

The Impact of Ar or N2 Atmosphere on the Structure of Bi-Fe-Carbon Xerogel Based Composites as Electrode Material for Detection of Pb2+ and H2O2
 , , , and
, , , and
Abstract
:1. Introduction
2. Results and Discussion
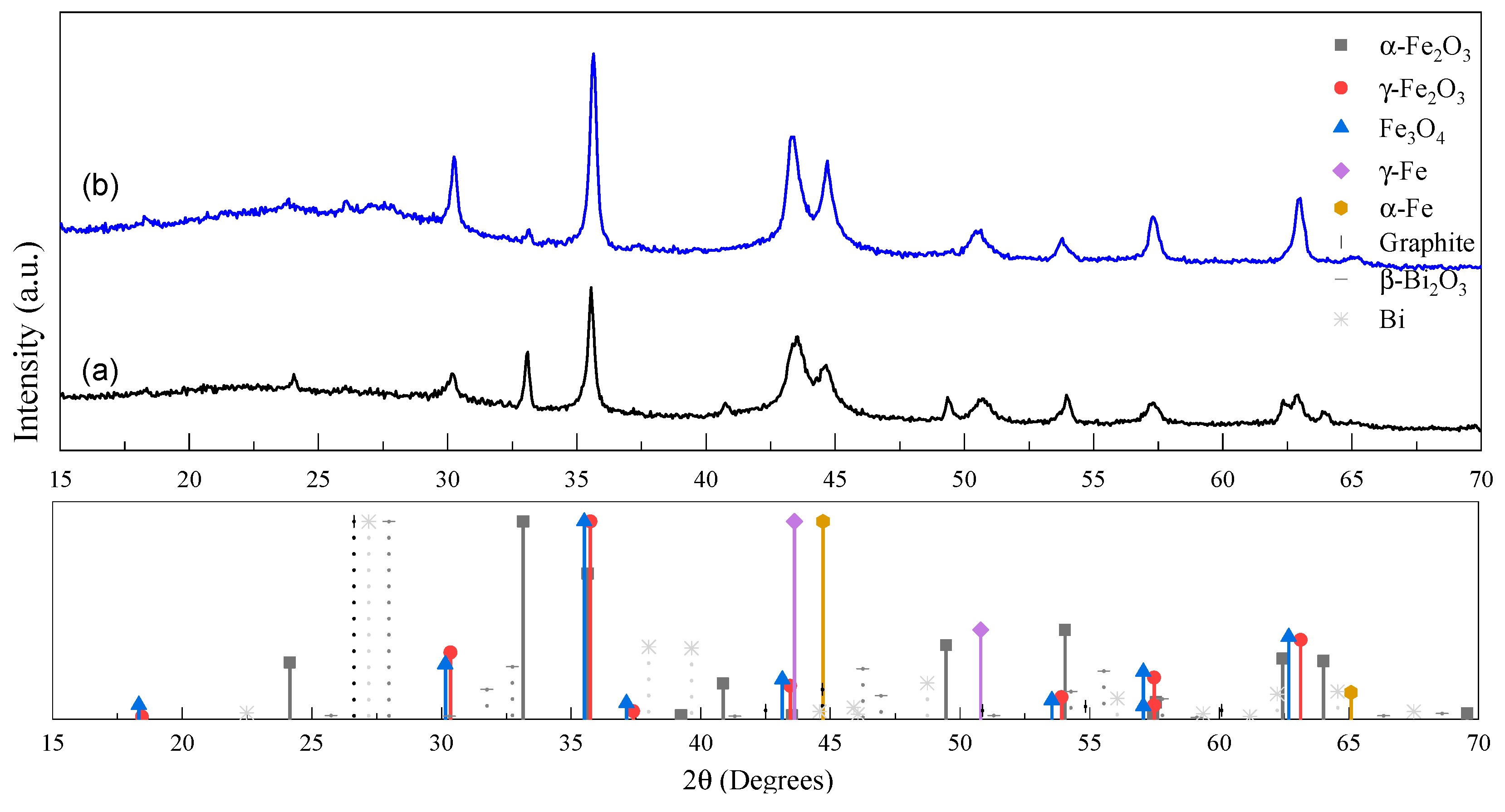
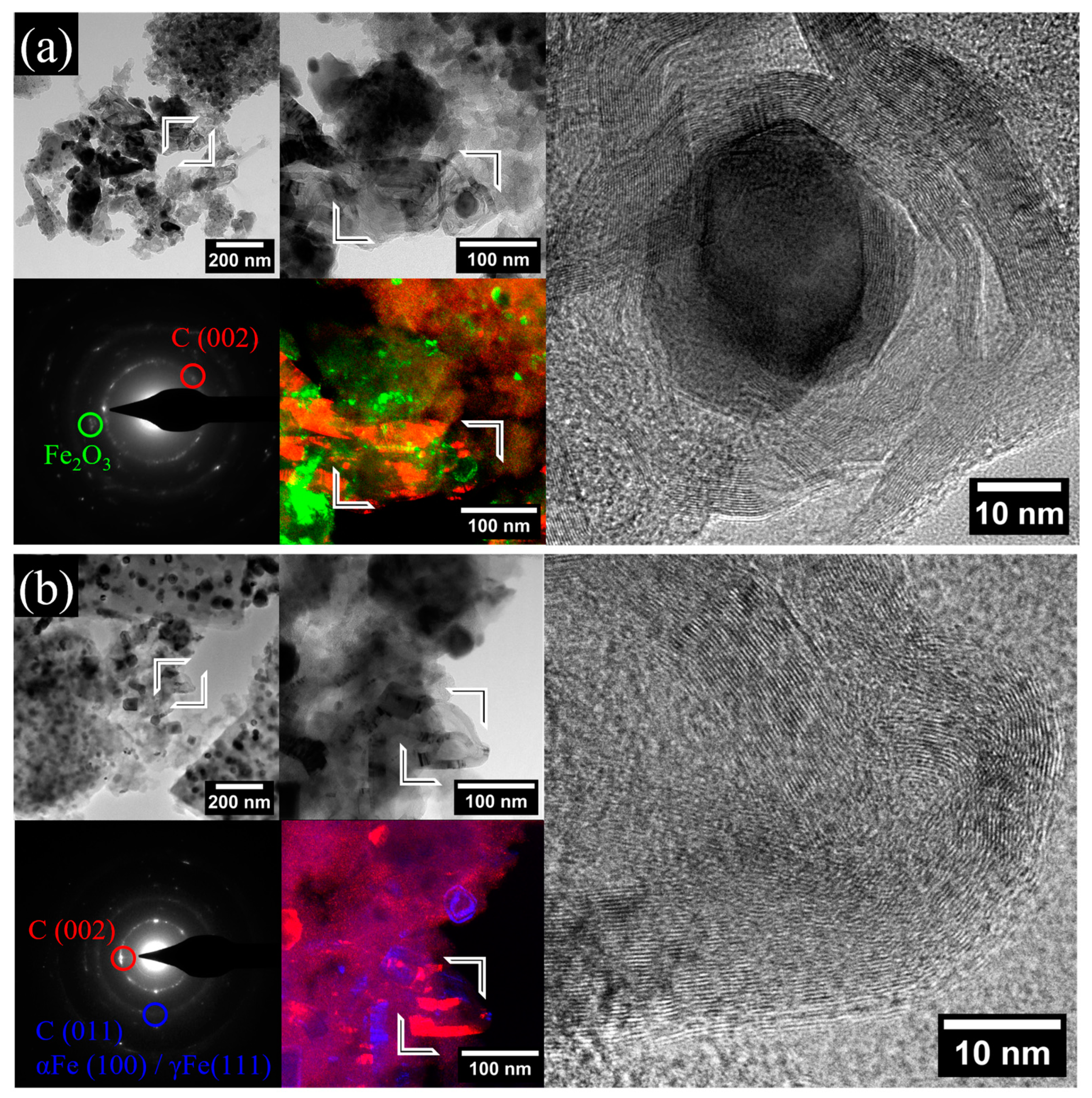
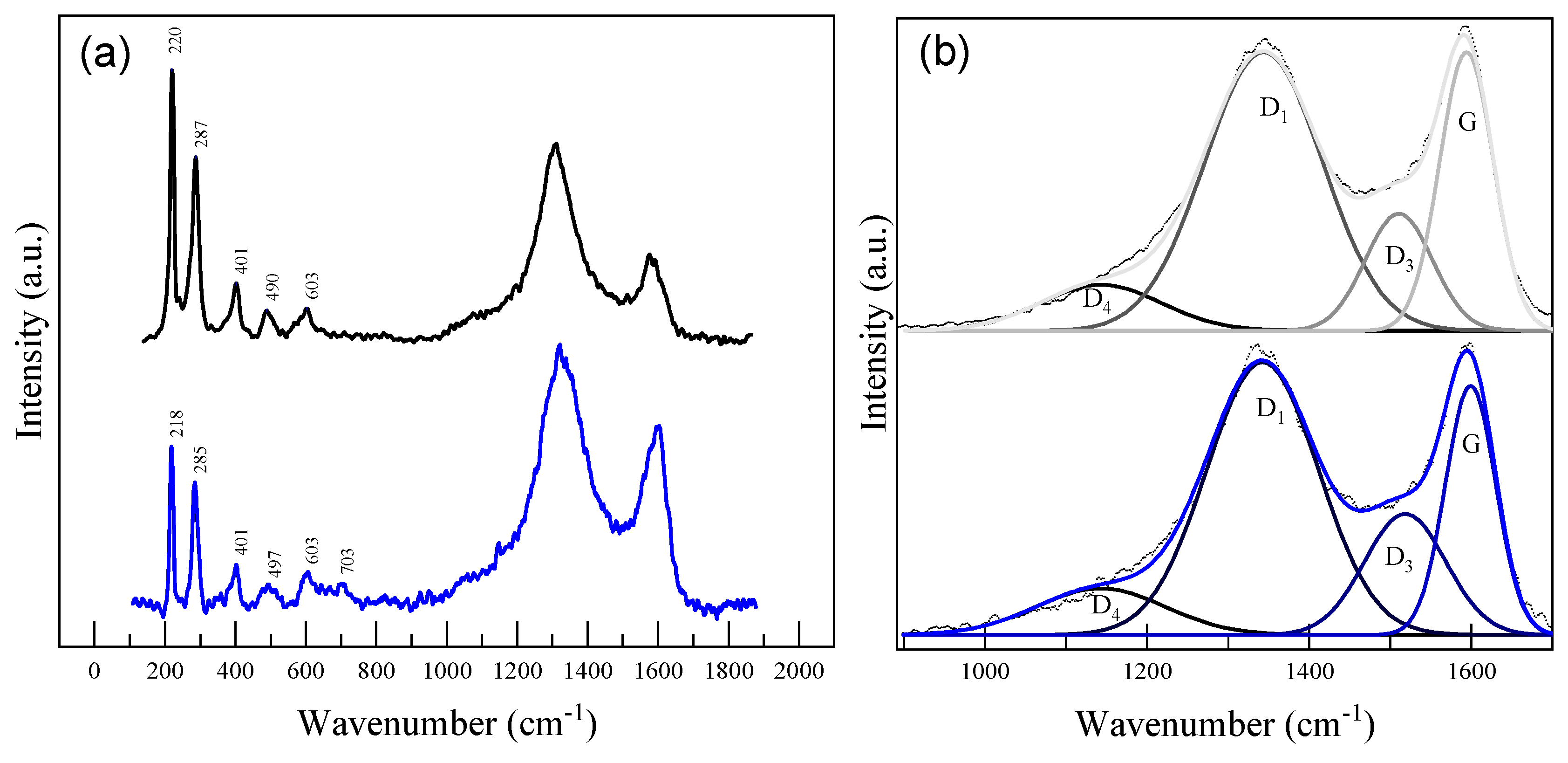
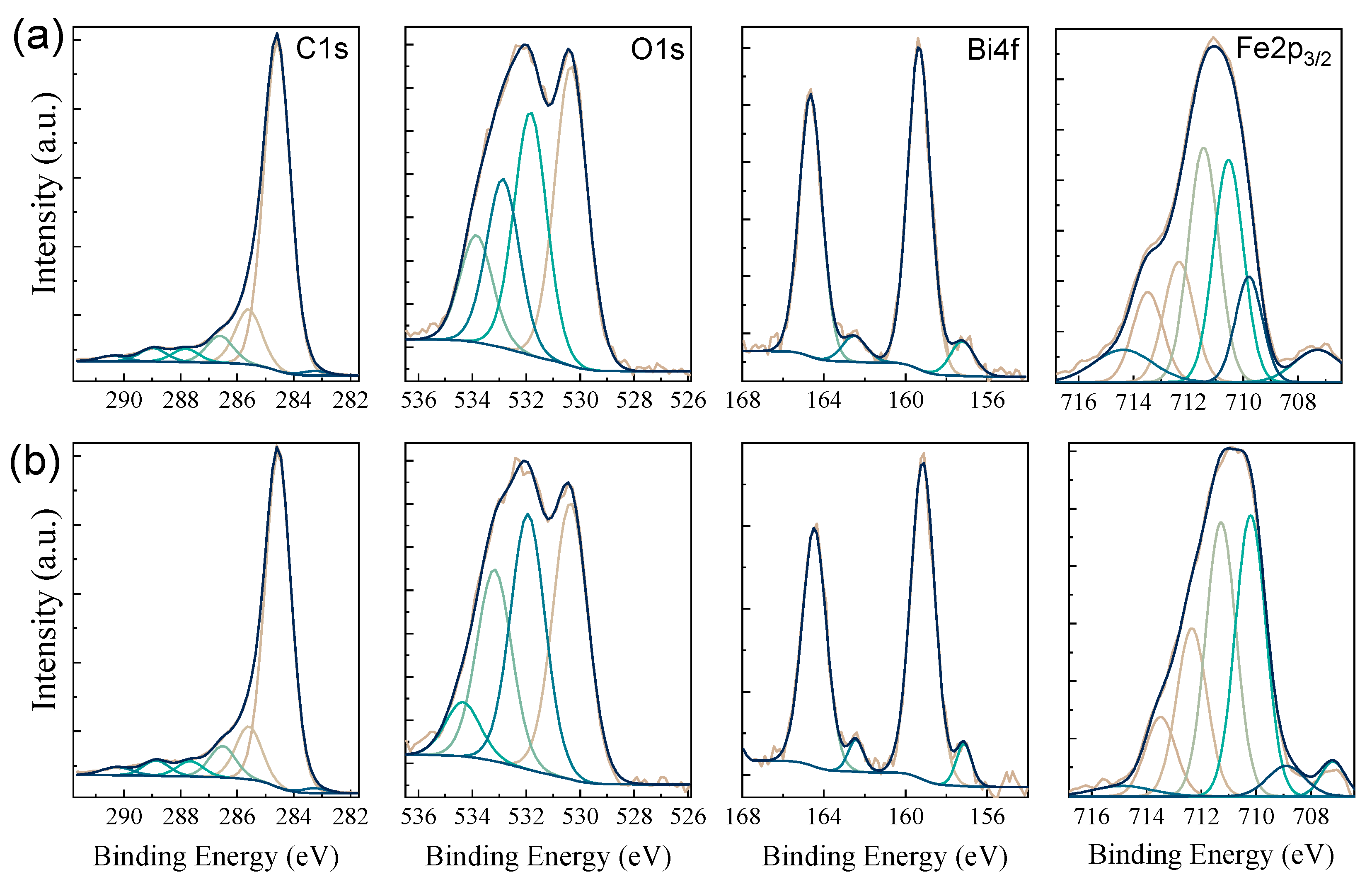
2.1. Morphological and Structural Analyses
2.2. Electrochemical Performances
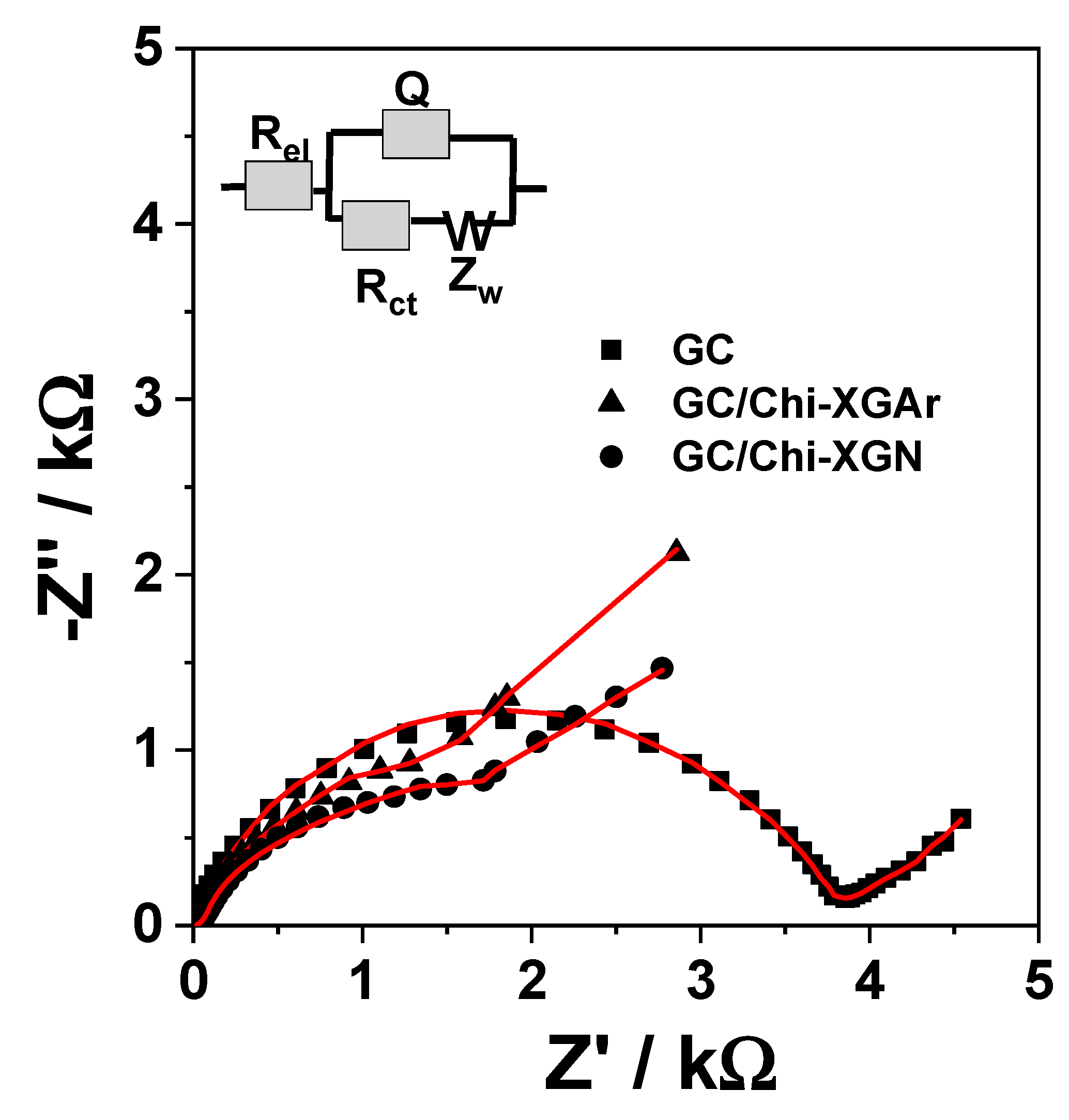
2.2.1. Electrochemical Characterization of XGAr- or XGN-Based Modified Electrodes
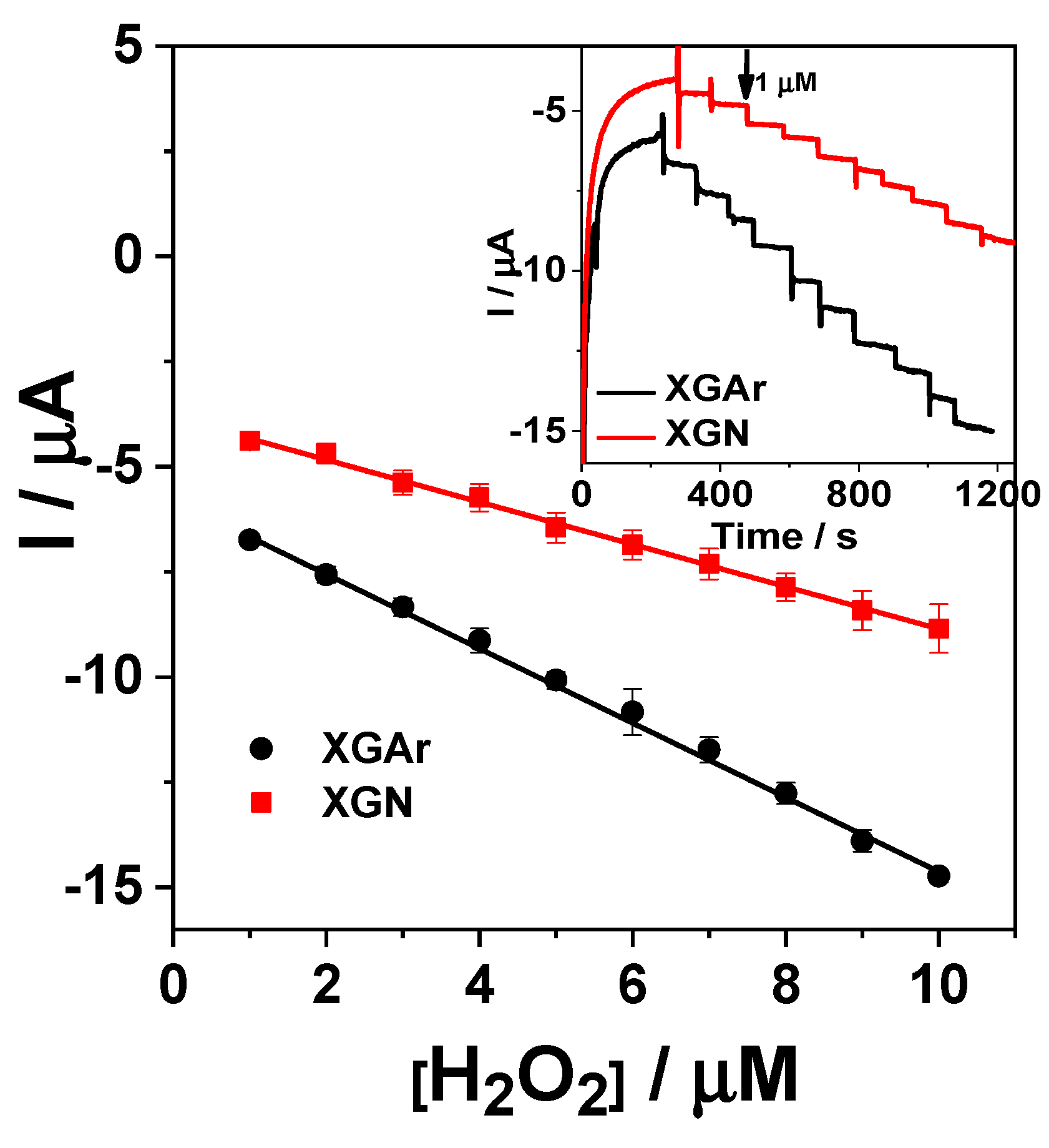
2.2.2. Amperometric Detection of H2O2
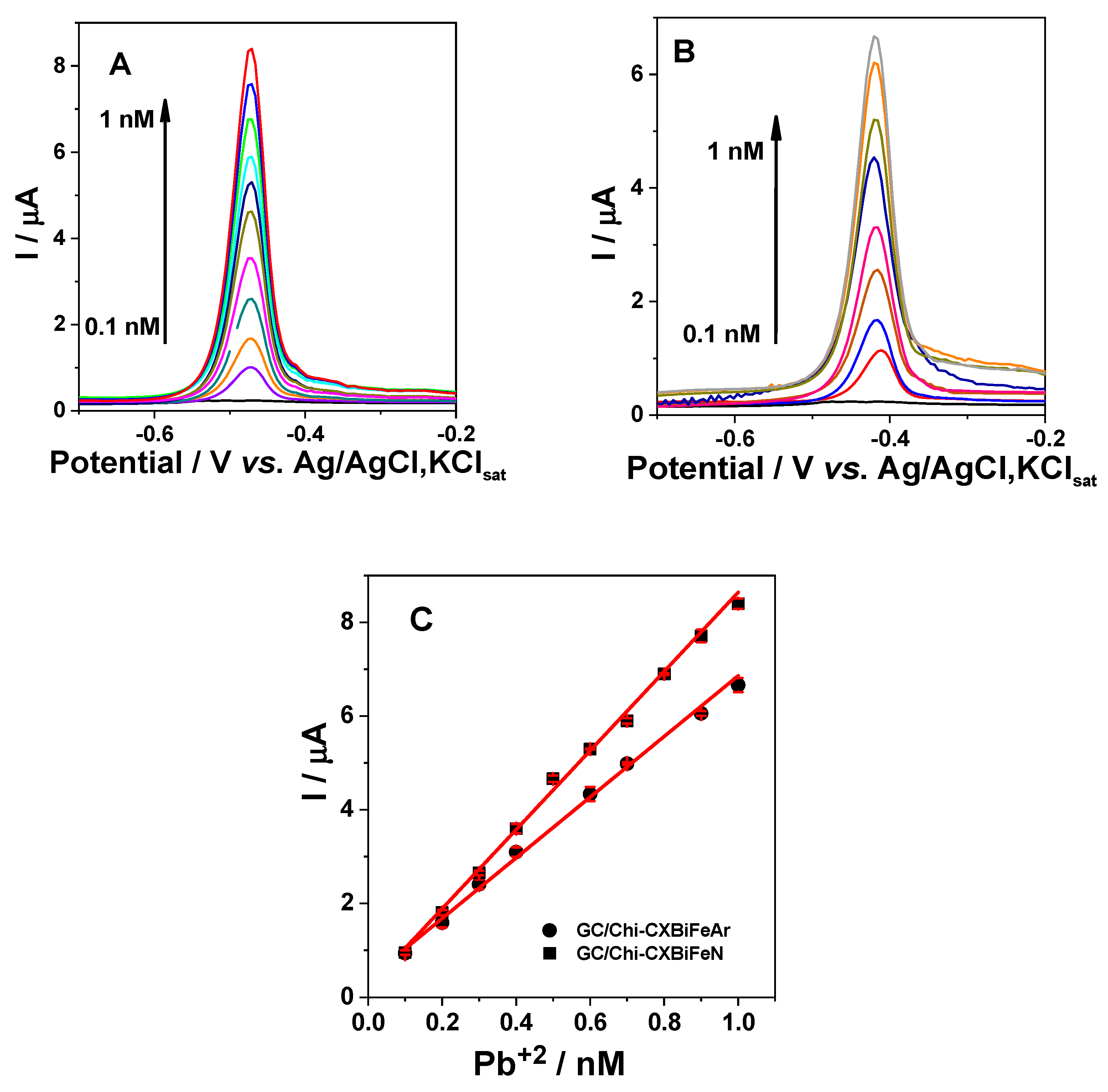
2.2.3. Voltammetric Detection of Pb2+
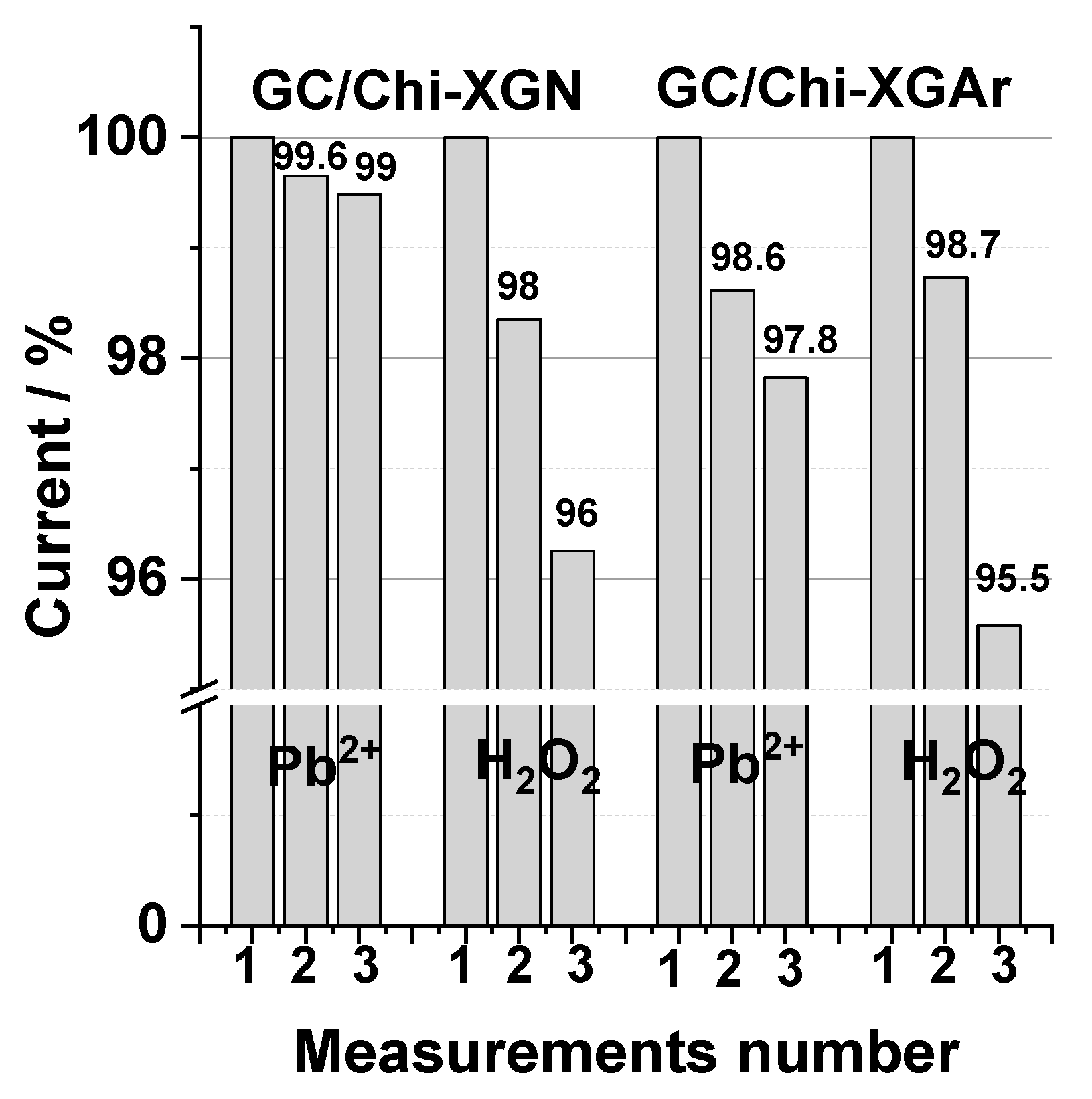
2.2.4. The Operational Stability (Reusability) of GC/Chi-XGAr and GC/Chi-XGN Electrodes
2.2.5. Time Stability
2.2.6. Real Sample Analysis
3. Conclusions
4. Materials and Methods
4.1. Reagents
4.2. Synthesis of Xerogel Ternary Composites
4.3. Characterization Methods
4.4. Preparation of the GC/Chi-XGArr and GC/Chi-XGN Electrodes
4.5. Electrochemical Measurements
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fort, C.I.; Pop, L.C. Heavy Metal and Metalloid Electrochemical Detection by Composite Nanostructures. In Advanced Nanostructures for Environmental Health; Elsevier: Amsterdam, The Netherlands, 2020; pp. 185–250. [Google Scholar]
- Lu, Y.; Liang, X.; Niyungeko, C.; Zhou, J.; Xu, J.; Tian, G. A review of the identification and detection of heavy metal ions in the environment by voltammetry. Talanta 2018, 178, 324–338. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, K.A.; Kazemeini, S.; Weber, D.C.; Cordero, P.A.; Garcia, E.M.; Rusinek, C.A. Electrochemical sensing of heavy metals in biological media: A review. Electroanalysis 2023, 35, e202300098. [Google Scholar] [CrossRef]
