
Supramolecular Self-Assembly of a Model Hydrogelator: Characterization of Fiber Formation and Morphology

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
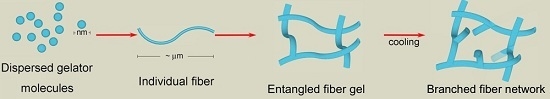
1. Introduction
2. Results and Discussion
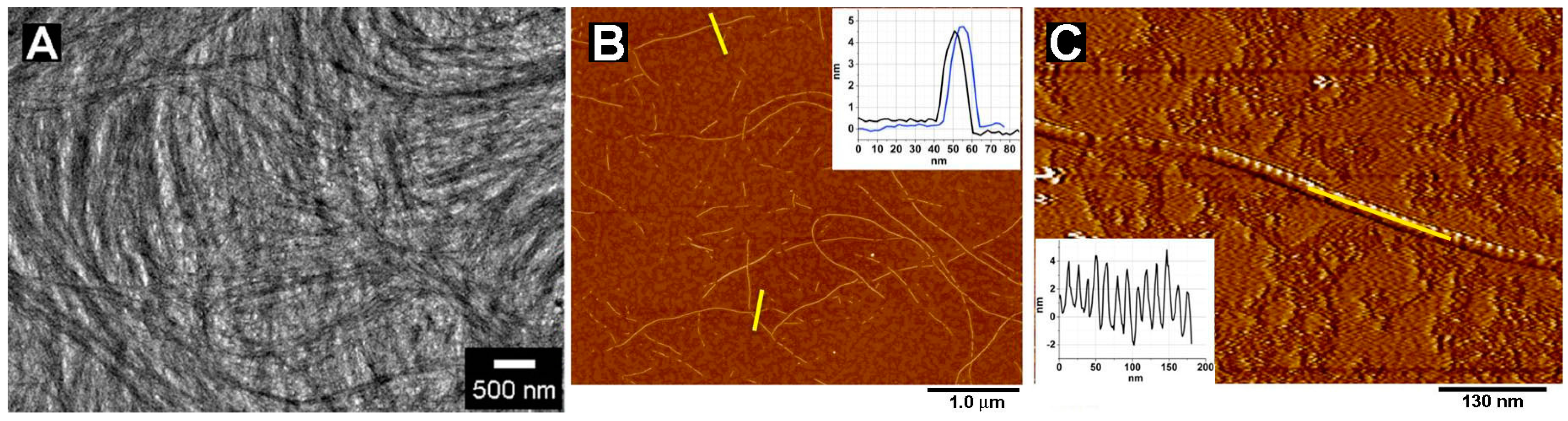
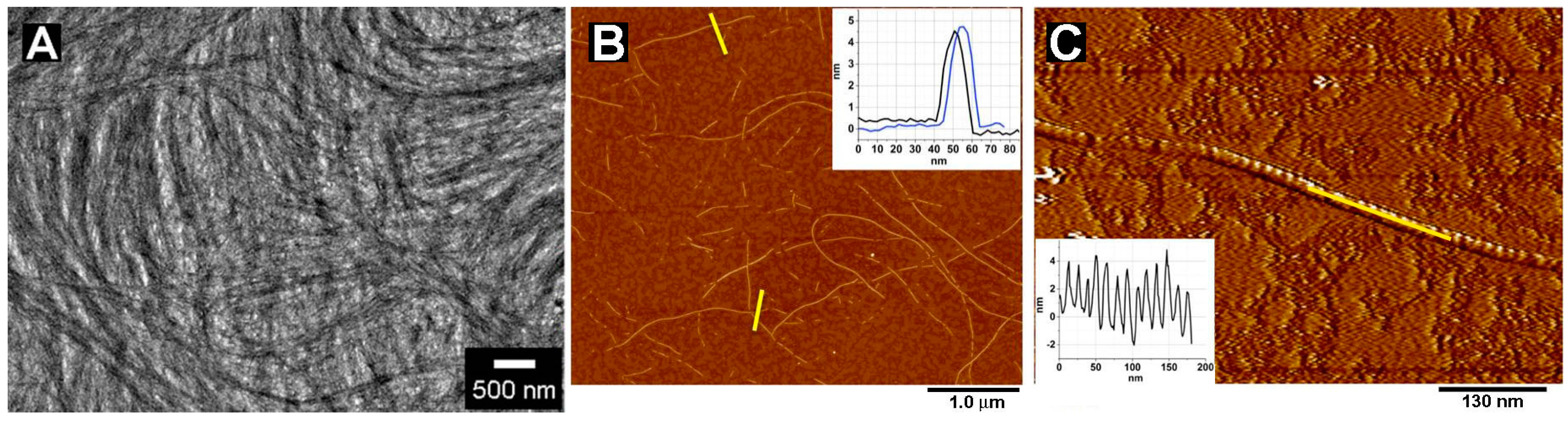
2.1. Morphology of Nanofibers
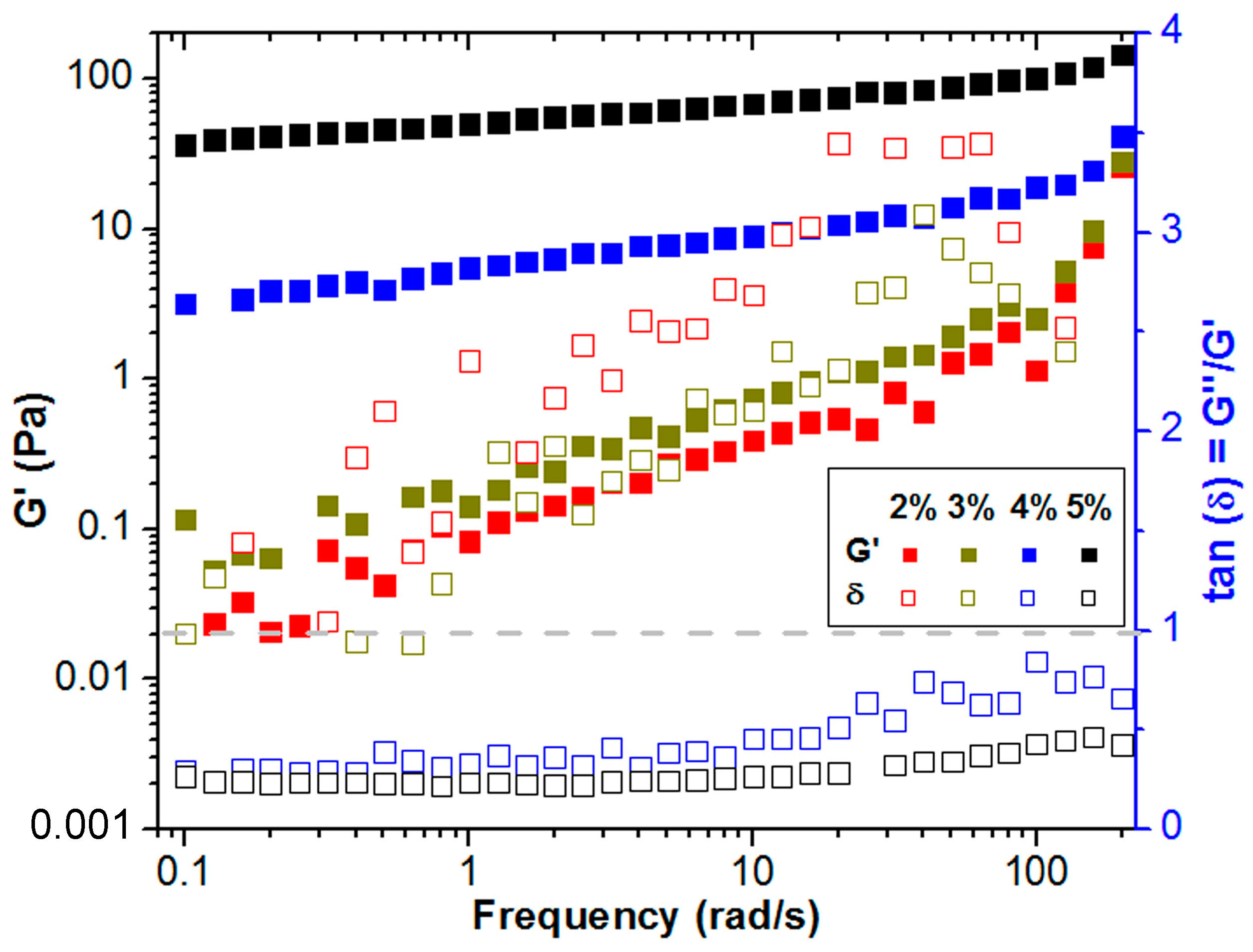
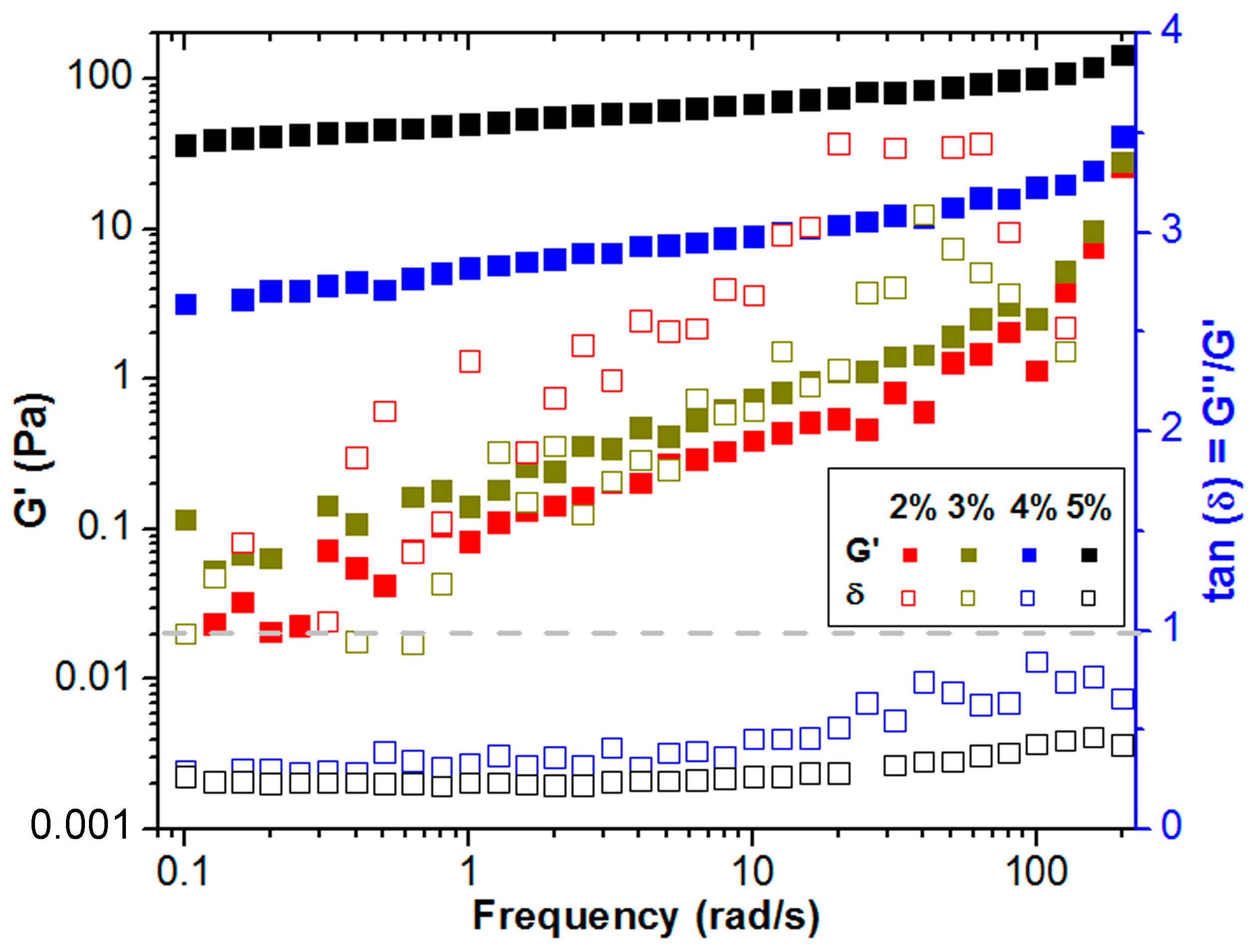
2.2. Characterization of the Rheological Behavior
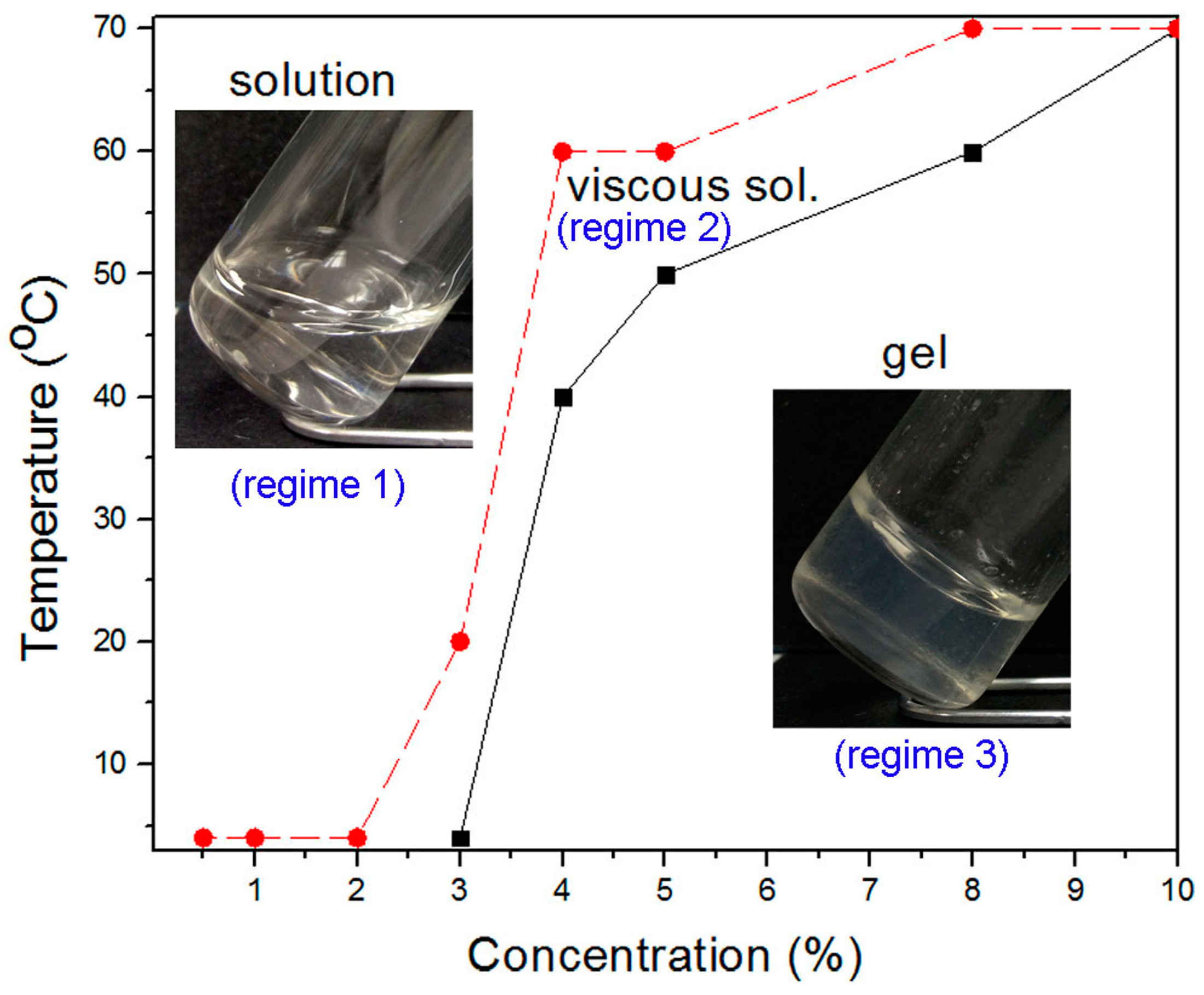
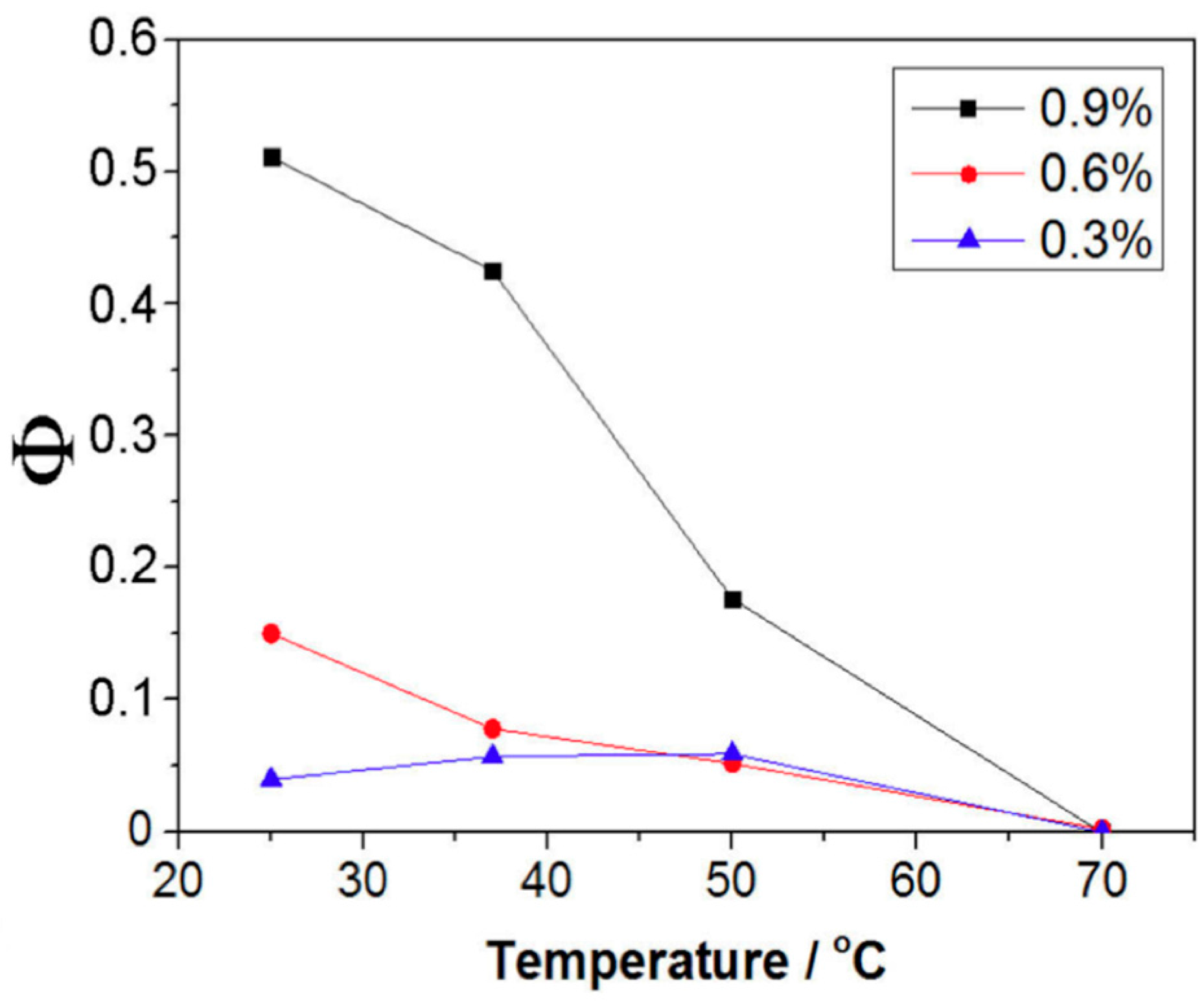
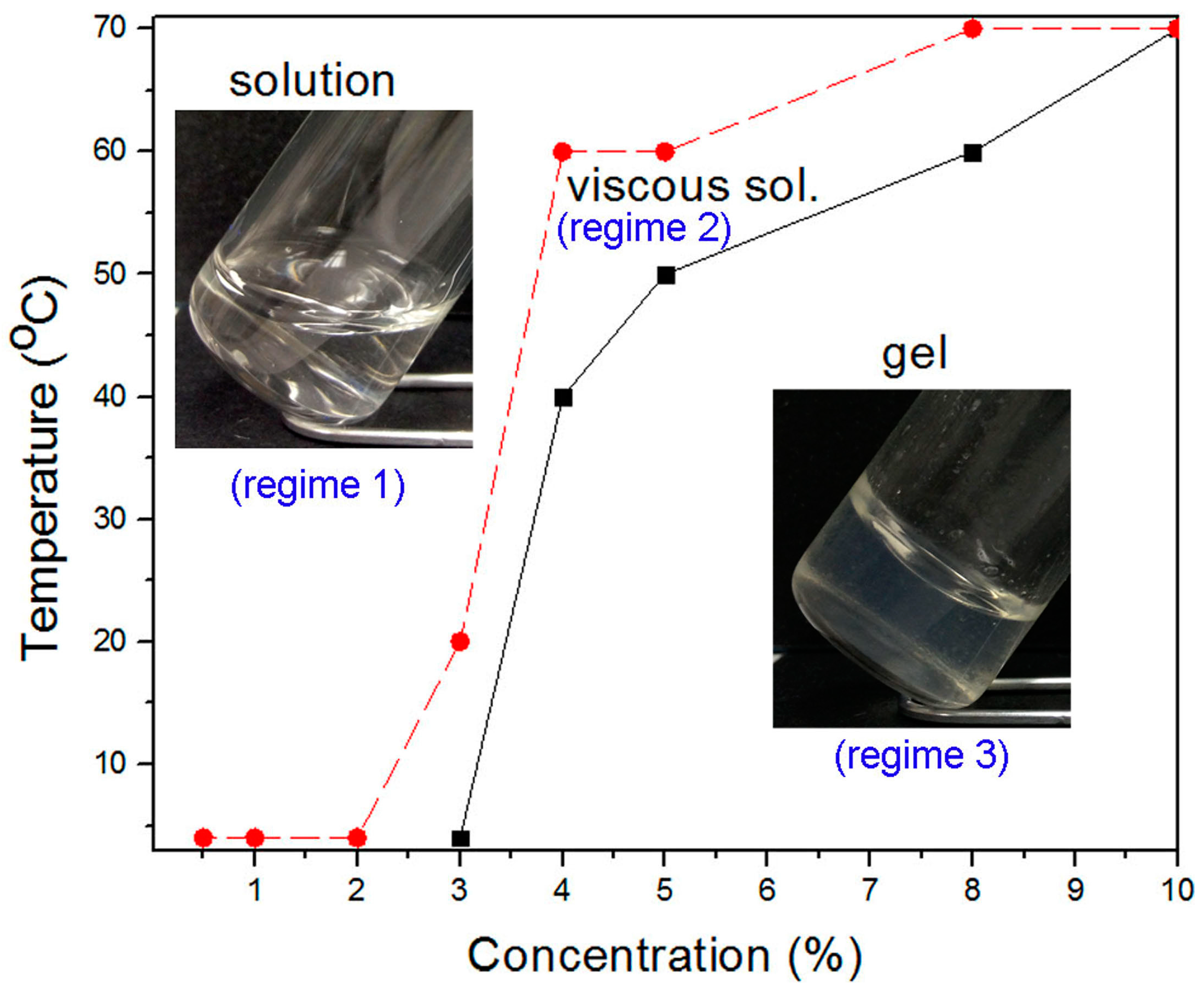
2.3. Characterization of Thermodynamic State
2.4. Rheological Characterization of Gelation Transition
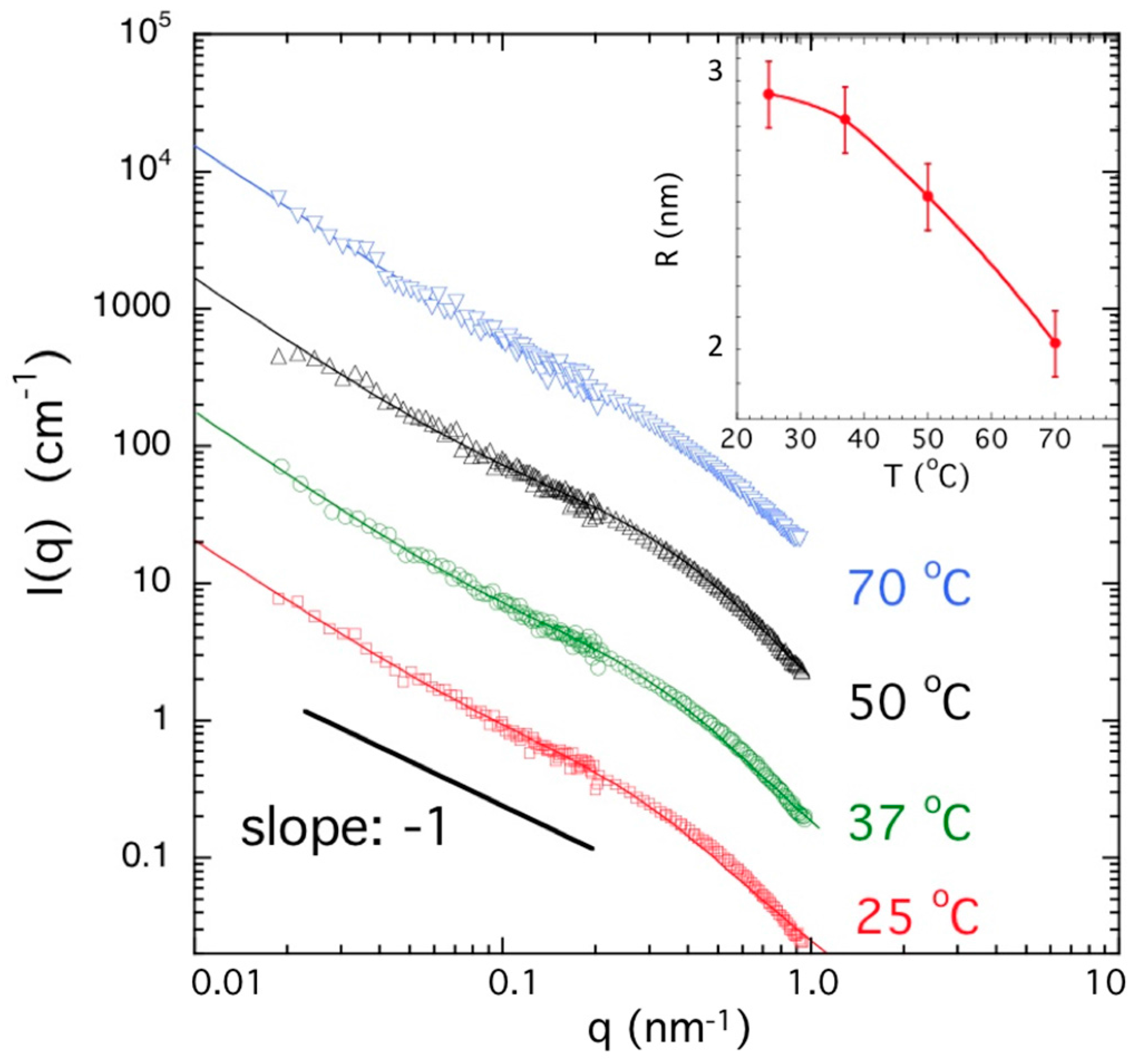
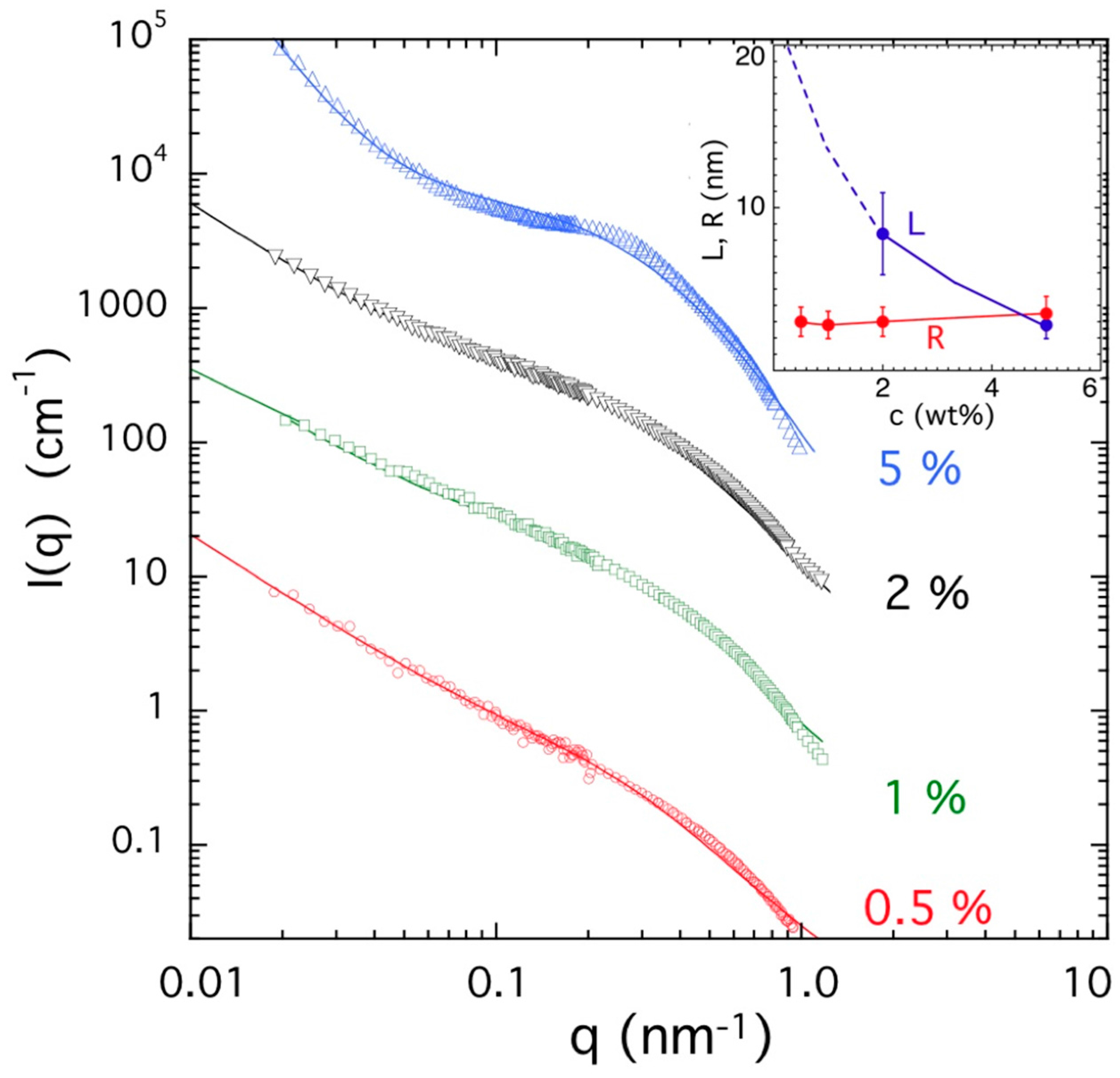
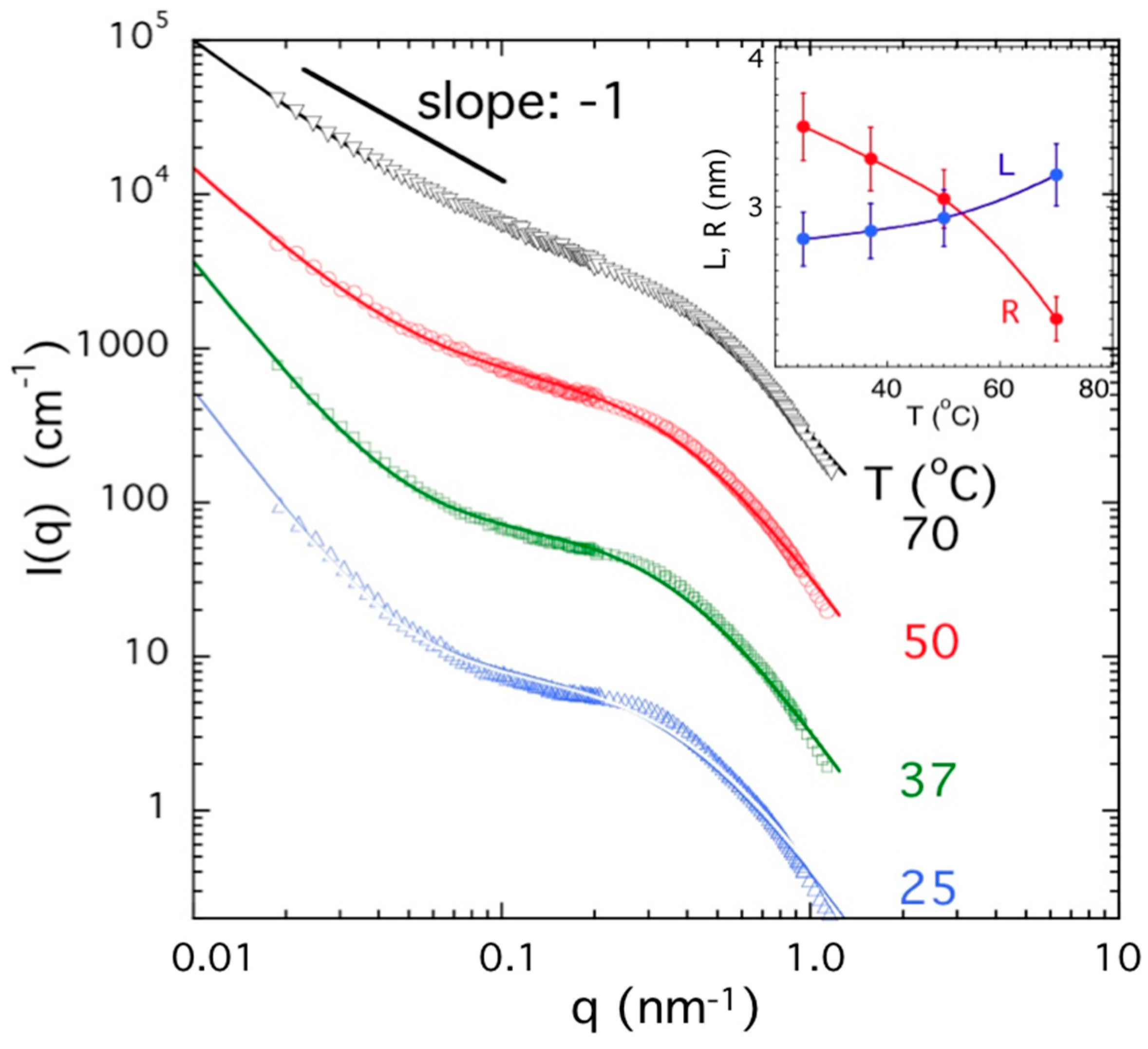
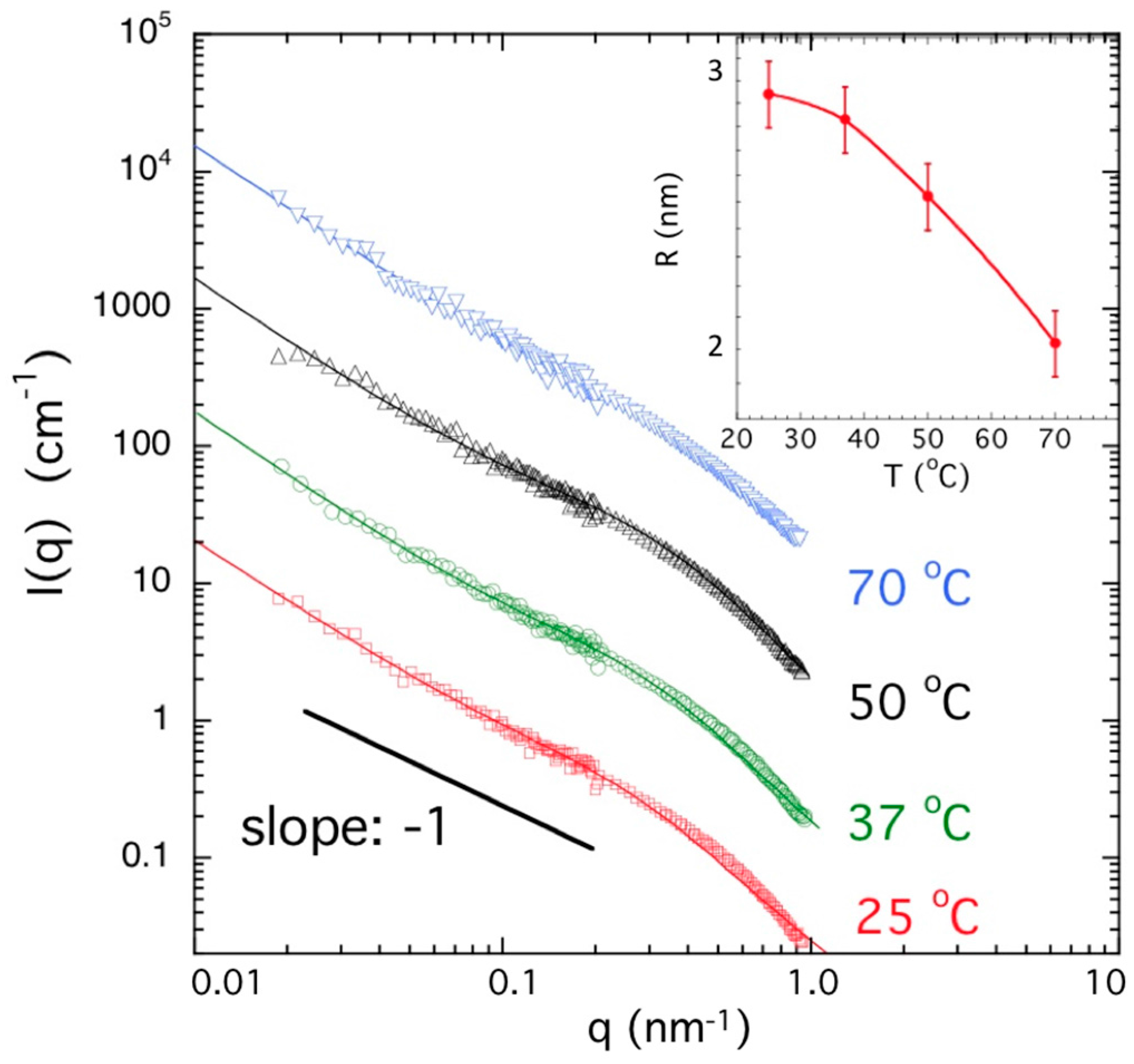
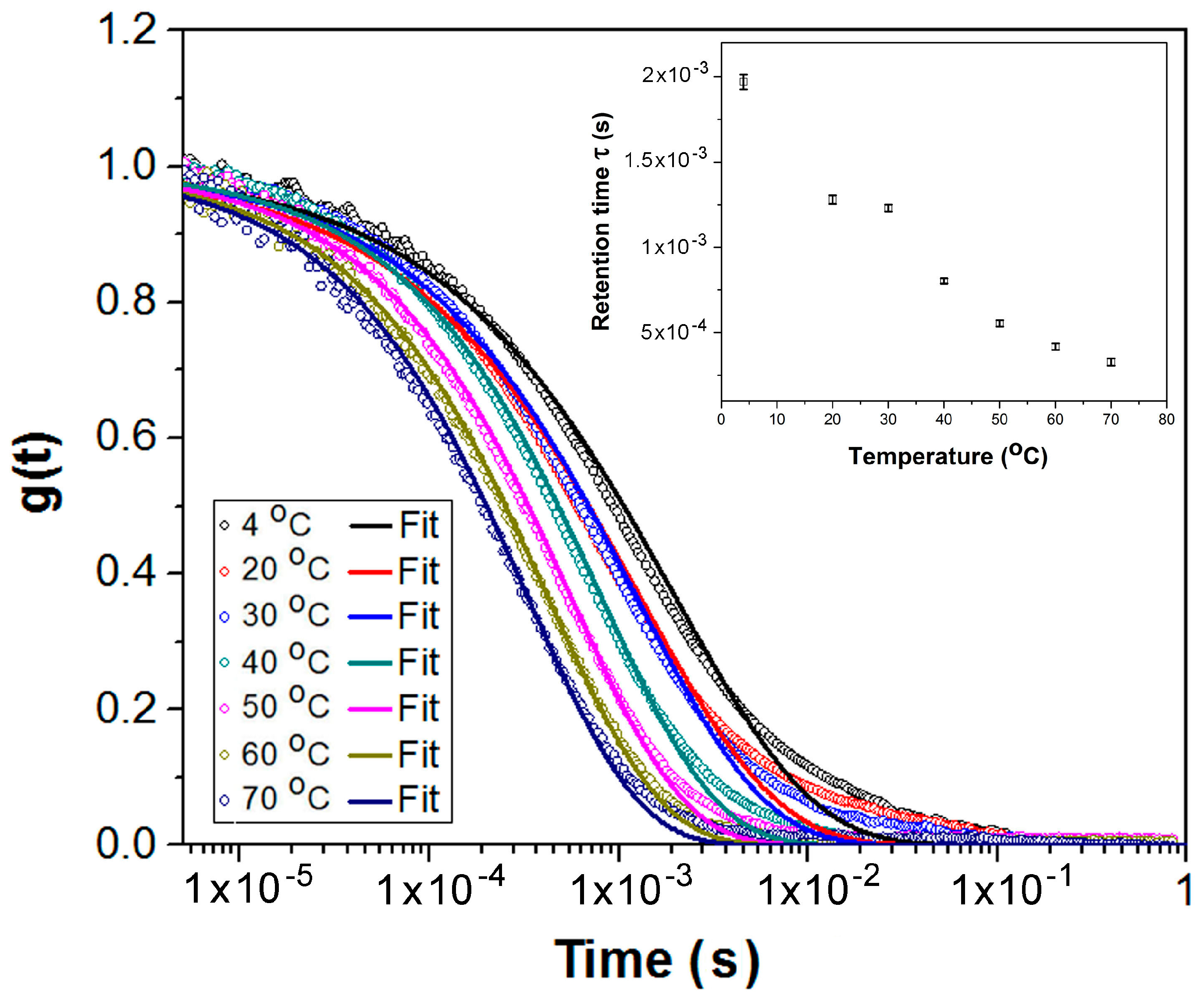
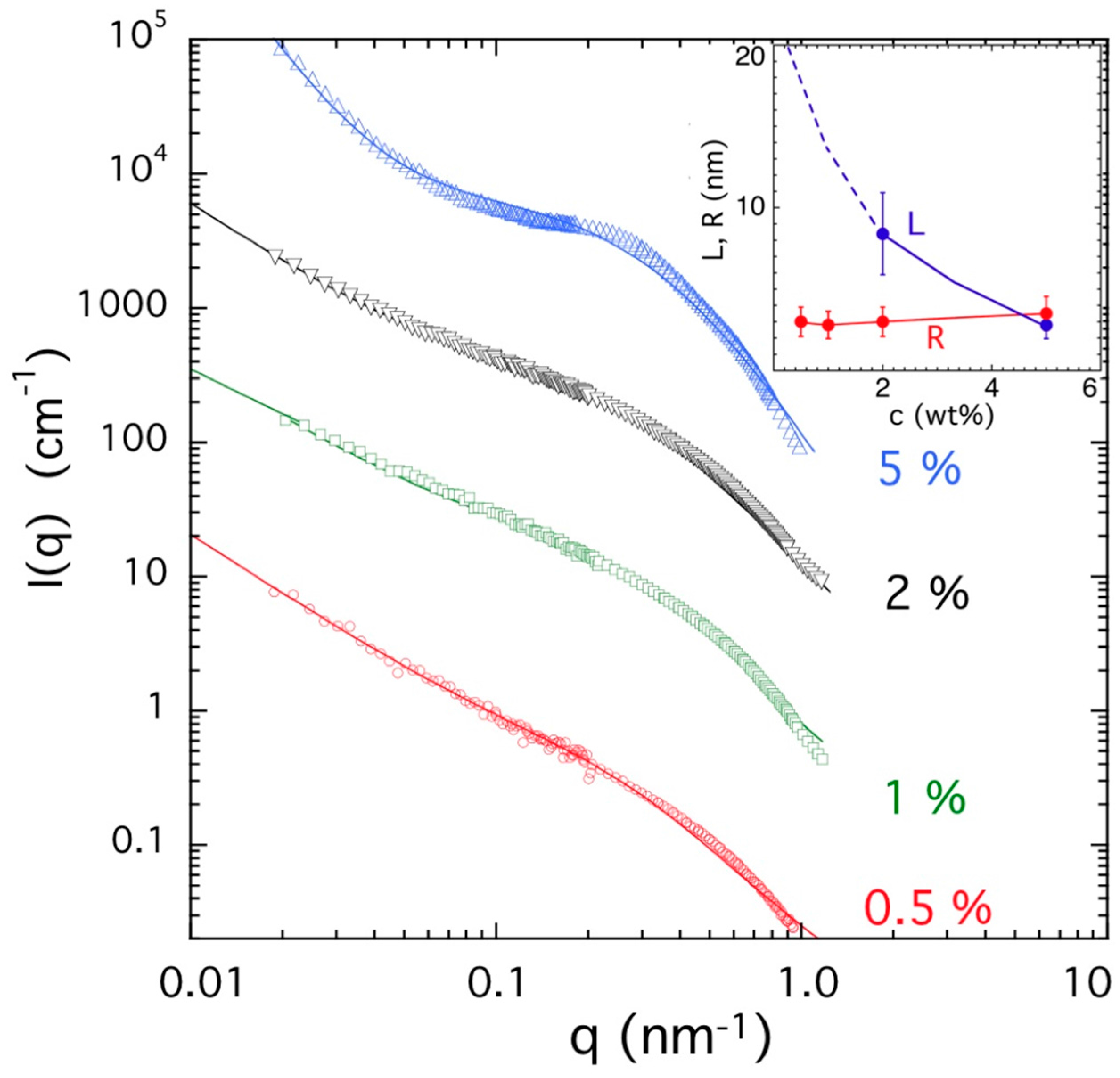
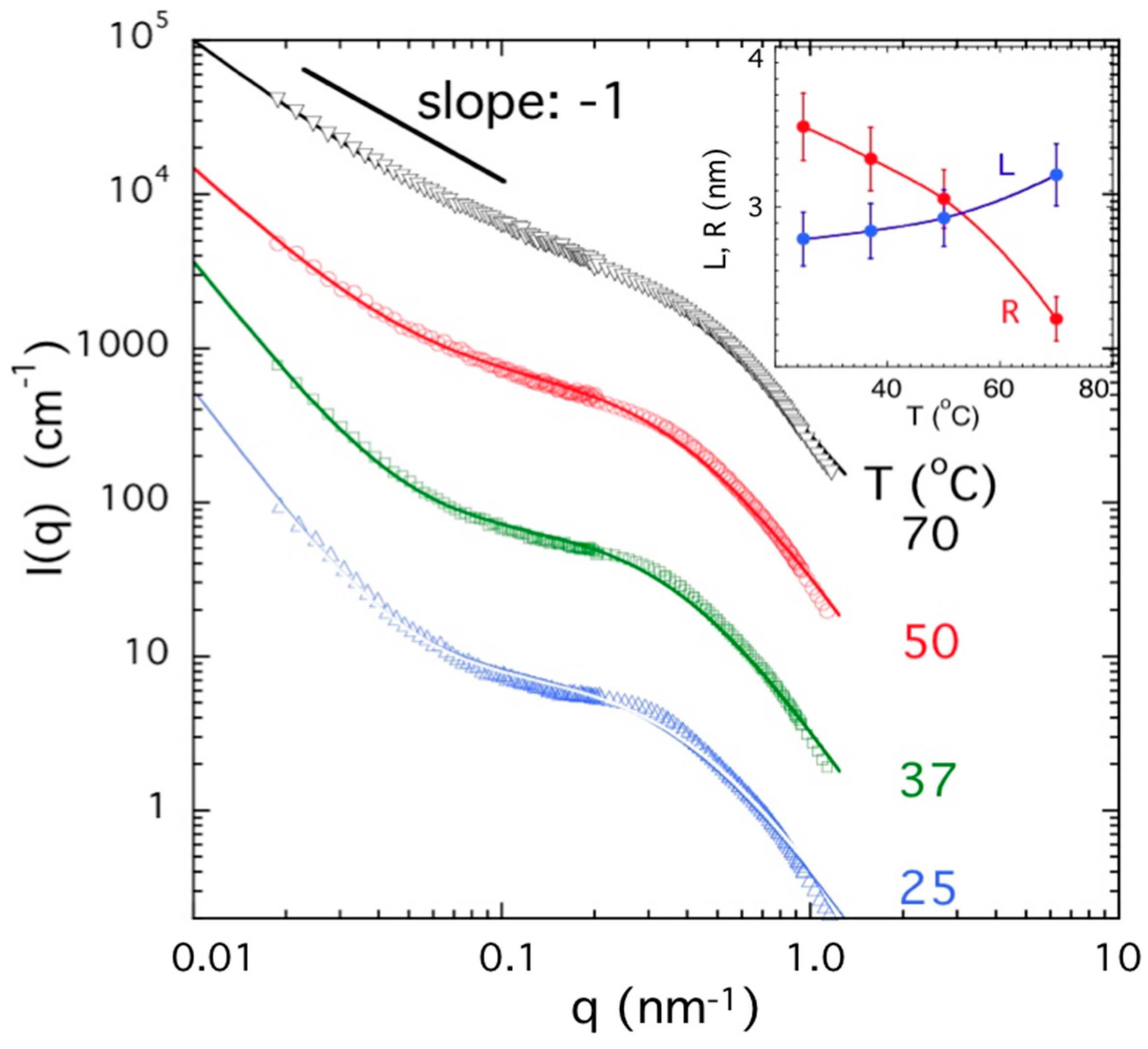
2.5. Neutron and Light Scattering Characterization of Sol and Gel States
3. Conclusions
4. Experimental
4.1. Peptide Synthesis
4.2. Hydrogel Preparation
4.3. Measurement Methods
4.3.1. Proton NMR
4.3.2. Dynamic Light Scattering (DLS)
4.3.3. Small Angle Neutron Scattering (SANS)
4.3.4. Transmission Electron Microscopy (TEM)
4.3.5. Atomic Force Microscopy (AFM)
4.3.6. Oscillatory Rheological Measurements
Acknowledgments
Author Contributions
Conflicts of Interest
References and Note
- Lehn, J.M. Supramolecular Chemistry—Scope and Perspectives Molecules, Supermolecules, and Molecular Devices. Angew. Chem. Int. Ed. 1988, 27, 89–112. [Google Scholar] [CrossRef]
