
[20(22)E]-Lanostane Triterpenes from the Fungus Ganoderma australe


1
School of Pharmaceutical Sciences, South-Central Minzu University, Wuhan 430074, China
2
National Center for Genetic Engineering and Biotechnology (BIOTEC), 113 Thailand Science Park, Phaholyothin Road, Klong Luang, Pathumthani 12120, Thailand
*
Authors to whom correspondence should be addressed.
†
These authors contributed equally to this work.
J. Fungi 2022, 8(5), 503; https://doi.org/10.3390/jof8050503
Submission received: 31 March 2022
/
Revised: 6 May 2022
/
Accepted: 11 May 2022
/
Published: 12 May 2022
(This article belongs to the Special Issue Secondary Metabolites from Fungi—in Honour of Prof. Dr. Ji-Kai Liu's 60th Birthday)
Abstract
:Twelve new lanostane triterpenoids (1–5, 7–13) were isolated from the fruiting bodies of the fungus Ganoderma australe. The structures of the new compounds were elucidated by extensive 1D and 2D NMR, and HRESIMS spectroscopic analysis. All the triterpenes are featured by 20(22)E configurations which are uncommon in the Ganoderma triterpene family. The absolute configuration of the C-25 of compounds 1, 2, and 6 were determined by the phenylglycine methyl ester (PGME) method. A postulated biosynthetic pathway for compound 1 was discussed. This study opens new insights into the secondary metabolites of the chemically underinvestigated fungus G. australe.
1. Introduction
Mushrooms are popular in the food market due to their delicious taste and nutrition values. Mushroom-derived secondary metabolites have contributed lots of lead compounds for medical and agricultural use. Psilocybin, a specialized compound from the genus Psilocybe, is a naturally occurring hallucinogenic prodrug for treating psychiatric disorders [1]. Strobilurins, firstly originated from the mushroom Strobilurus tenacellus, are a group of natural products and their synthetic analogs are used in agriculture as fungicides [2,3]. More and more attention has been paid to mining promising lead compounds from the mushroom natural product reservoir in recent years.
Ganoderma, called “lingzhi” in China, is a group of wood-decaying mushrooms with hard fruiting bodies which grow mostly in spare scatting sunshine, on the trees, and on open grounds [4]. It is a genus of notable medicinal fungi and traditional herbal medicine for the treatment of diseases such as hepatopathy, nephritis, neurasthenia, and asthma [5,6,7,8,9,10]. The Shennong Ben Cao Jing, an ancient Chinese medicinal book, documented that Ganoderma was effective for maintaining health, prolonging life, boosting memory, and relieving stress. Ganoderma lucidum and G. sinense are two registered species recorded in the Chinese Pharmacopoeia (2015). Many studies show that triterpenoids and polysaccharides are the main bioactive substances in Ganoderma [11,12,13,14,15,16,17]. Ganoderma australe is a species used in folk medicine as the alternative of G. lucidum. However, this fungus has rarely been chemically investigated compared to other Ganoderma species, such as G. lucidum, G. cochlear, and G. sinense. Previous studies on this fungus have led to the isolation of lanostane triterpenes [18,19,20,21], meroterpenoids [22,23], and alkaloids [22]. The lanostanoids from this species are over-oxygenated compared to the ones isolated from other species of Ganoderma, especially the position of C-20 [18,20]. The quaternary hydroxy group substituted at C-20 led to the introduction of an additional chiral carbon of which the stereochemistry was difficult to be assigned even by chemical derivatization. Moreover, this substituted pattern of the C-20 hydroxy group always triggered to dehydration between C-21 to produce the 20(22)-double bond, which always incorporated into an α,β-unsaturated ketone group with the C-23 carbonyl group. In this study, we have investigated the secondary metabolite profiles of G. australe, which led to the isolation of twelve new highly oxygenated lanostane triterpenes with uncommon 20(22)E configurations. We, herein, report the isolation and structural elucidation of the new compounds.
2. Experimental Section
2.1. General Experimental Procedures
Optical rotations were obtained on an Autopol IV-T digital polarimeter (Rudolph, Hackettstown, NJ, USA). UV spectra were recorded on a Hitachi UH5300 spectrophotometer (Hitachi, Tokyo, Japan). CD spectra were measured on a Chirascan Circular Dichroism spectrometer (Applied Photophysics Limited, Leatherhead, Surrey, UK). The 1D and 2D spectra were obtained on the Bruker Avance III 500 MHz and 600 MHz spectrometers (Bruker Corporation, Karlsruhe, Germany). HRESIMS spectra were measured on a Q Exactive Orbitrap mass spectrometer (Thermo Fisher Scientific, Waltham, MA, USA). Single-crystal X-ray diffraction data were recorded on the BRUKER D8 QUEST diffractometer (Bruker Corporation, Karlsruhe, Germany). Medium pressure liquid chromatography (MPLC) was performed on an Interchim system equipped with a column packed with RP-18 gel (40–75 μm, Fuji Silysia Chemical Co., Ltd., Kasugai, Japan). Preparative high performance liquid chromatography (prep-HPLC) was performed on an Agilent 1260 Infinity Ⅱ liquid chromatography system equipped with a Zorbax SB-C18 column (particle size 5 μm, dimensions 150 mm × i.d. 9.4 mm, flow rate 5 mL⋅min−1) and a DAD detector (Agilent Technologies, Santa Clara, CA, US). Sephadex LH-20 (GE Healthcare, Stockholm, Sweden) and silica gel (200–300 mesh, Qingdao Haiyang Chemical Co., Ltd., Qingdao, China) were used for column chromatography (CC). (S)- and (R)-phenylglycine methyl ester were bought from Sigma-Aldrich (St. Louis, MO, USA).
2.2. Fungal Material
The fruiting bodies of G. australe were collected in Tongbiguan Natural Reserve, Dehong, Yunnan Province, China, in 2016, and identified by Prof. Yu-Cheng Dai (Institute of Microbiology, Beijing Forestry University). A voucher specimen of G. australe was deposited in the Mushroom Bioactive Natural Products Research Group of South-Central University for Nationalities.
2.3. Extraction and Isolation
The dry fruiting bodies of G. australe (3.26 kg) were extracted four times with CHCl3:MeOH (1:1) at room temperature to obtain crude extract, which was further suspended in distilled water and partitioned against EtOAc to afford EtOAc layer extract (130 g). The EtOAc layer extract was eluted on MPLC with a stepwise gradient of MeOH−H2O (20:80−100:0) to afford eight fractions (A−H).
Fraction C was subjected to Sephadex LH-20 (MeOH) and obtained 16 subfractions (C1-C16), and C2 was separated by prep-HPLC (MeCN−H2O: 20:80−40:60, 25 min, 4 mL/min) to yield compound 2 (6.4 mg, tR = 20.5 min).
Fraction D was separated by Sephadex LH-20 (MeOH) to give eight subfractions (D1–D8). Subfraction D4 was subjected to silica gel CC (petroleum ether−acetone from v/v 6:1 to 1:1) and yielded eleven subfractions (D4a–D4k). Subfraction D4d was purified by prep-HPLC (MeCN−H2O: 20:80–40:60, 25 min, 4 mL/min) to yield compound 3 (2.0 mg, tR = 19.0 min).
Fraction E was separated by Sephadex LH-20 (CHCl3:MeOH = 1:1) to afford four subfractions (E1–E4). E2 was separated by CC on silica gel (petroleum ether−acetone from v/v 15:1 to 1:1) to obtain 10 subfractions (E2a–E2j). E2b was subjected to prep-HPLC (MeCN–H2O: 70:30–90:10, 25.0 min, 4 mL/min) to obtain compound 7 (3.8 mg, tR = 14.0 min) and 8 (3.7 mg, tR = 15.0 min). Compound 10 (3.7 mg, tR = 21.0 min) was purified from E2f by prep-HPLC (MeCN–H2O: 70:30–90:10, 25 min, 4 mL/min). E4 was subjected to CC on silica gel (petroleum ether–acetone from v/v 10:1 to 1:1) to obtain 12 fractions. Compound 12 (6.0 mg, tR = 18.0 min) was purified from E4c by prep-HPLC (MeCN–H2O: 40:60–60:80, 25 min, 4 mL/min). Compound 6 (5.1 mg, tR = 19.1 min) was purified from E4d by prep-HPLC (MeCN–H2O: 40:60–60:80, 25.2 min, 4 mL/min). Compound 9 (21.4 mg, tR = 20.0 min) was purified from E4f by prep-HPLC (MeCN–H2O: 40:60–60:80, 25 min, 4 mL/min). Compound 5 (7.3 mg, tR = 30.0 min) was purified from E4g by prep-HPLC (MeCN–H2O: 45:55–73:27, 35.0 min, 4 mL/min). Compound 11 (2.6 mg, tR = 27.0 min) was purified from E4h by prep-HPLC (MeCN–MeOH–H2O: 40:20:40, isocratic, 30 min, 4 mL/min). E7 was subjected to column chromatography (CC) on silica gel (petroleum ether–acetone from v/v 15:1 to 1:1) to obtain 13 fractions. E7j was purified by prep-HPLC (MeCN–H2O: 30:70–50:50, 25 min, 4 mL/min) to yield compound 1 (8.6 mg, tR = 14.0 min).