- Graham, N. Guidelines for Drinking-Water Quality, 2nd Edition, Addendum to Volume 1—Recommendations, World Health Organisation, Geneva, 1998, 36 Pages. Urban Water 1999, 1, 183. [Google Scholar] [CrossRef]
- European Parliment. European Parliament and the Council Directive 2013/35/EU. National Primary Drinking Water Regulations Contaminant—Directive 2013/35/EU; European Parliament: Strasbourg, France, 2009; Volume 1. [Google Scholar]
- Waheed, A.; Mansha, M.; Ullah, N. Nanomaterials-Based Electrochemical Detection of Heavy Metals in Water: Current Status, Challenges and Future Direction. TrAC Trends Anal. Chem. 2018, 105, 37–51. [Google Scholar] [CrossRef]
- Cadevall, M.; Ros, J.; Merkoçi, A. Bismuth Nanoparticles Integration into Heavy Metal Electrochemical Stripping Sensor. Electrophoresis 2015, 36, 1872–1879. [Google Scholar] [CrossRef]
- Musameh, M.M.; Hickey, M.; Kyratzis, I.L. Carbon Nanotube-Based Extraction and Electrochemical Detection of Heavy Metals. Res. Chem. Intermed. 2011, 37, 675–689. [Google Scholar] [CrossRef]
- Fort, C.I.; Cotet, L.C.; Danciu, V.; Turdean, G.L.; Popescu, I.C. Iron Doped Carbon Aerogel—New Electrode Material for Electrocatalytic Reduction of H2O2. Mater. Chem. Phys. 2013, 138, 893–898. [Google Scholar] [CrossRef]
- Rusu, M.M.; Fort, C.I.; Cotet, L.C.; Vulpoi, A.; Todea, M.; Turdean, G.L.; Danciu, V.; Popescu, I.C.; Baia, L. Insights into the Morphological and Structural Particularities of Highly Sensitive Porous Bismuth-Carbon Nanocomposites Based Electrochemical Sensors. Sens. Actuators B Chem. 2018, 268, 398–410. [Google Scholar] [CrossRef]
- Fort, C.I.; Cotet, L.C.; Vulpoi, A.; Turdean, G.L.; Danciu, V.; Baia, L.; Popescu, I.C. Bismuth Doped Carbon Xerogel Nanocomposite Incorporated in Chitosan Matrix for Ultrasensitive Voltammetric Detection of Pb(II) and Cd(II). Sens. Actuators B Chem. 2015, 220, 712–719. [Google Scholar] [CrossRef]
