphous SiC into crystalline SiC, which may feature better mechanical and chemical resistance.
Supplementary Materials
The following are available online at http://www.mdpi.com/2311-5629/3/3/27/s1.
Acknowledgments
The financial support by the German Research Foundation (DFG) within the DFG project BA2012/4-3 and AT24/16-3 is gratefully acknowledged.
Author Contributions
Burak Atakan and Dieter Bathen conceived and designed the experiments; Christian Bläker and Stefanie Heib performed the experiments; Christian Bläker, Stefanie Heib and Christoph Pasel analyzed the data; and, Christian Bläker, Stefanie Heib and Christoph Pasel wrote the paper.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Sircar, S.; Golden, T.C.; Rao, M.B. Activated carbon for gas separation and storage. Carbon 1996, 34, 1–12. [Google Scholar] [CrossRef]
- Rivera-Utrilla, J.; Sánchez-Polo, M.; Gómez-Serrano, V.; Álvarez, P.M.; Alvim-Ferraz, M.; Dias, J.M. Activated carbon modifications to enhance its water treatment applications. An overview. J. Hazard. Mater. 2011, 187, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Raso, R.A.; Zeltner, M.; Stark, W.J. Indoor Air Purification Using Activated Carbon Adsorbers: Regeneration Using Catalytic Combustion of Intermediately Stored VOC. Ind. Eng. Chem. Res. 2014, 53, 19304–19312. [Google Scholar] [CrossRef]
- Grande, C.A.; Blom, R.; Möller, A.; Möllmer, J. High-pressure separation of CH4/CO2 using activated carbon. Chem. Eng. Sci. 2013, 89, 10–20. [Google Scholar] [CrossRef]
- Nunes, D.L.; Oliveira, L.S.; Franca, A.S. Melanoidin Removal Mechanism in an Aqueous Adsorption System: An Equilibrium, Kinetic and Thermodynamic Study. Recent Pat. Food Nutr. Agric. 2015, 7, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Simaratanamongkol, A.; Thiravetyan, P. Decolorization of melanoidin by activated carbon obtained from bagasse bottom ash. J. Food Eng. 2010, 96, 14–17. [Google Scholar] [CrossRef]
- López, F.; Medina, F.; Prodanov, M.; Güell, C. Oxidation of activated carbon: Application to vinegar decolorization. J. Colloid Interface Sci. 2003, 257, 173–178. [Google Scholar] [CrossRef]
- Onuki, S.; Koziel, J.A.; Jenks, W.S.; Cai, L.; Rice, S.; van Leeuwen, J. Ethanol purification with ozonation, activated carbon adsorption, and gas stripping. Sep. Purif. Technol. 2015, 151, 165–171. [Google Scholar] [CrossRef]
- Juang, R.-S.; Lin, S.-H.; Cheng, C.-H. Liquid-phase adsorption and desorption of phenol onto activated carbons with ultrasound. Ultrason. Sonochem. 2006, 13, 251–260. [Google Scholar] [CrossRef] [PubMed]
- Bhatnagar, A.; Hogland, W.; Marques, M.; Sillanpää, M. An overview of the modification methods of activated carbon for its water treatment applications. Chem. Eng. J. 2013, 219, 499–511. [Google Scholar] [CrossRef]
- Sakoda, A.; Nomura, T.; Suzuki, M. Activated carbon membrane for water treatments: Application to decolorization of coke furnace wastewater. Adsorption 1997, 3, 93–98. [Google Scholar] [CrossRef]
- Hanigan, D.; Zhang, J.; Herckes, P.; Krasner, S.W.; Chen, C.; Westerhoff, P. Adsorption of N-Nitrosodimethylamine Precursors by Powdered and Granular Activated Carbon. Environ. Sci. Technol. 2012, 46, 12630–12639. [Google Scholar] [CrossRef]
- Choi, B.J.; Kim, D.R. Growth of silicon carbide by chemical vapour deposition. J. Mater. Sci. Lett. 1991, 10, 860–862. [Google Scholar] [CrossRef]
- Henry, A.; ul Hassan, J.; Bergman, J.P.; Hallin, C.; Janzén, E. Thick Silicon Carbide Homoepitaxial Layers Grown by CVD Techniques. Chem. Vap. Depos. 2006, 12, 475–482. [Google Scholar] [CrossRef]
- Pedersen, H.; Leone, S.; Kordina, O.; Henry, A.; Nishizawa, S.-I.; Koshka, Y.; Janzén, E. Chloride-Based CVD Growth of Silicon Carbide for Electronic Applications. Chem. Rev. 2012, 112, 2434–2453. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.W.; Qiu, H.P.; Jiao, J.; Li, X.Q.; Wang, Y.; Xie, W.J. Thermodynamic Analysis of Chemical Vapor Deposition Progress for SiC Coatings. Key Eng. Mater. 2015, 633, 183–188. [Google Scholar] [CrossRef]