- Roy, N.; Bruchmann, B.; Lehn, J.M. DYNAMERS: Dynamic Polymers as Self-healing Materials. Chem. Soc. Rev. 2015, 44, 3786–3807. [Google Scholar] [CrossRef] [PubMed]
- Chiappisi, L.; Simon, M.; Gradzielski, M. Toward Bioderived Intelligent Nanocarriers for Controlled Pollutant Recovery and pH-Sensitive Binding. ACS Appl. Mater. Interfaces 2015, 7, 6139–6145. [Google Scholar] [CrossRef] [PubMed]
- Caballero-Casero, N.; Ocak, M.; Ocak, U.; Rubio, S. Quick Supramolecular Solvent-based Microextraction for Quantification of Low Curcuminoid Content in Food. Anal. Bioanal. Chem. 2014, 406, 2179–2187. [Google Scholar] [CrossRef] [PubMed]
- Bae, Y.; Fukushima, S.; Harada, A.; Kataoka, K. Design of Environment-sensitive Supramolecular Assemblies for Intracellular Drug Delivery: Polymeric Micelles that are Responsive to Intracellular pH Change. Angew. Chem. Int. Ed. 2003, 42, 4640–4643. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Long, M.J.C.; Shi, J.F.; Hedstrom, L.; Xu, B. Using Supramolecular Hydrogels to Discover the Interactions between Proteins and Molecular Nanofibers of Small Molecules. Chem. Commun. 2012, 48, 8404–8406. [Google Scholar] [CrossRef] [PubMed]
- Estroff, L.A.; Hamilton, A.D. Water Gelation by Small Organic Molecules. Chem. Rev. 2004, 104, 1201–1217. [Google Scholar] [CrossRef] [PubMed]
- George, M.; Weiss, R.G. Molecular Organogels: Soft Matter Comprised of Low-molecular-mass Organic Gelators and Organic Liquids. Acc. Chem. Res. 2006, 39, 489–497. [Google Scholar] [CrossRef] [PubMed]
- Weiss, R.G. The Past, Present, and Future of Molecular Gels: What Is the Status of the Field, and Where Is It Going? J. Am. Chem. Soc. 2014, 136, 7519–7530. [Google Scholar] [CrossRef] [PubMed]