- Fort, C.I.; Rusu, M.M.; Cotet, L.C.; Vulpoi, A.; Florea, I.; Tuseau-Nenez, S.; Baia, M.; Baibarac, M.; Baia, L. Carbon Xerogel Nanostructures with Integrated Bi and Fe Components for Hydrogen Peroxide and Heavy Metal Detection. Molecules 2021, 26, 117. [Google Scholar] [CrossRef] [PubMed]
- Han, X.-J.; Zhou, S.-F.; Fan, H.-L.; Zhang, Q.-X.; Liu, Y.-Q. Mesoporous MnFe2O4 Nanocrystal Clusters for Electrochemistry Detection of Lead by Stripping Voltammetry. J. Electroanal. Chem. 2015, 755, 203–209. [Google Scholar] [CrossRef]
- Rusu, M.M.; Fort, C.I.; Vulpoi, A.; Barbu-Tudoran, L.; Baia, M.; Cotet, L.C.; Baia, L. Ultrasensitive Electroanalytical Detection of Pb2+ and H2O2 Using Bi and Fe—Based Nanoparticles Embedded into Porous Carbon Xerogel—The Influence of Nanocomposite Pyrolysis Temperatures. Gels 2023, 9, 868. [Google Scholar] [CrossRef]
- Rusu, M.M.; Vulpoi, A.; Maurin, I.; Cotet, L.C.; Pop, L.C.; Fort, C.I.; Baia, M.; Baia, L.; Florea, I. Thermal Evolution of C–Fe–Bi Nanocomposite System: From Nanoparticle Formation to Heterogeneous Graphitization Stage. Microsc. Microanal. 2022, 28, 317–329. [Google Scholar] [CrossRef] [PubMed]
- Najeh, I.; Mansour, N.B.E.N.; Dahman, H.; Alyamani, A.; Mir, L.E.L. Electrical Properties of Partial Carbonized Nanoporous Resin Based on Resorcinol-Formaldehyde. Sens. Transducers 2014, 27, 285–289. [Google Scholar]
- Liu, Z.; Lv, C.; Tan, X. One-Pot Synthesis of Fe, Co and Ni-Doped Carbon Xerogels and Their Magnetic Properties. J. Phys. Chem. Solids 2013, 74, 1275–1280. [Google Scholar] [CrossRef]
- Pauzon, C.; Hryha, E.; Forêt, P.; Nyborg, L. Effect of Argon and Nitrogen Atmospheres on the Properties of Stainless Steel 316 L Parts Produced by Laser-Powder Bed Fusion. Mater. Des. 2019, 179, 107873. [Google Scholar] [CrossRef]
- Su, J.; Lua, A.C. Effects of Carbonisation Atmosphere on the Structural Characteristics and Transport Properties of Carbon Membranes Prepared from Kapton® Polyimide. J. Membr. Sci. 2007, 305, 263–270. [Google Scholar] [CrossRef]