- Danielsson, Ö.; Forsberg, U.; Henry, A.; Janzén, E. Enlarging the Usable Growth Area in a Hot-Wall Silicon Carbide CVD Reactor by Using Simulation. Mater. Sci. Forum 2001, 353–356, 99–102. [Google Scholar] [CrossRef]
- Wellmann, P.; Pons, M. Editorial: Silicon Carbide CVD for Electronic Device Applications. Chem. Vap. Depos. 2006, 12, 463–464. [Google Scholar] [CrossRef]
- Moene, R.; Makkee, M.; Moulijn, J.A. Novel application of catalysis in the synthesis of catalysts. Catal. Lett. 1995, 34, 285–291. [Google Scholar] [CrossRef]
- Lee, Y.-J.; Choi, D.-J.; Park, J.-Y.; Hong, G.-W. The effect of diluent gases on the growth behavior of CVD SiC films with temperature. J. Mater. Sci. Lett. 2000, 35, 4519–4526. [Google Scholar] [CrossRef]
- Qiang, X.; Li, H.; Zhang, Y.; Wang, Z.; Ba, Z.; Zhang, X. Mechanical and oxidation protective properties of SiC nanowires-toughened SiC coating prepared in-situ by a CVD process on C/C composites. Surf. Coat. Technol. 2016, 307, 91–98. [Google Scholar] [CrossRef]
- Wang, C.; Huang, N.; Zhuang, H.; Zhai, Z.; Yang, B.; Liu, L.; Jiang, X. Growth of large-scale heteroepitaxial 3C-SiC films and nanosheets on silicon substrates by microwave plasma enhanced CVD at higher powers. Surf. Coat. Technol. 2016, 299, 96–103. [Google Scholar] [CrossRef]
- Walasek, E.; Jonas, S.; Stapinski, T.; Kluska, S.; Czyżewska, A. Chemical Vapour Infiltration on SiC in Porous Graphite Materials. Key Eng. Mater. 2001, 206–213, 567–570. [Google Scholar] [CrossRef]
- Liu, Y.; Men, J.; Feng, W.; Cheng, L.; Zhang, L. Catalyst-free growth of SiC nanowires in a porous graphite substrate by low pressure chemical vapor infiltration. Ceram. Int. 2014, 40, 11889–11897. [Google Scholar] [CrossRef]
- Moene, R.; Kramer, L.F.; Schoonman, J.; Makkee, M.; Moulijn, J.A. Synthesis of high surface area silicon carbide by fluidized bed chemical vapour deposition. Appl. Catal. A 1997, 162, 181–191. [Google Scholar] [CrossRef]
- Busch, M.; Kompch, A.; Suleiman, S.; Notthoff, C.; Bergmann, U.; Theissmann, R.; Atakan, B.; Winterer, M. NOx conversion properties of a novel material: Iron nanoparticles stabilized in carbon. Appl. Catal. B 2015, 166–167, 211–216. [Google Scholar] [CrossRef]
- Xie, Z.; Tao, D.; Wang, J. Synthesis of Silicon Carbide Nanotubes by Chemical Vapor Deposition. J. Nanosci. Nanotechnol. 2007, 7, 647–652. [Google Scholar] [CrossRef] [PubMed]
- Pflitsch, C.; Curdts, B.; Helmich, M.; Pasel, C.; Notthoff, C.; Bathen, D.; Atakan, B. Chemical vapor infiltration of activated carbon with tetramethylsilane. Carbon 2014, 79, 28–35. [Google Scholar] [CrossRef]
- Curdts, B.; Pflitsch, C.; Pasel, C.; Helmich, M.; Bathen, D.; Atakan, B. Novel silica-based adsorbents with activated carbon structure. Microporous Mesoporous Mater. 2015, 210, 202–205. [Google Scholar] [CrossRef]
- Curdts, B.; Helmich, M.; Pasel, C.; Bathen, D.; Atakan, B.; Pflitsch, C. Upscaling the Chemical Vapor Infiltration Process of Activated Carbon with TMS: EUROCVD 19—Nineteenth European Conference on Chemical Vapor Deposition. Phys. Procedia 2013, 46, 248–254. [Google Scholar] [CrossRef]
- Gregg, S.J.; Sing, K.S.W. Adsorption, Surface Area and Porosity, 2nd ed.; Acad Pr: London, UK, 1982. [Google Scholar]
- Lastoskie, C.; Gubbins, K.E.; Quirke, N. Pore size distribution analysis of microporous carbons: A density functional theory approach. J. Phys. Chem. 1993, 97, 4786–4796. [Google Scholar] [CrossRef]
- Treese, J.; Pasel, C.; Luckas, M.; Bathen, D. Chemical Surface Characterization of Activated Carbons by Adsorption Excess of Probe Molecules. Chem. Eng. Technol. 2016, 39, 1144–1150. [Google Scholar] [CrossRef]
- Bathen, D.; Graef, T.; Pasel, C. Adsorptive Entfernung von Monophenolethern aus primären und sekundären Alkoholen mit Aktivkohlen. Chem. Ing. Tech. 2009, 81, 1921–1930. [Google Scholar] [CrossRef]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).