Fraction G was separated by column chromatography (CC) on silica gel (petroleum ether–acetone from v/v 15:1 to 1:1) to obtain 10 subfractions (G1–G10). Compound 4 (13.3 mg, tR = 12.7 min) was purified from G7 by prep-HPLC (MeCN–H2O: 50:50–30:70, 25 min, 4 mL/min). Compound 13 (4.0 mg, tR = 13.2 min) was purified from G7 by prep-HPLC (MeCN–H2O: 50:50–30:70, 25 min, 4 mL/min).
2.4. Spectroscopic Data
Ganoaustralenone A (1): yellow oil. [α] +8.9 (c 0.09, MeOH); UV (MeOH) λmax (log ε) 250.0 (4.22); 1H NMR (600 MHz, CD3OD) data, see Table 1, 13C NMR (150 MHz, CD3OD) data, see Table 2 HRESIMS m/z 535.26685 [M + Na]+ (calcd for C30H40O7Na, 535.26717).
Ganoaustralenone B (2): white powder. [α] +81.3 (c 0.06, MeOH); UV (MeOH) λmax (log ε) 250.0 (4.00); 1H NMR (600 MHz, CDCl3) data, see Table 1, 13C NMR (150 MHz, CDCl3) data, see Table 2; HRESIMS m/z 551.26422 [M + Na]+ (calcd for C30H40O8Na, 551.26209).
Ganoaustralenone C (3): white powder. [α] +178.4 (c 0.05, MeOH); UV (MeOH) λmax (log ε) 250.0 (4.20); 1H NMR (500 MHz, CDCl3) data, see Table 1, 13C NMR (125 MHz, CDCl3) data, see Table 2; HRESIMS m/z 537.28180 [M + Na]+ (calcd for C30H42O7Na, 537.28227).
Ganoaustralenone D (4): yellow oil. [α] +102.2 (c 0.13, MeOH); UV (MeOH) λmax (log ε) 250.0 (4.24); 1H NMR (600 MHz, CD3COCD3) data, see Table 1, 13C NMR (150 MHz, CD3COCD3) data, see Table 2; HRESIMS m/z 565.27728 [M + Na]+ (calcd for C31H42O8Na, 565.27774).
Ganoaustralenone E (5): yellow oil. [α] +25.3 (c 0.07, MeOH); UV (MeOH) λmax (log ε) 250.0 (4.27); 1H NMR (600 MHz, CDCl3) data, see Table 1, 13C NMR (150 MHz, CDCl3) data, see Table 2; HRESIMS m/z 549.28210 [M + Na]+ (calcd for C31H42O7Na, 549.28282).
Ganoaustralenone F (7): yellow oil. [α] +5.7 (c 0.04, MeOH); UV (MeOH) λmax (log ε) 245.0 (3.53); 1H NMR (800 MHz, CDCl3) data, see Table 3, 13C NMR (150 MHz, CDCl3) data, see Table 2; HRESIMS m/z 547.30286 [M + Na]+ (calcd for C32H44O6Na, 547.30356).
Ganoaustralenone G (8): yellow oil. [α] +17.1 (c 0.04, MeOH); UV (MeOH) λmax (log ε) 245.0 (3.62); 1H NMR (800 MHz, CDCl3) data, see Table 3, 13C NMR (200 MHz, CDCl3) data, see Table 4; HRESIMS m/z 561.28223 [M + Na]+ (calcd for C32H42O7Na, 561.28282).
Ganoaustralenone H (9): yellow oil. [α] +31.3 (c 0.11, MeOH); UV (MeOH) λmax (log ε) 250.0 (3.39); 1H NMR (600 MHz, CDCl3) data, see Table 3, 13C NMR (150 MHz, CDCl3) data, see Table 4; HRESIMS m/z 513.32135 [M + H]+ (calcd for C31H45O6, 513.32161).
Ganoaustralenone I (10): pale yellow oil. [α] +67.7 (c 0.05, MeOH); UV (MeOH) λmax (log ε) 245.0 (4.02); 1H NMR (600 MHz, CDCl3) data, see Table 3, 13C NMR (150 MHz, CDCl3) data, see Table 4; HRESIMS m/z 563.29749 [M + Na]+ (calcd for C32H44O7Na, 563.29847).
Ganoaustralenone J (11): yellow oil. [α] +119.56 (c 0.05, MeOH); UV (MeOH) λmax (log ε) 245.0 (4.17); 1H NMR (600 MHz, CDCl3) data, see Table 5, 13C NMR (150 MHz, CDCl3) data, see Table 4; HRESIMS m/z 549.28210 [M + Na]+ (calcd for C31H42O7Na, 549.28282).
2.5. Synthesis of the Phenylglycine Methyl Ester (PGME) Derivatives
To a solution of 1 (2.0 mg, 3.9 μmol) in DMF (0.5 mL) on ice add PyBOP (2.5 mg, 4.8 μmol), HBTU (1.9 mg, 5.0 μmol), N-methylmorpholine 100 μL, and (S)-PGME (1.0 mg, 4.9 μmol). The reaction mixture was stirred at room temperature for 3 h. The reaction was stopped by adding 1 mL of EtOAc and then washed with H2O. The EtOAc layer was concentrated under reduced pressure to obtain a pale yellow oil sample, which was purified by HPLC to furnish (S)-PGME amide product 1a. Similarly, (R)-PGME amide product 1b was prepared from 1 (2.0 mg) and (R)-PGME (1.0 mg). NMR assignments of the protons for 1a and 1b were achieved by analysis of the 1H-1H COSY spectra.
Similarly, 2a was prepared from 2 (0.5 mg) and (S)-PGME, 2b was prepared from 2 (0.5 mg) and (R)-PGME. 3a was prepared from 3 (1 mg) and (S)-PGME, 3b was prepared from 3 (1 mg) and (R)-PGME. NMR assignments of the protons for 2a and 2b were achieved by analysis of the 1H-1H COSY spectra.
To a solution of 6 (2.5 mg) in THF (1 mL) was added 1 mL of LiOH (1 mol/L). The reaction mixture was stirred at room temperature overnight. The reaction was stopped by concentrating under reduced pressure to obtain a pale yellow oil sample, which was purified by HPLC to obtain 6H (0.4 mg). Then 6Ha was prepared from 6H (0.2 mg) with (S)-PGME, 6Hb was prepared from 6H (0.2 mg) with (R)-PGME. NMR assignments of the protons for 6Ha and 6Hb were achieved by analysis of the 1H-1H COSY spectra.
1a: 1H NMR (600 MHz, CDCl3) 2.824 (1H, m, H-1a), 1.814 (1H, m, H-1b), 1.887 (1H, m H-2a), 2273 (1H, m, H-2b), 2.393 (1H, m, H-5), 2.247 (1H, m, H-6a), 2.530 (1H, m, H-6b), 2.480 (1H, m, H-12a), 2.770 (1H, m, H-12b), 2.500 (1H, m, H-15a), 2.923 (1H, m, H-15b), 2.608 (1H, m, H-16a), 1.737 (1H, m H-16b), 2.962 (1H, m, H-17), 0.667 (3H, s, H-18), 1.266 (3H, s, H-19), 2.141 (3H, s, H-21), 6.321 (1H, s, H-22), 4.123 (1H, dd, J = 6.0, 3.3 Hz, H-24), 2.924 (1H, m, H-25), 1.377 (3H, d, J = 7.2 Hz, H-27), 1.133 (3H, s, H-28), 1.108 (3H, s, H-29), 1.291 (1H, s, H-30), 6.892 (1H, d, J = 7.0 Hz, NH), 5.423 (1H, d, J = 7.0 Hz, H-2′ of PGME), 7.343–7.307 (5H, overlapped, phenyl group), 3.709 (3H, s, OCH3). HRESIMS m/z 682.33380 [M + Na]+ (calcd for C39H49O8NNa, 682.33559).
1b: 1H NMR (600 MHz, CDCl3) 2.818 (1H, m, H-1a), 1.855 (1H, m, H-1b), 1.910 (1H, m H-2a), 2265 (1H, m, H-2b), 2.388 (1H, m, H-5), 2.267 (1H, m, H-6a), 2.522 (1H, m, H-6b), 2.500 (1H, m, H-12a), 2.743 (1H, d, J = 16.6 Hz, H-12b), 2.515 (1H, m, H-15a), 2.927 (1H, m, H-15b), 2.613 (1H, m, H-16a), 1.744 (1H, m H-16b), 2.972 (1H, m, H-17), 0.536 (3H, s, H-18), 1.266 (3H, s, H-19), 2.098 (3H, s, H-21), 6.308 (1H, s, H-22), 4.132 (1H, dd, J = 6.0, 3.3 Hz, H-24), 2.927 (1H, m, H-25), 1.387 (3H, d, J = 7.2 Hz, H-27), 1.133 (3H, s, H-28), 1.110 (3H, s, H-29), 1.290 (1H, s, H-30), 7.061 (1H, d, J = 6.7 Hz, NH), 5.428 (1H, d, J = 6.7 Hz, H-2′ of PGME), 7.352–7.309 (5H, overlapped, phenyl group), 3.699 (3H, s, OCH3). HRESIMS m/z 682.33392 [M + Na]+ (calcd for C39H49O8NNa, 682.33559).