- Shebanova, O.N.; Lazor, P. Raman Study of Magnetite (Fe3O4): Laser-Induced Thermal Effects and Oxidation. J. Raman Spectrosc. 2003, 34, 845–852. [Google Scholar] [CrossRef]
- Bokobza, L.; Bruneel, J.-L.; Couzi, M. Raman Spectra of Carbon-Based Materials (from Graphite to Carbon Black) and of Some Silicone Composites. C 2015, 1, 77–94. [Google Scholar] [CrossRef]
- Alegre, C.; Sebastián, D.; Lázaro, M.J. Carbon Xerogels Electrochemical Oxidation and Correlation with Their Physico-Chemical Properties. Carbon 2019, 144, 382–394. [Google Scholar] [CrossRef]
- Bazhan, Z.; Ghodsi, F.E.; Mazloom, J. Effect of Phase Transition Induced by Annealing Temperature on Wettability, Optical and Photocatalytic Properties of Nanostructured Iron Oxide Thin Film. J. Mater. Sci. Mater. Electron. 2018, 29, 11489–11497. [Google Scholar] [CrossRef]
- Grosvenor, A.P.; Kobe, B.A.; Biesinger, M.C.; McIntyre, N.S. Investigation of Multiplet Splitting of Fe 2p XPS Spectra and Bonding in Iron Compounds. Surf. Interface Anal. 2004, 36, 1564–1574. [Google Scholar] [CrossRef]
- Yamashita, T.; Hayes, P. Analysis of XPS Spectra of Fe 2+ and Fe 3+ Ions in Oxide Materials. Appl. Surf. Sci. 2008, 254, 2441–2449. [Google Scholar] [CrossRef]
- Zhang, L.; Ni, Y.; Wang, X.; Zhao, G. Direct Electrocatalytic Oxidation of Nitric Oxide and Reduction of Hydrogen Peroxide Based on α-Fe2O3 Nanoparticles-Chitosan Composite. Talanta 2010, 82, 196–201. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-Y.; Chang, C.-T. Iron Oxide Nanorods Array in Electrochemical Detection of H2O2. Sens. Actuators B Chem. 2015, 220, 695–704. [Google Scholar] [CrossRef]
- Chang, C.-T.; Lin, C.-Y. Electrochemical Reduction of Hydrogen Peroxide by Nanostructured Hematite Modified Electrodes. RSC Adv. 2016, 6, 67428–67434. [Google Scholar] [CrossRef]
- Teymourian, H.; Salimi, A.; Khezrian, S. Fe3O4 Magnetic Nanoparticles/Reduced Graphene Oxide Nanosheets as a Novel Electrochemical and Bioeletrochemical Sensing Platform. Biosens. Bioelectron. 2013, 49, 1–8. [Google Scholar] [CrossRef]