- Silva, G.A.; Czeisler, C.; Niece, K.L.; Beniash, E.; Harrington, D.A.; Kessler, J.A.; Stupp, S.I. Selective Differentiation of Neural Progenitor Cells by High-epitope Density Nanofibers. Science 2004, 303, 1352–1355. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.G. Fabrication of Novel Biomaterials Through Molecular Self-assembly. Nat. Biotechnol. 2003, 21, 1171–1178. [Google Scholar] [CrossRef] [PubMed]
- Hirst, A.R.; Escuder, B.; Miravet, J.F.; Smith, D.K. High-Tech Applications of Self-Assembling Supramolecular Nanostructured Gel-Phase Materials: From Regenerative Medicine to Electronic Devices. Angew. Chem. Int. Ed. 2008, 47, 8002–8018. [Google Scholar] [CrossRef] [PubMed]
- Dankers, P.Y.W.; Harmsen, M.C.; Brouwer, L.A.; van Luyn, M.J.A.; Meijer, E.W. A Modular and Supramolecular Approach to Bioactive Scaffolds for Tissue Engineering. Nat. Mater. 2005, 4, 568–574. [Google Scholar] [CrossRef] [PubMed]
- Mart, R.J.; Osborne, R.D.; Stevens, M.M.; Ulijn, R.V. Peptide-based Stimuli-responsive Biomaterials. Soft Matter 2006, 2, 822–835. [Google Scholar] [CrossRef]
- Banerjee, S.; Das, R.K.; Maitra, U. Supramolecular Gels ‘in action’. J. Mater. Chem. 2009, 19, 6649–6687. [Google Scholar] [CrossRef]
- Yan, C.Q.; Pochan, D.J. Rheological Properties of Peptide-based Hydrogels for Biomedical and Other Applications. Chem. Soc. Rev. 2010, 39, 3528–3540. [Google Scholar] [CrossRef] [PubMed]
- Chow, D.; Nunalee, M.L.; Lim, D.W.; Simnick, A.J.; Chilkoti, A. Peptide-based Biopolymers in Biomedicine and Biotechnology. Mater. Sci. Eng. R Rep. 2008, 62, 125–155. [Google Scholar] [CrossRef] [PubMed]