2a: 1H NMR (600 MHz, CDCl3) 2.782 (1H, m, H-1a), 1.838 (1H, m, H-1b), 2.324 (1H, m H-2a), 2720 (1H, m, H-2b), 2.297 (1H, d, J = 13.5, H-5), 4.408 (1H, dd, J = 13.5, 3.1 Hz, H-6), 2.505 (1H, d, J = 17.0 Hz, H-12a), 2.784 (1H, m, H-12b), 1.708 (1H, m, H-15a), 2.249 (1H, m, H-15b), 1.906 (1H, m, H-16a), 2.835 (1H, m H-16b), 1.932 (1H, m, H-17), 0.658 (3H, s, H-18), 1.219 (3H, s, H-19), 2.131 (3H, s, H-21), 6.328 (1H, s, H-22), 4.115 (1H, dd, J = 6.0, 3.4 Hz, H-24), 2.932 (1H, dd, J = 7.0, 3.4 Hz, H-25), 1.378 (3H, d, J = 7.0 Hz, H-27), 1.343 (3H, s, H-28), 1.438 (3H, s, H-29), 1.362 (1H, s, H-30), 6.875 (1H, d, J = 7.0 Hz, NH), 5.417 (1H, d, J = 6.8 Hz, H-2′ of PGME), 7.352–7.301 (5H, overlapped, phenyl group), 3.704 (3H, s, OCH3). HRESIMS m/z 698.33087 [M + Na]+ (calcd for C39H49O9NNa, 698.33050).
2b: 1H NMR (600 MHz, CDCl3) 2.873 (1H, m, H-1a), 1.838 (1H, m, H-1b), 2.328 (1H, m H-2a), 2722 (1H, m, H-2b), 2.293 (1H, d, J = 13.5, H-5), 4.400 (1H, dd, J = 13.5, 3.1 Hz, H-6), 2.399 (1H, d, J = 17.0 Hz, H-12a), 2.754 (1H, m, H-12b), 1.700 (1H, m, H-15a), 2.243 (1H, m, H-15b), 1.906 (1H, m, H-16a), 2.839 (1H, m H-16b), 1.958 (1H, m, H-17), 0.519 (3H, s, H-18), 1.221 (3H, s, H-19), 2.093 (3H, s, H-21), 6.315 (1H, s, H-22), 4.128 (1H, dd, J = 5.0, 3.5 Hz, H-24), 2.935 (1H, dd, J = 7.3, 3.5 Hz, H-25), 1.386 (3H, d, J = 7.2 Hz, H-27), 1.343 (3H, s, H-28), 1.440 (3H, s, H-29), 1.348 (1H, s, H-30), 7.047 (1H, d, J = 6.6 Hz, NH), 5.419 (1H, d, J = 6.6 Hz, H-2′ of PGME), 7.347–7.277 (5H, overlapped, phenyl group), 3.696 (3H, s, OCH3). HRESIMS m/z 698.32990 [M + Na]+ (calcd for C39H49O9NNa, 698.33050).
3a: 1H NMR (600 MHz, CDCl3) 2.976 (1H, m, H-1a), 1.709 (1H, m, H-1b), 2.474 (1H, m H-2a), 2595 (1H, m, H-2b), 2.084 (1H, overlapped H-5), 2.902 (1H, overlapped, H-6a), 1.687 (1H, overlapped, H-6b), 4.459 (1H, overlapped, H-7), 2.749 (1H, d, J = 17.3 Hz, H-12a), 2.375 (1H, d, J = 17.3 Hz, H-12b), 1.701 (1H, m, H-15a), 2.439 (1H, m, H-15b), 1.978 (1H, m, H-16a), 2.026 (1H, m H-16b), 2.981 (1H, m, H-17), 0.642 (3H, s, H-18), 1.022 (3H, s, H-19), 2.137 (3H, s, H-21), 6.343 (1H, s, H-22), 4.137 (1H, dd, J = 6.0, 3.4 Hz, H-24), 2.939 (1H, dd, J = 7.3, 3.4 Hz, H-25), 1.385 (3H, d, J = 7.3 Hz, H-27), 1.147 (3H, s, H-28), 1.073 (3H, s, H-29), 1.344 (1H, s, H-30), 6.903 (1H, d, J = 6.9 Hz, NH), 5.426 (1H, d, J = 6.9 Hz, H-2′ of PGME), 7.343–7.306 (5H, overlapped, phenyl group), 3.703 (3H, s, OCH3). HRESIMS m/z 684.35034 [M + Na]+ (calcd for C39H51O8NNa, 684.35124).
3b: 1H NMR (600 MHz, CDCl3) 2.976 (1H, m, H-1a), 1.692 (1H, m, H-1b), 2.609 (1H, m H-2a), 2.458 (1H, m, H-2b), 2.073 (1H, overlapped H-5), 1.691 (1H, overlapped, H-6a), 1.638 (1H, overlapped, H-6b), 4.438 (1H, t, J = 4.2 Hz, H-7), 2.711 (1H, d, J = 16.9 Hz, H-12a), 2.692 (1H, d, J = 16.9 Hz, H-12b), 1.809 (1H, m, H-15a), 2.021 (1H, m, H-15b), 1.977 (1H, m, H-16a), 1.911 (1H, m H-16b), 2.904 (1H, m, H-17), 0.463 (3H, s, H-18), 1.025 (3H, s, H-19), 2.096 (3H, s, H-21), 6.316 (1H, s, H-22), 4.136 (1H, dd, J = 4.5, 3.7 Hz, H-24), 2.948 (1H, dd, J = 7.3, 3.7 Hz, H-25), 1.402 (3H, d, J = 7.3 Hz, H-27), 1.145 (3H, s, H-28), 1.076 (3H, s, H-29), 1.326 (1H, s, H-30), 7.116 (1H, d, J = 6.9 Hz, NH), 5.420 (1H, d, J = 6.9 Hz, H-2′ of PGME), 7.333–7.265 (5H, overlapped, phenyl group), 3.697 (3H, s, OCH3). HRESIMS m/z 684.35022 [M + Na]+ (calcd for C39H51O8NNa, 684.35124).
6Ha: 1H NMR (600 MHz, CDCl3) 2.260 (1H, m, H-1a), 2.015 (1H, m, H-1b), 2.953 (1H, m H-2a), 2.347 (1H, m, H-2b), 1.861 (1H, overlapped H-5), 2.300 (1H, overlapped, H-6a), 2.295 (1H, overlapped, H-6b), 6.510 (1H, overlapped, H-7), 5.666 (1H, overlapped, H-11), 4.585 (1H, m, H-15), 2.500 (1H, m, H-16a), 1.812 (1H, m H-16b), 3.308 (1H, m, H-17), 0.907 (3H, s, H-18), 1.095 (3H, s, H-19), 2.273 (3H, s, H-21), 6.365 (1H, s, H-22), 2.540 (1H, m H-24a), 2.540 (1H, m H-24b), 2.937 (1H, m, H-25), 1.139 (3H, d, J = 6.8 Hz, H-27), 1.122 (3H, s, H-28), 1.161 (3H, s, H-29), 1.298 (1H, s, H-30), 6.914 (1H, d, J = 7.0 Hz, NH), 5.487 (1H, d, J = 7.0 Hz, H-2′ of PGME), 7.355–7.338 (5H, overlapped, phenyl group), 3.686 (3H, s, OCH3). HRESIMS m/z 666.33997 [M + Na]+ (calcd for C39H49O7NNa, 666.34067).
6Hb: 1H NMR (600 MHz, CDCl3) 2.267 (1H, m, H-1a), 1.790 (1H, m, H-1b), 2.790 (1H, m H-2a), 2.875 (1H, m, H-2b), 1.875 (1H, overlapped H-5), 2.286 (1H, overlapped, H-6a), 2.301 (1H, overlapped, H-6b), 6.494 (1H, overlapped, H-7), 5.662 (1H, s, H-11), 4.553 (1H, m, H-15), 2.453 (1H, m, H-16a), 1.477 (1H, m H-16b), 3.267 (1H, m, H-17), 1.295 (3H, s, H-18), 1.080 (3H, s, H-19), 2.197 (3H, s, H-21), 6.289 (1H, s, H-22), 2.510 (1H, m H-24a), 2.510 (1H, m H-24b), 2.914 (1H, m, H-25), 1.200 (3H, d, J = 7.2 Hz, H-27), 1.120 (3H, s, H-28), 1.160 (3H, s, H-29), 0.821 (1H, s, H-30), 6.803 (1H, d, J = 7.2 Hz, NH), 5.516 (1H, d, J = 7.2 Hz, H-2′ of PGME), 7.360–7.293 (5H, overlapped, phenyl group), 3.722 (3H, s, OCH3). HRESIMS m/z 666.33978 [M + Na]+ (calcd for C39H49O7NNa, 666.34067).
2.6. Biological Activity Assays
Biological activity assays, including the cytotoxicity against five human cancer cell lines [24], α-glucosidase inhibition [25], protein tyrosine phosphatase 1 β (PTP1B) [26], dipeptidyl peptidase 4 (DDP4) [27], and angiotensin converting enzyme 2 (ACE2) [28], were screened according to the protocols in the Supplementary Materials.