- Heydaryan, K.; Almasi Kashi, M.; Sharifi, N.; Ranjbar-Azad, M. Efficiency Improvement in Non-Enzymatic H2O2 Detection Induced by the Simultaneous Synthesis of Au and Ag Nanoparticles in an RGO/Au/Fe3O4/Ag Nanocomposite. New J. Chem. 2020, 44, 9037–9045. [Google Scholar] [CrossRef]
- Lee, S.; Oh, J.; Kim, D.; Piao, Y. Talanta A Sensitive Electrochemical Sensor Using an Iron Oxide/Graphene Composite for the Simultaneous Detection of Heavy Metal Ions. Talanta 2016, 160, 528–536. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; You, Y.; Liu, Y.; Yang, Z. A Lead(II) Sensor Based on a Glassy Carbon Electrode Modified with Fe3O4 Nanospheres and Carbon Nanotubes. Microchim. Acta 2013, 180, 379–385. [Google Scholar] [CrossRef]
- Sun, Y.; Zhang, W.; Yu, H.; Hou, C.; Li, D.; Zhang, Y.; Liu, Y. Controlled Synthesis Various Shapes Fe3O4 Decorated Reduced Graphene Oxide Applied in the Electrochemical Detection. J. Alloys Compd. 2015, 638, 182–187. [Google Scholar] [CrossRef]
- Nguyen, L.D.; Doan, T.C.D.; Huynh, T.M.; Nguyen, V.N.P.; Dinh, H.H.; Dang, D.M.T.; Dang, C.M. An electrochemical sensor based on polyvinyl alcohol/chitosan-thermally reduced graphene composite modified glassy carbon electrode for sensitive voltammetric detection of lead. Sens. Actuators B Chem. 2021, 345, 130443. [Google Scholar] [CrossRef]
- Hall-Wallace, M.; Downs, R.T. The American Mineralogist Crystal Structure Database. Am. Mineral. 2003, 88, 247–250. [Google Scholar]
- Lutterotti, L.; Matthies, S.; Wenk, H.R. MAUD: A Friendly Java Program for Material Analysis Using Diffraction. IUCr Newsl. CPD 1999, 21, 14–15. [Google Scholar]
- Gražulis, S.; Daškevič, A.; Merkys, A.; Chateigner, D.; Lutterotti, L.; Quirós, M.; Serebryanaya, N.R.; Moeck, P.; Downs, R.T.; Le Bail, A. Crystallography Open Database (COD): An Open-Access Collection of Crystal Structures and Platform for World-Wide Collaboration. Nucleic Acids Res. 2012, 40, D420–D427. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Fe Crystalline Phase Analysis (wt%) | ID/IG | <DTEM> (nm) | C/O/Bi/Fe (at%) | |||