- George, M.; Weiss, R.G. Molecular Gels; Weiss, G.R., Terech, P., Eds.; Springer: Dordrecht, The Netherlands, 2006; pp. 449–553. [Google Scholar]
- Caran, K.; Lee, D.-C.; Weiss, R. Molecular Gels and Their Fibrillar Networks. In Soft Fibrillar Materials: Fabrication and Applications; Liu, X.Y., Li, J.-L., Eds.; Wiley-VCH: Weinheim, Germany, 2013; pp. 3–75. [Google Scholar]
- Gao, Y.; Kuang, Y.; Guo, Z.F.; Guo, Z.H.; Krauss, I.J.; Xu, B. Enzyme-Instructed Molecular Self-assembly Confers Nanofibers and a Supramolecular Hydrogel of Taxol Derivative. J. Am. Chem. Soc. 2009, 131, 13576–13577. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Shi, J.F.; Yuan, D.; Xu, B. Imaging enzyme-triggered self-assembly of small molecules inside live cells. Nat. Commun. 2012, 3, 1033. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Berciu, C.; Kuang, Y.; Shi, J.F.; Nicastro, D.; Xu, B. Probing Nanoscale Self-Assembly of Nonfluorescent Small Molecules inside Live Mammalian Cells. ACS Nano 2013, 7, 9055–9063. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Du, X.W.; Gao, Y.; Shi, J.F.; Xu, B. Aromatic-Aromatic Interactions Enhance Interfiber Contacts for Enzymatic Formation of a Spontaneously Aligned Supramolecular Hydrogel. J. Am. Chem. Soc. 2014, 136, 2970–2973. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.; Gao, Y.; Lin, J.; Hu, H.; Liao, H.S.; Yan, X.F.; Tang, Y.X.; Jin, A.; Song, J.B.; Niu, G.; et al. Tumor-Specific Formation of Enzyme-Instructed Supramolecular Self-Assemblies as Cancer Theranostics. ACS Nano 2015, 9, 9517–9527. [Google Scholar] [CrossRef] [PubMed]