3. Results and Discussion
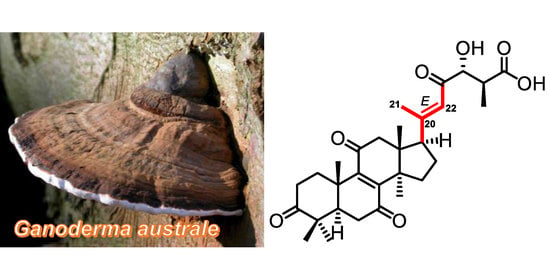
Compound 1 (Figure 1), obtained as a yellow oil, gave an [M + Na]+ ion peak at m/z 535.26685 in the HRESIMS (calcd for C30H40O7Na, 535.26717). The 1H NMR spectroscopic data (Table 1) displayed six methyl singlets at δH 1.28 (Me-19), δH 0.75 (Me-18), δH 2.16 (Me-21), δH 1.13 (Me-28), δH 1.10 (Me-29), and δH 1.35 (Me-30), one methyl doublet at δH 1.18 (d, J = 7.3 Hz, Me-27), an olefinic proton at δH 6.51 (s, H-22), and an oxygenated methine proton at δH 4.21 (d, J = 4.9 Hz, H-24). The 13C NMR and DEPT spectroscopic data (Table 2) of 1 showed 30 carbon resonances which were ascribed to seven methyl carbons at δC 19.1 (C-18), 18.4 (C-19), 22.2 (C-21), 13.8 (C-27), 27.7 (C-28), 20.7 (C-29), and 26.4 (C-30), six methylenes at δC 36.1 (C-1), 35.0 (C-2), 37.7 (C-6), 50.9 (C-12), 33.5 (C-15), and 24.2 (C-16), four methines at δC 50.8 (C-5), 54.6 (C-17), 88.0 (C-24), and 44.6 (C-25), two pairs of olefinic carbons at δC 151.3 (C-8), 152.4 (C-9), 161.3 (C-20), and 122.0 (C-22), four sp3-quaternary carbons at δC 47.8 (C-4), 40.1 (C-10), 49.7 (C-13, C-14), and four carbonyls at δC 218.6 (C-3), 202.7 (C-7), 202.8 (C-11), 202.0 (C-23), and 176.9 (C-27). The chemical shifts of 1D NMR of 1 indicated that it was a lanostane triterpenoid similar to resinacein N, except for the substitutions at C-3, C-7, and C-15 [29]. In the HMBC spectrum of 1, the correlations from Me-29 to the carbonyl C-3, from Me-30 to the methylene carbon C-15, and from H-5 (δH 2.38) and H-6 (δH 2.36, 2.68) to the carbonyl C-7, along with the 1H-1H COSY correlation of H-15 (δH 2.24, 1.87)/H-16 (δH 2.04, 1.91) (Figure 2), suggested that C-3 and C-7 were ketone carbons and C-15 was a methylene instead of being a hydroxylated methine in resinacein N. Therefore, the planar structure of 1 was elucidated as shown in Figure 1.
The key ROESY correlations between H-22 (δH 6.51) and H-16a/b (δH 2.04, 1.91) allowed the assignment of the E configuration of the C-20–C-22 double bond (Figure 3). The absolute configuration of the chiral center C-25 was determined by the PGME method (Figure 4). The (R)- and (S)-PGME amide derivatives were chemically synthesized, and the ΔδH (δS − δR) values indicated that C-25 was the S configuration. The attempt to assign the absolute configuration of C-24 by Mosher’s method failed, probably due to the bulky groups around the hydroxy group. Therefore, the configuration of C-24 remained unassigned. Compound 1 was elucidated as [20(22)E,24R,25R]-24-hydroxy-3,7,11,23-tetraoxolanosta-8,20-dien-26-oic acid, and was given the trivial name ganoaustralenone A.
Compound 2, obtained as a white powder, displayed an [M + Na]+ peak at m/z 551.26422 in the HRESIMS (calcd for C30H40O8Na, 551.26209). The 1D NMR data of 2 (Table 1 and Table 2) showed a resemblance to those of compound 1, implying the analogous structures of the two compounds. Analysis of the 1D NMR data suggested that the only difference between 1 and 2 was C-6. The HMBC correlation from H-5 to a hydroxymethine at δC 72.2 (C-6), as well as the 1H-1H COSY correlation of H-5 and the proton at δH 4.44 (H-6) (Figure 2), revealed that the C-6 in 2 attached to a hydroxy group compared to that of 1. These assignments are consistent with the HRESIMS result. The absolute configuration of C-25 was determined by the PGME method, as in the case of compound 1 (Figure 4). Therefore, compound 2 was determined as shown in Figure 1, and trivially named ganoaustralenone B.
Compound 3, obtained as a yellow oil, displayed an [M + Na]+ ion peak at m/z 537.28180 in the HRESIMS analysis (calcd for C30H42O7Na, 537.28227). The 1H NMR and 13C NMR data of 3 (Table 1 and Table 2) highly resemble those of 1, except for the chemical shift of C-7. The key 1H-1H COSY correlations H-5 (δH 2.11)/H-6 (δH 1.70)/H-7 (δH 4.47), as well as the HMBC correlation from H-7 (δH 4.47) and C-8 (δC 160.3) (Figure 2), implied the presence of a hydroxyl group at C-7. The key ROESY correlations of H-7/H-15β/H3-18 indicated the α orientation of 7-OH (Figure 3). The absolute configurations of C-25 were determined by the PGME method, as in the case of compound 1 (Figure 4). Therefore, compound 3 was determined as shown in Figure 1, and identified as ganoaustralenone C.
The yellowish oil compounds 4 and 5 gave the sodium adduct ion peaks of m/z 565.27728 and m/z 549.28210 in the HRESIMS analysis, corresponding to the molecular formulas of C31H42O8 and C31H42O7 (calcd for C31H42O8Na 565.27774; C31H42O7Na, 549.28282), respectively. The 1D NMR spectra of the two compounds (Table 1 and Table 2) showed characteristic signals of triterpene, indicating the same skeletons of 1–5. Analysis of the 1D NMR spectra of 4 and 5 suggested that the two compounds were highly similar to those of 1 and 2, respectively. The differences between these two pairs of compounds (1 vs. 4, 2 vs. 5) were the status of C-26 carboxylic group. The correlations from the methoxy singlets to the carbonyl group (C-26) in the HMBC spectra of 4 and 5 (Figure 2) indicated that C-26 of 4 and 5 have been methyl esterified instead of being free carboxylic groups in 1 and 2. Therefore, compounds 4 and 5 were elucidated as the C-26 methyl ester derivatives of 1 and 2, respectively. However, these changes hampered the absolute configuration determination of C-25 of 4 and 5 by the PGME method. The relative configurations of C-24 and C-25 were assigned as R* and S*, respectively, by analysis of the Newman projections of C-24–C-25 and the coupling constants of H-24 (4.0 Hz, and 3.1 Hz). Therefore, compounds 4 and 5 were named ganoaustralenones D and E, respectively.
Compound 6 was determined to be methyl gibbosate O by comparison with the NMR spectroscopic data (Supplementary Materials) [30,31]. However, the chemical shifts of C-13 and C-14 of methyl gibbosate O have been erroneously assigned previously [30]. The key HMBC correlation of H-11 (δH 5.66, s) to an sp3-quaternary carbon at δC 58.0, together with the HMBC correlation from H-7 (δH 6.50, m) to an sp3-quaternary carbon at δC 52.5 enabled the correct assignment of the chemical shifts of C-13 (58.0 ppm) and C-14 (52.5 ppm). Moreover, the absolute configuration of C-25 of gibbosic acid O was assigned as S without any evidence [31], while for methyl gibbosate O, the C-25 configuration was assigned to be same with gibbosic acid O only by comparison with the chemical shifts [30]. However, C-25 is far away from any other chiral centers in the structure, so the chemical shift deviation is inadequate to discriminate the S and R configuration of C-25. Therefore, more solid evidence should be presented to corroborate the real configuration of C-25. In order to determine the absolute configuration of the chiral center C-25, compound 6 was firstly hydrolyzed by LiOH to obtain the previously reported compound gibbosic acid O (6H). Then, the (R)- and (S)-PGME amide derivatives of 6H were chemically synthesized (Scheme 1), and the ΔδH (δS − δR) values indicated that C-25 was the S configuration (Figure 4). Therefore, the absolute configuration of compound 6 has been fully assigned.
The HRESIMS analysis of 7, a yellow oily compound, gave a sodium adduct ion peak at m/z 547.30286, corresponding to the molecular formula of C32H44O6 (calcd for C32H44O6Na, 547.30356) with 11 double bond equivalences. Comparing the 1D NMR data of 7 (Table 2 and Table 3) with those of 6 suggested that 7 differed from 6 with the presence of an oxygenated methylene and a triplet methyl group with the absence of the methoxy group. These signals were assigned to be ethyl ester moiety of the C-26 carbonyl group instead of the methyl ester moiety in 6. The 1H-1H COSY correlation of OCH2CH3 (δH 4.13)/OCH2CH3 (δH 1.25), and the HMBC correlation from OCH2CH3 (δH 4.13) to C-27 (δC 176.3) (Figure 2), confirmed the above assignments. Notably, 15-OH was assigned to be β orientation by the key ROESY correlation of H-15 (δH 4.31)/Me-30 (δH 1.00) (Figure 3). Therefore, compound 7 was named ganoaustralenone F.
Compound 8 had an [M + Na]+ ion peak at m/z 561.28223 (C32H42O7Na) in the HRESIMS analysis (calcd for C32H42O7Na, 561.28282). The molecular formula of 8 is two oxygen atoms more than that of 7, indicating the existence of more oxygenated carbons in 8 than those of 7. The 1H NMR spectra of 8 (Table 3) displayed six methyl singlets (δH 1.12, 1.08, 2.26, 1.12, 1.09, and 1.29). The 13C NMR (Table 4) and DEPT spectra of 8 exhibited signals for eight methyls, six methylenes (one was oxygenated at δC 60.7), six methines including two sp2-ones and four sp3-ones (one was oxygenated, δC 58.9), and eleven quaternary carbons (four carbonyls, five sp3-ones, and two sp2-ones). Further analysis of the 1D NMR data (Table 3 and Table 4) allowed the assignment of 8 to be an analog of 7, except for the positions of C-7, C-8, and C-15. The 13C NMR chemical shifts of these three positions (δC 58.9, C-7; δC 62.7, C-8; δC 209.9, C-15) of 8 implied that an epoxy ring was located at C-7 and C-8, while C-15 was a ketone compared to that of 7. These assignments were corroborated by the 1H-1H COSY correlations of H-5/H-6/H-7 and the HMBC correlations from H-7 to C-8, and from Me-30 to C-15 (Figure 2). Thus, compound 8 was trivially named ganoaustralenone G.