|---|---|---|---|---|---|---|---|
| α-Fe2O3 | γ-Fe2O3/Fe3O4 | α-Fe | γ-Fe | ||||
| XGAr | 27 | 6/13 | 17 | 37 | 1.00 | 17 | 88/10.5/0.3/1.2 |
| XGN | 3 | 2.2/44 | 18 | 33 | 1.09 | 26 | 87.8/10.3/0.2/1.7 |
| Iron Oxide | ||||||
|---|---|---|---|---|---|---|
| Sample | Reference Signals [18] | |||||
| XGAr | XGN | α-Fe2O3 | γ-Fe2O3 | Fe3O4 | ||
| 220 | 218 | 220 A1g | 200 T2g | |||
| 287 | 285 | <300 Eg(3) | 306 Eg | |||
| 401 | 401 | 410 | 400 | |||
| 490 | 497 | 500 T2g(2) | ||||
| 603 | 603 | 600 Eg(5) | 600 | |||
| 703 | 700 | 665 A1g | ||||
| Carbon | ||||||
| Sample | Spectral Band | Peak Type | Area Fit (%) | Position (cm−1) | Max Height (Normalized) | FWHM (cm−1) |
| XGAr | D4 | Gaussian | 9.04 | 1142 | 15.12 | 175 |
| D1 | 53.56 | 1342 | 91.53 | 171 | ||
| D3 | 12.81 | 1511 | 38.43 | 97 | ||
| G | 24.59 | 1594 | 91.51 | 78 | ||
| XGN | D4 | Gaussian | 10.18 | 1143 | 15.27 | 187 |
| D1 | 51.73 | 1342 | 89.59 | 162 | ||
| D3 | 16.72 | 1518 | 39.74 | 118 | ||
| G | 21.37 | 1599 | 81.77 | 73 | ||
| C1s | |||||||||
|---|---|---|---|---|---|---|---|---|---|
 | Sample | Component | Defects | C=C sp2 | C-C sp3 | C-OH C-O-C | C=O | O=C-OH | π- π* Shake-Up |
| XGAr | B.E. (eV) | 283.3 | 284.5 | 285.6 | 286.6 | 287.8 | 288.9 | 290.3 | |
| % | 1 | 73.5 | 12.3 | 6 | 3 | 3 | 1.3 | ||
| XGN | B.E. (eV) | 283.3 | 284.5 | 285.5 | 286.5 | 287.7 | 288.9 | 290.3 | |
| % | 1.1 | 72 | 11.4 | 6.8 | 3.3 | 3.3 | 2.1 | ||
| O1s | |||||||||
| Sample | Component | Fe-O, Bi-O | OH | C-O | O-C=O | ||||
| XGAr | B.E. (eV) | 530.3 | 531.8 | 532.9 | 533.8 | ||||
| % | 36.6 | 29.7 | 20.6 | 13.1 | |||||
| XGN | B.E. (eV) | 530.4 | 531.9 | 533.1 | 534.3 | ||||
| % | 35.5 | 32.8 | 24.7 | 7 | |||||
| Bi4f | |||||||||
| Sample | Bi4f Component | Bi3+ | Bi0 | ||||||
| XGAr | B.E. (eV) | 4f7/2 | 4f5/2 | 4f7/2 | 4f5/2 | 4f5/2 | |||