- Raghavan, S.R.; Douglas, J.F. The Conundrum of Gel Formation by Molecular Nanofibers, Wormlike Micelles, and Filamentous Proteins: Gelation without Cross-links? Soft Matter 2012, 8, 8539–8546. [Google Scholar] [CrossRef]
- Nagarkar, R.P.; Hule, R.A.; Pochan, D.J.; Schneider, J.P. De novo Design of Strand-swapped Beta-hairpin Hydrogels. J. Am. Chem. Soc. 2008, 130, 4466–4474. [Google Scholar] [CrossRef] [PubMed]
- Carretti, E.; Bonini, M.; Dei, L.; Berrie, B.H.; Angelova, L.V.; Baglioni, P.; Weiss, R.G. New Frontiers in Materials Science for Art Conservation: Responsive Gels and Beyond. Acc. Chem. Res. 2010, 43, 751–760. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Kuang, Y.; Du, X.W.; Zhou, J.; Chandran, P.; Horkay, F.; Xu, B. Imaging Self-Assembly Dependent Spatial Distribution of Small Molecules in a Cellular Environment. Langmuir 2013, 29, 15191–15200. [Google Scholar] [CrossRef] [PubMed]
- Hirst, A.R.; Coates, I.A.; Boucheteau, T.R.; Miravet, J.F.; Escuder, B.; Castelletto, V.; Hamley, I.W.; Smith, D.K. Low-molecular-weight Gelators: Elucidating the Principles of Gelation Based on Gelator Solubility and a Cooperative Self-assembly Model. J. Am. Chem. Soc. 2008, 130, 9113–9121. [Google Scholar] [CrossRef] [PubMed]
- Van Workum, K.; Douglas, J.F. Equilibrium Polymerization in the Stockmayer Fluid as a Model of Supermolecular Self-organization. Phys. Rev. E 2005, 71, 031502. [Google Scholar] [CrossRef] [PubMed]
- Dudowicz, J.; Freed, K.F.; Douglas, J.F. Lattice Model of Equilibrium Polymerization. IV. Influence of Activation, Chemical Initiation, Chain Scission and Fusion, and Chain Stiffness on Polymerization and Phase Separation. J. Chem. Phys. 2003, 119, 12645–12666. [Google Scholar] [CrossRef]