Compound 9 showed an [M + H]+ peak at m/z 513.32135 in the HRESIMS, indicating the molecular formula C31H44O6 (calcd for C31H45O6, 513.32161). The 1D NMR data of 9 (Table 3 and Table 4) displayed thirty-one carbon resonances, which were categorized into seven methyl carbons at δC 17.7 (C-19), 18.6 (C-18), 21.4 (C-21), 17.3 (C-27), 27.8 (C-28), 20.6 (C-29), and 27.8 (C-30), one methoxy carbon at δC 52.0, seven methylenes at δC 34.2 (C-1), 34.9 (C-2), 29.4 (C-6), 50.3 (C-12), 30.4 (C-15), 23.2 (C-16), and 47.9 (C-24), five methines at δC 45.2 (C-5), 67.3 (C-7), 54.1 (C-17), 123.8 (C-22), and 35.0 (C-25), and eleven proton-free carbons at δC 218.0 (C-3), 46.5 (C-4), 159.5 (C-8), 140.2 (C-9), 38.0 (C-10), 195.5 (C-11), 48.6 (C-13), 50.6 (C-14), 158.0 (C-20), 198.4 (C-23), and 176.7 (C-26). The above data suggested that 9 was a similar structure to 7β-hydroxy-3,11,15,23-tetraoxolanosta-8,20E(22)-diene-26-oic acid methyl ester, except for the position at C-15 and the configuration of 7-OH [32]. The 1H-1H COSY correlations of H-15/H-16/H-17, as well as the HMBC correlation from Me-30 (δH 1.35, s) to the methylene carbon at δC 30.4 (C-15) (Figure 2), suggested that C-15 in 9 was a methylene instead of being a ketone carbon in 7β-hydroxy-3,11,15,23-tetraoxolanosta-8,20E(22)-diene-26-oic acid methyl ester. In addition, the key ROESY correlations of H-7 (δH 4.47)/H-15β (δH 2.04)/Me-18 (δH 0.67) (Figure 3) suggested that 7-OH was an α orientation. Therefore, compound 9 was named ganoaustralenone H.
Compound 10, a pale yellow oil, gave an [M + Na]+ ion peak at m/z 563.29749 (C32H44O7Na) in the HRESIMS (calcd for C32H44O7Na, 563.29847). The 1H and 13C NMR spectroscopic data of 10 (Table 3 and Table 4) showed high similarity to those of the structure 15α-hydroxy-3,11,23-trioxolanosta-8,20E(22)-dien-26-oic acid methyl ester, a lanostane triterpenoid isolated from the G. lucidum [33]. Further analysis of the 2D NMR spectra revealed that the only difference between these two structures was C-7. The diagnostic HMBC correlations from the protons at δH 2.26 (H-5), 2.49 (H-6a), 2.62 (H-6b) to a carbonyl group at δC 204.4 (Figure 2) suggested that C-7 was a carbonyl group in 10 instead of being a methylene group in 15α-hydroxy-3,11,23-trioxolanosta-8,20E(22)-dien-26-oic acid methyl ester. In addition, the alcohol for forming the C-26 ester group was ethanol in 10 instead of methanol in the reported structure, as supported by the two chemical shifts at δC 60.6 (-OCH2CH3) and 14.2 (-OCH2CH3). Therefore, compound 10 was identified as ethyl 20(22)E-15α-hydroxy-3,7,11,23-tetraoxolanosta-8,20(22)-dien-26-oate, and was trivially named ganoaustralenone I.
Compound 11, obtained as a yellow oil, displayed an [M + Na]+ ion peak at m/z 549.28210 in the HRESIMS analysis (calcd for C31H42O7Na, 549.28282), revealing the molecular formula of C31H42O7. The 1D NMR data of 11 (Table 4 and Table 5) showed 30 carbon resonances with high resemblance to those of compound 10. Further analysis of the 2D NMR data (Figure 2 and Figure 3) suggested that 11 differed from 10 by the presence of the methyl ester group. The significant HMBC correlation from the methoxy group (δH 3.68) to the carbonyl group C-26 (δC 176.6) (Figure 2) verified the terminal carboxylic group in 11 has been methyl esterified instead of being ethyl esterified in 10. Therefore, compound 11 was identified as ganoaustralenone J.
The pale yellow oil compound 12 exhibited an [M + Na]+ ion peak at m/z 565.31287 in the HRESIMS analysis (calcd for C32H46O7Na, 565.31412). The NMR spectroscopic data of 12 (Table 4 and Table 5) highly resemble those of 9, except for the chemical shifts of C-24 and the alcoholic part of the C-26 ester. The important HMBC correlations from Me-27 (δH 1.30) to a hydroxymethine at δC 78.6 (C-24) (Figure 2), together with the chemical shifts of the alcoholic part [δC 61.0 (-OCH2CH3) and 14.3 (-OCH2CH3)], indicated that a hydroxy group situated at C-24 and the presence of ethyl ester of C-26 in 12 compared to those of 9. Therefore, compound 12 was identified as ganoaustralenone K.
Compound 13, obtained as a yellow oil, displayed an [M + Na]+ ion peak at m/z 563.29688 in the HRESIMS analysis (calcd for C32H44O7Na, 563.29792). Analysis of the 1H and 13C NMR data (Table 4 and Table 5) revealed that this compound was a structural analog to 12. The main difference between the NMR data of the two analogs was the position C-7 (δC 201.2), which indicated that C-7 was a carbonyl carbon. In the HMBC spectrum of compound 13, significant correlations from H2-6 (δH 2.48, 2.34) to C-7 (δC 201.2) (Figure 2) indicated that C-7 was a carbonyl carbon. Therefore compound 13 was identified as ganoaustralenone L.
The identification of a series of 20(22)E-lanostanes from this species of Ganoderma inspired a proposal of the possible biosynthetic pathways. Take compound 1 as an example, as shown in Scheme 2, the common precursor squalene, which was derived from two molecules of farnesyl pyrophosphate, which was oxygenated and followed by function migration to give the lanostane scaffold. The lanosterol was oxygenated at the positions of C-3, C-7, C-11, C-20, C-23, and C-26 to give the key intermediate A, which underwent an elimination reaction by the E1cb mechanism to yield B. Finally, a nucleophilic attack at C-24 by a hydroxy group produced compound 1.
All the isolates were subjected to evaluate their cytotoxicity against the five human cancer cell lines (the HL-60 (ATCC CCL-240), the human myeloid leukemia cell line, the SMMC-7721 human hepatocellular carcinoma cell line, the A549 (ATCC CCL-185) lung cancer cell line, the MCF-7 (ATCC HTB-22) breast cancer cell line, and the SW-480 (ATCC CCL-228) human colon cancer cell line, as well as the inhibitory activity against α-glucosidase, protein tyrosine phosphate 1 β (PTP1B), dipeptidyl peptidase 4 (DDP4), and angiotensin-converting enzyme 2 (ACE2). However, no significant bioactivity was observed.
4. Conclusions
In conclusion, twelve previously undescribed lanostane-type triterpenes were obtained from the medicinal mushroom Ganoderma australe. By using the NMR and HRESIMS techniques for structural elucidation, the structures of twelve triterpenes were determined, and the absolute configurations of 1, 2, and 6 were assigned by the phenylglycine methyl ester (PGME) method. Ganoderma triterpenes have been reported to have more than 400 chemical entities to date [15]. Most of them were oxygenated at the positions of C-3, C-7, C-11, C-15, and C-26. Interestingly, more and more studies have revealed that there was an oxygenated position bias that differed from species to species. The triterpenes described here are featured by an unusual 20(22)-trans double bond, which has rarely been found in the Ganoderma lanostanoid family. Although no significant biological activities were found in this study, the results have also initiated the understanding of the structural diversity of Ganoderma-derived triterpenoids.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/jof8050503/s1, including the NMR, HRMS data of compounds 1–13.
Author Contributions
H.-P.C. and Z.-H.L. designed and supervised the project; L.Z. and L.-L.G. performed the experiments; L.Z., L.-L.G., M.I., Z.-H.L. and H.-P.C. analyzed the data, discussed the results, and wrote the paper; L.Z. and L.-L.G. contributed equally to this work. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Natural Science Foundation of China (21961142008, 81903512) and the Thailand Research Fund (DBG6280008).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available in this manuscript and can be requested from the corresponding author.