| % | 159.3 | 164.6 | 157.2 | 162.5 | 162.5 | ||||
| XGN | B.E. (eV) | 50.2 | 40.7 | 5.2 | 3.9 | 4.2 | |||
| % | 159.2 | 164.5 | 157.1 | 162.4 | 162.4 | ||||
| Fe2p3/2 | |||||||||
| Sample | Component | Fe0 | Peak1 | Peak2 | Peak3 | Peak4 | Peak5 | Surface Structures | |
| XGAr | B.E. (eV) | 707.28 | 709.78 | 710.53 | 711.44 | 712.34 | 713.48 | 714.37 | |
| % | 5.6 | 10.77 | 25.30 | 27 | 13.85 | 10.36 | 7.09 | ||
| XGN | B.E. (eV) | 707.20 | 708.90 | 710.20 | 711.28 | 712.34 | 713.48 | 714.87 | |
| % | 3.14 | 4.20 | 31.66 | 30.89 | 18.93 | 8.93 | 2.24 | ||
| Electrode | |||
|---|---|---|---|
| GC | GC/Chi-XGN | GC/Chi-XGAr | |
| Rel (Ω·cm2) | 5.40 ± 1.23 | 10.72 ± 6.67 | 7.58 ± 3.30 |
| CPEdl (µS·sn/cm2) | 14.83 ± 5.6 | 327.9 ± 1.26 | 662.5 ± 6.82 |
| n | 0.80 ± 0.94 | 0.63 ± 3.38 | 0.72 ± 1.99 |
| Rct (Ω·cm2) | 3706 ± 1.88 | 2432 ± 2.25 | 2624 ± 2.31 |
| W (mS·s1/2/cm2) | 1.53 ± 2.36 | 1.51 ± 2.70 | 1.70 ± 3.15 |
| C (µF/cm2) | 7.18 | 287.1 | 821.4 |
| χ2 | 0.005918 | 0.003435 | 0.00148 |
| Electrode Type | Applied Potential V vs. Ag|AgCl,KClsat | Linear Range | Detection Limit (µM) | Ref. |
|---|---|---|---|---|
| (Fe-CA)-CPE | −0.3 | 1–50 mM | 500 | [9] |
| GC/Chi-CXBiFe0 | −0.3 | 1–10 mM | 842.24 | [12] |
| GC/Chi-CXBiFe0.01 | −0.3 | 3–30 µM | 0.85 | [12] |
| GC/Chi-CXBiFe0.12 | −0.3 | 3–30 µM | 0.43 | [12] |
| GC/Chi-CXBiFe1.2 | −0.3 | 3–30 µM | 0.24 | [12] |
| α-Fe2O3NS|FePO4 | −0.3 | 6–270 µM | 3.4 | [28] |
| Fe3O4/r-GO/GC | −0.3 | 20–280 nM | 0.006 | [29] |
| GC/RGO/Au/Fe3O4/Ag | 0.55 | 0.002–1.2 mM | 1.43 | [30] |
| GC/Chi-XGAr | −0.3 | 1–10 µM | 0.17 | This work |
| GC/Chi-XGN | −0.3 | 1–10 µM | 0.36 |
| Electrode Type | Peak Potential V vs. Ag|AgCl,KClsat | Linear Range | Sensitivity (µA/µM) | Detection Limit (pM) | Ref. |
|---|---|---|---|---|---|
| GC/Chi-(Bi-CX) | −0.55 | 1–10 pM | 1.15·106 | 0.36 | [11] |
| GC/Chi-(Bi-CX) | −0.56 | 1–10 pM | 1.37·106 | 0.28 | [10] |
| GC/Chi-(Bi-CA) | −0.44 | 1–10 pM | 2.3·105 | 0.48 | [10] |