- Sasselli, I.R.; Halling, P.J.; Ulijn, R.V.; Tuttle, T. Supramolecular Fibers in Gels Can Be at Thermodynamic Equilibrium: A Simple Packing Model Reveals Preferential Fibril Formation versus Crystallization. ACS Nano 2016, 10, 2661–2668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roos, W.H.; Bruinsma, R.; Wuite, G.J.L. Physical Virology. Nat. Phys. 2010, 6, 733–743. [Google Scholar] [CrossRef]
- Pochan, D.J.; Schneider, J.P.; Kretsinger, J.; Ozbas, B.; Rajagopal, K.; Haines, L. Thermally Reversible Hydrogels via Intramolecular Folding and Consequent Self-assembly of a de Novo Designed Peptide. J. Am. Chem. Soc. 2003, 125, 11802–11803. [Google Scholar] [CrossRef] [PubMed]
- Ozbas, B.; Kretsinger, J.; Rajagopal, K.; Schneider, J.P.; Pochan, D.J. Salt-triggered Peptide Folding and Consequent Self-assembly into Hydrogels with Tunable Modulus. Macromolecules 2004, 37, 7331–7337. [Google Scholar] [CrossRef]
- Schneider, J.P.; Pochan, D.J.; Ozbas, B.; Rajagopal, K.; Pakstis, L.; Kretsinger, J. Responsive Hydrogels from the Intramolecular Folding and Self-assembly of a Designed Peptide. J. Am. Chem. Soc. 2002, 124, 15030–15037. [Google Scholar] [CrossRef] [PubMed]
- Ozbas, B.; Rajagopal, K.; Schneider, J.P.; Pochan, D.J. Semiflexible Chain Networks Formed via Self-assembly of Beta-hairpin Molecules. Phys. Rev. Lett. 2004, 93, 268106. [Google Scholar] [CrossRef] [PubMed]
- Greer, S.C. Physical Chemistry of Equilibrium Polymerization. J. Phys. Chem. B 1998, 102, 5413–5422. [Google Scholar] [CrossRef]