Acknowledgments
We thank the Analytical and Measuring Center, School of Pharmaceutical Sciences, South-Central University for Nationalities for the spectra measurement. We thank the Bioactivity Screening Center of Natural Products, Kunming Institute of Botany, Chinese Academy of Sciences for the biological activity tests of the isolated compounds.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Dinis-Oliveira, R.J. Metabolism of psilocybin and psilocin: Clinical and forensic toxicological relevance. Drug Metab. Rev. 2017, 49, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Anke, T.; Oberwinkler, F.; Steglich, W.; Schramm, G. The strobilurins-New antifungal antibiotics from the basidiomycete Strobilurus tenacellus. J. Antibiot. 1977, 30, 806–810. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Guo, X.F.; Fan, Z.J.; Zhang, N.L.; Zhu, Y.J.; Zhang, Z.M.; Khazhieva, I.; Yurievich, M.Y.; Belskaya, N.P.; Bakulev, V.A. Synthesis and fungicidal activity of 3,4-dichloroisothiazole based strobilurins as potent fungicide candidates. RSC Adv. 2017, 7, 3145–3151. [Google Scholar] [CrossRef] [Green Version]
- Richter, C.; Wittstein, K.; Kirk, P.M.; Stadler, M. An assessment of the taxonomy and chemotaxonomy of Ganoderma. Fungal Divers. 2014, 71, 1–15. [Google Scholar] [CrossRef]
- Fatmawati, S.; Shimizu, K.; Kondo, R. Ganoderic acid Df, a new triterpenoid with aldose reductase inhibitory activity from the fruiting body of Ganoderma lucidum. Fitoterapia 2010, 81, 1033–1036. [Google Scholar] [CrossRef]
- Mizushinaa, Y.; Hanashimaa, L.; Yamaguchib, T.; Takemurac, M.; Sugawaraa, F.; Saneyoshib, M.; Matsukaged, A.; Yoshidac, S.; Sakaguchi, K. A Mushroom Fruiting Body-Inducing Substance Inhibits Activities of Replicative DNA Polymerases. Biochem. Biophys. Res. Commun. 1998, 249, 17–22. [Google Scholar] [CrossRef]
- Nishitoba, T.; Oda, K.; Sato, H.; Sakamura, S. Novel triterpenoids from the fungus Ganoderma lucidum. Agric. Biol. Chem. 1988, 52, 367–372. [Google Scholar] [CrossRef]
- Wasser, S.P. Reishi or ling zhi (Ganoderma lucidum). Encycl. Diet. Suppl. 2005, 1, 603–622. [Google Scholar] [CrossRef]
- Wasser, S.P.; Weis, A.L. Therapeutic effects of substances occurring in higher Basidiomycetes mushrooms: A modern perspective. Crit. Rev. Immunol. 1999, 19, 65–96. [Google Scholar] [CrossRef]
- Peng, X.-R.; Liu, J.-Q.; Wang, C.-F.; Li, X.-Y.; Shu, Y.; Zhou, L.; Qiu, M.-H. Hepatoprotective effects of triterpenoids from Ganoderma cochlear. J. Nat. Prod. 2014, 77, 737–743. [Google Scholar] [CrossRef]
- Boh, B.; Berovic, M.; Zhang, J.; Zhi-Bin, L. Ganoderma lucidum and its pharmaceutically active compounds. Biotechnol. Annu. Rev. 2007, 13, 265–301. [Google Scholar] [CrossRef] [PubMed]
- Amaral, A.E.; Carbonero, E.R.; Simão, R.D.C.G.; Kadowaki, M.K.; Sassaki, G.L.; Osaku, C.A.; Gorin, P.A.; Iacomini, M. An unusual water-soluble β-glucan from the basidiocarp of the fungus Ganoderma resinaceum. Carbohydr. Polym. 2008, 72, 473–478. [Google Scholar] [CrossRef]
- Peng, X.-R.; Liu, J.-Q.; Han, Z.-H.; Yuan, X.-X.; Luo, H.-R.; Qiu, M.-H. Protective effects of triterpenoids from Ganoderma resinaceum on H2O2-induced toxicity in HepG2 cells. Food Chem. 2013, 141, 920–926. [Google Scholar] [CrossRef] [PubMed]
- Zengin, G.; Sarikurkcu, C.; Gunes, E.; Uysal, A.; Ceylan, R.; Uysal, S.; Gungor, H.; Aktumsek, A. Two Ganoderma species: Profiling of phenolic compounds by HPLC–DAD, antioxidant, antimicrobial and inhibitory activities on key enzymes linked to diabetes mellitus, Alzheimer’s disease and skin disorders. Food Funct. 2015, 6, 2794–2802. [Google Scholar] [CrossRef] [PubMed]
- Baby, S.; Johnson, A.J.; Govindan, B. Secondary metabolites from Ganoderma. Phytochemistry 2015, 114, 66–101. [Google Scholar] [CrossRef] [PubMed]
- Xia, Q.; Zhang, H.; Sun, X.; Zhao, H.; Wu, L.; Zhu, D.; Yang, G.; Shao, Y.; Zhang, X.; Mao, X.; et al. A Comprehensive Review of the Structure Elucidation and Biological Activity of Triterpenoids from Ganoderma spp. Molecules 2014, 19, 17478–17535. [Google Scholar] [CrossRef]
- Paterson, R.R.M. Ganoderma–A therapeutic fungal biofactory. Phytochemistry 2006, 67, 1985–2001. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.-C.; Yang, L.; Ma, Q.-Y.; Ge, Y.-Z.; Kong, F.-D.; Zhou, L.-M.; Zhang, F.; Xie, Q.-Y.; Yu, Z.-F.; Dai, H.-F.; et al. Triterpenoids and meroterpenoids with α-glucosidase inhibitory activities from the fruiting bodies of Ganoderma australe. Bioorganic Chem. 2021, 117, 105448. [Google Scholar] [CrossRef]
- León, F.; Valencia, M.; Rivera, A.; Nieto, I.; Quintana, J.; Estévez, F.; Bermejo, J. Novel Cytostatic Lanostanoid Triterpenes from Ganoderma australe. Helvetica Chim. Acta 2003, 86, 3088–3095. [Google Scholar] [CrossRef]
- Isaka, M.; Chinthanom, P.; Mayteeworakoon, S.; Laoteng, K.; Choowong, W.; Choeyklin, R. Lanostane triterpenoids from cultivated fruiting bodies of the basidiomycete Ganoderma australe. Nat. Prod. Res. 2017, 32, 1044–1049. [Google Scholar] [CrossRef]
- Jain, A.C.; Gupta, S.K. The isolation of lanosta-7,9(11),24-trien-3β,21-diol from the fungus Ganoderma australe. Phytochemistry 1984, 23, 686–687. [Google Scholar] [CrossRef]
- Zhang, J.-J.; Dong, Y.; Qin, F.-Y.; Yan, Y.-M.; Cheng, Y.-X. Meroterpenoids and alkaloids from Ganoderma australe. Nat. Prod. Res. 2019, 35, 3226–3232. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.-J.; Dong, Y.; Qin, F.-Y.; Cheng, Y.-X. Australeols A−F, neuroprotective meroterpenoids from Ganoderma australe. Fitoterapia 2019, 134, 250–255. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.-P.; Zhao, Z.-Z.; Li, Z.-H.; Dong, Z.-J.; Wei, K.; Bai, X.; Zhang, L.; Wen, C.-N.; Feng, T.; Liu, J.-K. Novel Natural Oximes and Oxime Esters with a Vibralactone Backbone from the Basidiomycete Boreostereum vibrans. ChemistryOpen 2016, 5, 142–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Li, Z.-H.; Yao, J.-N.; Peng, Y.-L.; Huang, R.; Feng, T.; Liu, J.-K. Isoindolinone-containing meroterpenoids with α-glucosidase inhibitory activity from mushroom Hericium caput-medusae. Fitoterapia 2017, 122, 107–114. [Google Scholar] [CrossRef]
- Ren, Y.-M.; Zhang, R.; Feng, Z.; Ke, C.-Q.; Yao, S.; Tang, C.; Lin, L.; Ye, Y. Macrocephatriolides A and B: Two Guaianolide Trimers from Ainsliaea macrocephala as PTP1B Inhibitors and Insulin Sensitizers. J. Org. Chem. 2021, 86, 17782–17789. [Google Scholar] [CrossRef]
- Zhu, Y.; Zhang, P.; Yu, H.; Li, J.; Wang, M.W.; Zhao, W. Anti-Helicobacter pylori and Thrombin Inhibitory Components from Chinese Dragon’s Blood, Dracaena cochinchinensis. J. Nat. Prod. 2007, 70, 1570–1577. [Google Scholar] [CrossRef]
- Liao, W.; Bhullar, K.S.; Chakrabarti, S.; Davidge, S.T.; Wu, J. Egg White-Derived Tripeptide IRW (Ile-Arg-Trp) Is an Activator of Angiotensin Converting Enzyme 2. J. Agric. Food Chem. 2018, 66, 11330–11336. [Google Scholar] [CrossRef]
- Chen, X.-Q.; Zhao, J.; Chen, L.-X.; Wang, S.-F.; Wang, Y.; Li, S.-P. Lanostane triterpenes from the mushroom Ganoderma resinaceum and their inhibitory activities against α-glucosidase. Phytochemistry 2018, 149, 103–115. [Google Scholar] [CrossRef]
- Peng, X.-R.; Wang, Q.; Su, H.-G.; Zhou, L.; Xiong, W.-Y.; Qiu, M.-H. Anti-Adipogenic Lanostane-Type Triterpenoids from the Edible and Medicinal Mushroom Ganoderma applanatum. J. Fungi 2022, 8, 331. [Google Scholar] [CrossRef]
- Pu, D.; Li, X.; Lin, J.; Zhang, R.; Luo, T.; Wang, Y.; Gao, J.; Zeb, M.A.; Zhang, X.; Li, X.; et al. Triterpenoids from Ganoderma gibbosum: A Class of Sensitizers of FLC-Resistant Candida albicans to Fluconazole. J. Nat. Prod. 2019, 82, 2067–2077. [Google Scholar] [CrossRef] [PubMed]
- Shim, S.H.; Ryu, J.; Kim, J.S.; Kang, S.S.; Xu, Y.; Jung, S.H.; Lee, Y.S.; Lee, S.; Shin, K.H. New Lanostane-Type Triterpenoids from Ganoderma applanatum. J. Nat. Prod. 2004, 67, 1110–1113. [Google Scholar] [CrossRef] [PubMed]
- Su, H.-G.; Peng, X.-R.; Shi, Q.-Q.; Huang, Y.-J.; Zhou, L.; Qiu, M.-H. Lanostane triterpenoids with anti-inflammatory activities from Ganoderma lucidum. Phytochemistry 2020, 173, 112256. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Chemical structures of compounds 1–13. (Red: known compounds.)