| GC/Chi-CXBiFe0 | −0.53 | 1–10 pM | 1.17·106 | 0.36 | [12] |
| GC/Chi-CXBiFe0.01 | −0.58 | 1–10 pM | 1.01·106 | 0.54 | [12] |
| GC/Chi-CXBiFe0.12 | −0.56 | 1–10 pM | 3.77·105 | 0.77 | [12] |
| GC/Chi-CXBiFe1.2 | −0.51 | 1–10 pM | 6.39·105 | 1.24 | [12] |
| Bi/Fe2O3/G/GC | −0.6 | 4.8–480 nM | 0.08 | 338.16 | [31] |
| Fe3O4/MWCNTs/GC | −0.63 | 20 pM–1.6 nM | 1.14·104 | 6 | [32] |
| Fe3O4/rGO GCE | −0.6 | 0.4–1.5 µM | 13.6 | 169·103 | [33] |
| PVA/chitosan-TRG/GC | −0.58 | 4.8–241.3 nM | - | 241 | [34] |
| GC/Chi-XGN | −0.46 | 0.1–1 nM | 8.44·103 | 33.05 | This work |
| GC/Chi- XGAr | −0.42 | 0.1–1 nM | 6.47·103 | 46.36 |
| Electrode | Analyte | Peak Potential | Current | ||
|---|---|---|---|---|---|
| V vs. Ag/AgCl,KClsat | RSD (%) | µA | RSD (%) | ||
| GC/Chi-XGAr | Pb2+ | −0.414 ± 0.007 * | 1.84 * | 4.97 ± 0.02 * | 0.42 * |
| H2O2 | - | - | 2.807 ± 0.05 ** | 1.88 ** | |
| GC/Chi-XGN | Pb2+ | −0.464 ± 0.005 * | 1.26 * | 5.84 ± 0.01 * | 0.26 * |
| H2O2 | - | - | 1.396 ± 0.03 ** | 2.32 ** | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fort, C.I.; Rusu, M.M.; Cotet, L.C.; Vulpoi, A.; Todea, M.; Baia, M.; Baia, L. The Impact of Ar or N2 Atmosphere on the Structure of Bi-Fe-Carbon Xerogel Based Composites as Electrode Material for Detection of Pb2+ and H2O2. Gels 2024, 10, 230. https://doi.org/10.3390/gels10040230
Fort CI, Rusu MM, Cotet LC, Vulpoi A, Todea M, Baia M, Baia L. The Impact of Ar or N2 Atmosphere on the Structure of Bi-Fe-Carbon Xerogel Based Composites as Electrode Material for Detection of Pb2+ and H2O2. Gels. 2024; 10(4):230. https://doi.org/10.3390/gels10040230
Chicago/Turabian StyleFort, Carmen I., Mihai M. Rusu, Liviu C. Cotet, Adriana Vulpoi, Milica Todea, Monica Baia, and Lucian Baia. 2024. "The Impact of Ar or N2 Atmosphere on the Structure of Bi-Fe-Carbon Xerogel Based Composites as Electrode Material for Detection of Pb2+ and H2O2" Gels 10, no. 4: 230. https://doi.org/10.3390/gels10040230