- Niranjan, P.S.; Yim, P.B.; Forbes, J.G.; Greer, S.C.; Dudowicz, J.; Freed, K.F.; Douglas, J.F. The Polymerization of Actin: Thermodynamics Near the Polymerization Line. J. Chem. Phys. 2003, 119, 4070–4084. [Google Scholar] [CrossRef]
- George, M.; Tan, G.; John, V.T.; Weiss, R.G. Urea and Thiourea Derivatives as Low Molecular-mass Organogelators. Chemistry 2005, 11, 3243–3254. [Google Scholar] [CrossRef] [PubMed]
- Yucel, T.; Micklitsch, C.M.; Schneider, J.P.; Pochan, D.J. Direct Observation of Early-time Hydrogelation in Beta-hairpin Peptide Self-assembly. Macromolecules 2008, 41, 5763–5772. [Google Scholar] [CrossRef] [PubMed]
- Douglas, J.F. Theoretical Issues Relating to Thermally Reversible Gelation by Supermolecular Fiber Formation. Langmuir 2009, 25, 8386–8391. [Google Scholar] [CrossRef] [PubMed]
- Stukalin, E.B.; Douglas, J.F.; Freed, K.F. Multistep Relaxation in Equilibrium Polymer Solutions: A Minimal Model of Relaxation in “Complex” Fluids. J. Chem. Phys. 2008, 129, 094901. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Tanaka, T. Phase-Transitions of Gels. Annu. Rev. Mater. Sci. 1992, 22, 243–277. [Google Scholar] [CrossRef]
- Horkay, F.; Basser, P.J.; Hecht, A.M.; Geissler, E. Chondroitin Sulfate in Solution: Effects of Mono- and Divalent Salts. Macromolecules 2012, 45, 2882–2890. [Google Scholar] [CrossRef] [PubMed]
- Ottinger, E.A.; Shekels, L.L.; Bernlohr, D.A.; Barany, G. Synthesis of Phosphotyrosine-Containing Peptides and Their Use as Substrates for Protein-Tyrosine Phosphatases. Biochemistry 1993, 32, 4354–4361. [Google Scholar] [CrossRef] [PubMed]
- Certain commercial equipment or suppliers are identified in this paper to specify our procedures adequately. Such identification does not imply recommendation or endorsement by the National Institute of Standards and Technology, nor does it imply that the material or equipment identified are necessarily the best available for the purpose.