Figure 2.
Key 1H-1H COSY and HMBC correlations of compounds 1−5, 7−13.

Figure 3.
Key ROESY correlations of 1–5, 7–13.

Figure 4.
The structures and ΔδH (δS − δR) of (S)/(R)-PGME derivative of 1, 2, 3, and 6H.

Scheme 1.
Alkaline hydrolysis and PGME derivatization of compound 6.

Scheme 2.
Plausible biosynthetic pathways to 1.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
1H NMR Spectroscopic Data of Compounds 1−5.
| No. | 1 ad | 2 ac | 3 bc | 4 ae | 5 ac |
|---|---|---|---|---|---|
| 1 | 2.87, ddd (14.3, 8.5, 5.8) | 2.85, ddd (14.2, 9.8, 7.6) | 2.93, overlapped | 2.75, ddd (13.8, 9.7, 7.8) | 2.95, ddd (14.2, 8.4, 6.2) |
| 1.82, ddd (14.3, 9.7, 6.2) | 1.84, ddd (14.2, 11.9, 2.1) | 1.70, overlapped | 1.94, ddd (13.8, 12.0, 2.5) | 1.73, ddd (14.2, 9.2, 6.7) | |
| 2 | 2.71, ddd (15.2, 9.7, 5.8) | 2.74, ddd (14.4, 11.9, 7.6) | 2.60, ddd (15.5, 9.8, 6.2) | 2.86, ddd (14.5, 12.0, 7.8) | 2.60, ddd (15.5, 9.2, 6.2) |
| 2.45, ddd (15.2, 8.5, 6.2) | 2.33, ddd (14.4, 9.8, 2.1) | 2.44, ddd (15.0, 8.3, 6.2) | 2.16, ddd (14.5, 9.7, 2.5) | 2.50, ddd (15.5, 8.4, 6.7) | |
| 5 | 2.38, dd (14.7, 2.4) | 2.31, d (13.7) | 2.11, dd (9.9, 5.5) | 2.46, d (13.6) | 2.25, dd (15.3, 2.6) |
| 6 | 2.68, t (14.7) | 4.44, d (13.7) | 1.70, overlapped, 2H | 4.57, d (13.6) | 2.53, dd (15.3, 14.7) |
| 2.36, dd (14.7, 2.4) | 2.40, dd (14.7, 2.6) | ||||
| 7 | 4.47, t (2.3) | ||||
| 12 | 2.96, d (16.3) | 2.83, d (17.0) | 2.77, d (17.3) | 3.02, d (16.9) | 2.81, d (16.2) |
| 2.41, d (16.3) | 2.51, d (17.0) | 2.38. d (17.3) | 2.45, d (16.9) | 2.47, d (16.2) | |
| 15 | 2.24, m | 2.29, overlapped | 1.86, m | 2.22, overlapped | 2.30, ddd (12.4, 8.3, 2.7) |
| 1.87, overlapped | 1.74, m | 2.05, m | 1.81, td (12.0, 7.0) | 1.83, m | |
| 16 | 2.04, m | 1.95, overlapped, 2H | 1.98, m, 2H | 2.07, overlapped | 1.92, overlapped, 2H |
| 1.91, dt (11.3, 2.6) | 1.93, overlapped | ||||
| 17 | 2.99, overlapped | 2.91, t (9.2) | 2.95, overlapped | 3.06, overlapped | 2.87, t (9.1) |
| 18 | 0.75, s | 0.69, s | 0.67, s | 0.77, s | 0.70, s |
| 19 | 1.28, s | 1.23, s | 1.00, s | 1.23, s | 1.27, s |
| 21 | 2.16, s | 2.18, s | 2.17, s | 2.18, s | 2.19, s |
| 22 | 6.51, s | 6.23, br.s | 6.25, s | 6.56, s | 6.22, br.s |
| 24 | 4.21, d (4.9) | 4.29, d (2.9) | 4.33, d (3.1) | 4.22, d (4.0) | 4.23, dd (5.7, 3.2) |
| 25 | 2.93, dd (7.3, 4.9) | 3.01, dd (7.2, 2.9) | 3.00, dd (7.2, 3.1) | 3.04, dd (7.1, 4.0) | 2.99, dd (7.2, 3.2) |
| 27 | 1.18, d (7.3) | 1.28, d (7.2) | 1.24, d (7.2) | 1.18, d (7.1) | 1.28, d (7.2) |
| 28 | 1.13, s | 1.34, s | 1.13, s | 1.30, s | 1.13, s |
| 29 | 1.10, s | 1.42, s | 1.06, s | 1.38, s | 1.11, s |
| 30 | 1.35, s | 1.38, s | 1.35, s | 1.42, s | 1.31, s |
| OCH3 | 3.57, s | 3.63, s | |||
| 6-OH | 4.17, br.s | ||||
| 24-OH | 3.89, d (5.7) |
a Measured at 600 MHz; b Measured at 500 MHz; c Measured in CDCl3; d Measured in CD3OD; e Measured in acetone-d6.
Table 2.
13C NMR Spectroscopic Data of Compounds 1−5, and 7.
| No. | 1 be | 2 bd | 3 ad | 4 bf | 5 bd | 7 cd |
|---|---|---|---|---|---|---|
| 1 | 36.1, CH2 | 35.3, CH2 | 34.9, CH2 | 35.8, CH2 | 35.0, CH2 | 36.0, CH2 |
| 2 | 35.0, CH2 | 33.4, CH2 | 34.2, CH2 | 33.7, CH2 | 34.1, CH2 | 34.5, CH2 |
| 3 | 218.6, C | 216.8, C | 218.6, C | 216.3, C | 215.6, C | 214.7, C |
| 4 | 47.8, C | 47.7, C | 46.5, C | 48.1, C | 46.8, C | 47.9, C |
| 5 | 50.8, CH | 54.9, CH | 45.0, CH | 54.8, CH | 49.8, CH | 49.9, CH |
| 6 | 37.7, CH2 | 72.2, CH | 29.3, CH2 | 72.8, CH | 37.1, CH2 | 24.3, CH2 |
| 7 | 202.7, C | 202.6, C | 67.1, CH | 203.4, C | 201.1, C | 131.2, CH |
| 8 | 151.3, C | 148.3, C | 160.3, C | 149.2, C | 150.9, C | 138.3, C |
| 9 | 152.4, C | 150.0, C | 140.1, C | 150.1, C | 150.2, C | 160.4, C |
| 10 | 40.1, C | 39.9, C | 37.9, C | 40.5, C | 39.0, C | 38.2, C |
| 11 | 202.8, C | 200.1, C | 200.1, C | 200.7, C | 200.8, C | 119.1, CH |
| 12 | 50.9, CH2 | 49.8, CH2 | 50.1, CH2 | 50.5, CH2 | 50.0, CH2 | 203.3, C |
| 13 | 49.7, C | 48.2, C | 48.9, C | 48.9, C | 48.5, C | 57.8, C |
| 14 | 49.7, C | 48.1, C | 50.8, C | 48.9, C | 48.4, C | 56.4, C |
| 15 | 33.5, CH2 | 31.8, CH2 | 30.4, CH2 | 32.6, CH2 | 32.4, CH2 | 77.0, CH |
| 16 | 24.2, CH2 | 23.4, CH2 | 23.3, CH2 | 23.8, CH2 | 23.4, CH2 | 36.9, CH2 |
| 17 | 54.6, CH | 53.5, CH | 54.4, CH | 53.9, CH | 53.6, CH | 47.8, CH |
| 18 | 19.1, CH3 | 18.7, CH3 | 18.8, CH3 | 18.9, CH3 | 18.6, CH3 | 17.3, CH3 |
| 19 | 18.4, CH3 | 19.3, CH3 | 17.7, CH3 | 19.7, CH3 | 18.0, CH3 | 21.3, CH3 |
| 20 | 161.3, C | 161.7, C | 162.6, C | 160.1, C | 161.5, C | 157.9, C |
| 21 | 22.2, CH3 | 22.2, CH3 | 21.9, CH3 | 22.0, CH3 | 22.3, CH3 | 21.0, CH3 |
| 22 | 122.0, CH | 120.0, CH | 119.6, CH | 121.4, CH | 120.1, CH | 126.2, CH |
| 23 | 202.0, C | 198.7, C | 199.0, C | 200.3, C | 198.8, C | 199.0, C |
| 24 | 80.0, CH | 78.2, CH | 78.2, CH | 79.4, CH | 78.5, CH | 47.6, CH2 |
| 25 | 44.6, CH | 43.1, CH | 43.3, CH | 44.0, CH | 43.3, CH | 35.2, CH |
| 26 | 176.9, C | 177.1, C | 177.1, C | 173.6, C | 173.5, C | 176.3, C |
| 27 | 13.8, CH3 | 13.3, CH3 | 13.0, CH3 | 13.9, CH3 | 13.6, CH3 | 20.0, CH3 |
| 28 | 27.7, CH3 | 31.2, CH3 | 27.7, CH3 | 31.2, CH3 | 27.6, CH3 | 25.2, CH3 |
| 29 | 20.7, CH3 | 19.5, CH3 | 20.6, CH3 | 19.6, CH3 | 20.5, CH3 | 22.6, CH3 |
| 30 | 26.4, CH3 | 26.4, CH3 | 27.6, CH3 | 26.4, CH3 | 26.3, CH3 | 26.5, CH3 |
| -OCH3 | 51.8, CH3 | 52.1, CH3 | ||||
| -OCH2CH3 | 60.6, CH2 | |||||
| -OCH2CH3 | 14.3, CH3 |
a Measured at 500 MHz; b Measured at 600 MHz; c Measured at 800 MHz; d Measured in CDCl3; e Measured in CD3OD; f Measured in acetone-d6.