- Horkay, F.; Hammouda, B. Small-angle Neutron Scattering from Typical Synthetic and Biopolymer Solutions. Colloid Polym. Sci. 2008, 286, 611–620. [Google Scholar] [CrossRef]
- Hule, R.A.; Nagarkar, R.P.; Hammouda, B.; Schneider, J.P.; Pochan, D.J. Dependence of Self-Assembled Peptide Hydrogel Network Structure on Local Fibril Nanostructure. Macromolecules 2009, 42, 7137–7145. [Google Scholar] [CrossRef] [PubMed]
- Horkay, F.; Hecht, A.M.; Mallam, S.; Geissler, E.; Rennie, A.R. Macroscopic and Microscopic Thermodynamic Observations in Swollen Poly(Vinyl Acetate) Networks. Macromolecules 1991, 24, 2896–2902. [Google Scholar] [CrossRef]


© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, Y.; Nieuwendaal, R.; Dimitriadis, E.K.; Hammouda, B.; Douglas, J.F.; Xu, B.; Horkay, F. Supramolecular Self-Assembly of a Model Hydrogelator: Characterization of Fiber Formation and Morphology. Gels 2016, 2, 27. https://doi.org/10.3390/gels2040027
Gao Y, Nieuwendaal R, Dimitriadis EK, Hammouda B, Douglas JF, Xu B, Horkay F. Supramolecular Self-Assembly of a Model Hydrogelator: Characterization of Fiber Formation and Morphology. Gels. 2016; 2(4):27. https://doi.org/10.3390/gels2040027
Chicago/Turabian StyleGao, Yuan, Ryan Nieuwendaal, Emilios K. Dimitriadis, Boualem Hammouda, Jack F. Douglas, Bing Xu, and Ferenc Horkay. 2016. "Supramolecular Self-Assembly of a Model Hydrogelator: Characterization of Fiber Formation and Morphology" Gels 2, no. 4: 27. https://doi.org/10.3390/gels2040027