Table 3.
1H NMR Spectroscopic Data of Compounds 7−10.
| No. | 7 ac | 8 ac | 9 bc | 10 bc |
|---|---|---|---|---|
| 1 | 2.28, overlapped | 2.10, ddd (13.9, 10.9, 6.2) | 2.61, ddd (15.5, 9.7, 6.0) | 2.94, ddd (14.4, 8.3, 6.1) |
| 1.86, m | 1.95, ddd (13.9, 9.2, 5.0) | 2.46, ddd (15.5, 8.5, 6.5) | 1.77, ddd (14.4, 9.1, 7.0) | |
| 2 | 2.81, td (14.6, 5.6) | 2.69, overlapped | 2.97, ddd (14.3, 8.5, 6.0) | 2.62, ddd (15.3, 9.1, 6.1) |
| 2.43, overlapped | 2.48, overlapped | 1.70, ddd (14.3, 9.7, 6.5) | 2.52, ddd (15.3, 8.3, 7.0) | |
| 5 | 1.70, dd (12.1, 3.6) | 2.75, dd (13.0, 3.0) | 2.09, dd (11.8, 3.9) | 2.26, dd (15.3, 2.8) |
| 6 | 2.44, overlapped | 2.17, dt (15.0, 3.0) | 1.70, overlapped | 2.62, t (15.3) |
| 2.28, overlapped | 1.75, dd (15.0, 13.0) | 2.49, dd (15.3, 2.8) | ||
| 7 | 6.27, d (6.7) | 4.68, d (3.0) | 4.47, m | |
| 11 | 5.69, s | 6.05, s | ||
| 12 | 2.76, d (17.3) | 2.87, d (16.9) | ||
| 2.38, d (17.3) | 2.44, d (16.9) | |||
| 15 | 4.31, d (6.2) | 2.04, overlapped | 4.40, ddd (10.2, 5.6, 1.8) | |
| 1.84, m | ||||
| 16 | 2.52, ddd (15.4, 8.8, 6.2) | 2.66, dd (12.7, 9.3) | 2.03, overlapped | 2.49, overlapped |
| 2.04, dd (15.4, 8.8) | 2.46, dd (12.7, 9.3) | 1.94, overlapped | 1.82, ddd (15.0, 10.2, 5.6) | |
| 17 | 3.23, t (8.8) | 3.51, t (9.3) | 2.88, t (8.7) | 2.92, overlapped |
| 18 | 1.17, s | 1.13, s | 0.67, s | 0.73, s |
| 19 | 1.36, s | 1.09, s | 1.02, s | 1.28, s |
| 21 | 2.28, s | 2.26, s | 2.11, s | 2.10, s |
| 22 | 6.44, s | 6.34, s | 6.12, s | 6.09, s |
| 24 | 2.94, overlapped | 2.95, overlapped | 2.95, ddd (20.5, 14.2) | 2.94, overlapped |
| 2.55, dd (20.4, 8.6) | 2.54, dd (20.5, 8.5) | 2.52, dd (20.5, 8.6) | 2.51, overlapped | |
| 25 | 2.94, overlapped | 2.94, overlapped | 2.96, ddd (14.2, 8.6, 6.8) | 2.94, overlapped |
| 27 | 1.18, s | 1.19, d (6.9) | 1.19, d (6.8) | 1.19, d (6.8) |
| 28 | 1.12, s | 1.12, s | 1.07, s | 1.15, s |
| 29 | 1.17, s | 1.08, s | 1.14, s | 1.12, s |
| 30 | 1.00, s | 1.29, s | 1.35, s | 1.26, s |
| -OCH3 | 3.68, s | |||
| -OCH2CH3 | 4.13, overlapped, 2H | 4.13, overlapped, 2H | 4.13, m, 2H | |
| -OCH2CH3 | 1.25, overlapped | 1.25, overlapped | 1.25, t (7.1) | |
| 15-OH | 4.48, d (1.8) |
a Measured at 800 MHz; b Measured at 600 MHz; c Measured in CDCl3.
Table 4.
13C NMR Spectroscopic Data of Compounds 8−13.
| No. | 8 cd | 9 bd | 10 bd | 11 bd | 12 bd | 13 ad |
|---|---|---|---|---|---|---|
| 1 | 36.1, CH2 | 34.2, CH2 | 35.1, CH2 | 35.2, CH2 | 34.9, CH2 | 35.1, CH2 |
| 2 | 33.7, CH2 | 34.9, CH2 | 34.0, CH2 | 37.0, CH2 | 34.2, CH2 | 34.1, CH2 |
| 3 | 216.4, C | 218.0, C | 214.9, C | 215.1, C | 218.0, C | 215.6, C |
| 4 | 45.9, C | 46.5, C | 46.6, C | 46.7, C | 46.5, C | 46.8, C |
| 5 | 40.7, CH | 45.2, CH | 49.2, CH | 49.3, CH | 45.1, CH | 49.8, CH |
| 6 | 22.8, CH2 | 29.4, CH2 | 36.9, CH2 | 34.1, CH2 | 29.4, CH2 | 37.1, CH2 |
| 7 | 58.9, CH | 67.3, CH | 204.4, C | 204.5, C | 67.3, CH | 201.2, C |
| 8 | 62.7, C | 159.5, C | 150.4, C | 150.5, C | 159.5, C | 150.9, C |
| 9 | 164.3, C | 140.2, C | 152.8, C | 153, C | 140.2, C | 150.2, C |
| 10 | 40.4, C | 38.0, C | 39.2, C | 39.3, C | 38.0, C | 39.0, C |
| 11 | 129.9, CH | 199.5, C | 200.3, C | 200.4, C | 199.3, C | 200.7, C |
| 12 | 200.7, C | 50.3, CH2 | 50.5, CH2 | 50.6, CH2 | 50.3, CH2 | 50.0, CH2 |
| 13 | 57.7, C | 48.6, C | 48.9, C | 49.0, C | 48.9, C | 48.6, C |
| 14 | 55.2, C | 50.6, C | 52.3, C | 52.4, C | 50.7, C | 48.5, C |
| 15 | 209.9, C | 30.4, CH2 | 72.4, CH | 72.5, CH | 30.4, CH2 | 32.4, CH2 |
| 16 | 38.5, CH2 | 23.2, CH2 | 32.0, CH2 | 32.1, CH2 | 23.3, CH2 | 23.4, CH2 |
| 17 | 42.9, CH | 54.1, CH | 51.9, CH | 52.0, CH | 54.5, CH | 53.9, CH |
| 18 | 18.3, CH3 | 18.6, CH3 | 18.9, CH3 | 19.0, CH3 | 18.8, CH3 | 18.6, CH3 |
| 19 | 25.0, CH3 | 17.7, CH3 | 17.7, CH3 | 17.8, CH3 | 17.7, CH3 | 18.1, CH3 |
| 20 | 154.0, C | 158.0, C | 155.8, C | 156.1, C | 162.0, C | 162.9, C |
| 21 | 20.6, CH3 | 21.4, CH3 | 21.1, CH3 | 21.2, CH3 | 22.2, CH3 | 22.5, CH3 |
| 22 | 127.0, CH | 123.8, CH | 124.5, CH | 124.5, CH | 119.6, CH | 119.6, CH |
| 23 | 198.8, C | 198.4, C | 198.2, C | 198.3, C | 198.8, C | 198.7, C |
| 24 | 47.9, CH2 | 47.9, CH2 | 47.7, CH2 | 47.9, CH2 | 78.6, CH | 77.3, CH |
| 25 | 35.1, CH | 35.0, CH | 35.0, CH | 34.9, CH | 43.2, CH | 42.8, CH |
| 26 | 176.1, C | 176.7, C | 176.0, C | 176.6, C | 173.0, C | 173.4, C |
| 27 | 17.3, CH3 | 17.3, CH3 | 17.2, CH3 | 17.3, CH3 | 13.8, CH3 | 9.5, CH3 |
| 28 | 28.8, CH3 | 27.8, CH3 | 27.4, CH3 | 27.5, CH3 | 27.7, CH3 | 27.6, CH3 |
| 29 | 21.7, CH3 | 20.6, CH3 | 20.4, CH3 | 20.5, CH3 | 20.6, CH3 | 20.5, CH3 |
| 30 | 17.8, CH3 | 27.8, CH3 | 20.7, CH3 | 20.9, CH3 | 27.7, CH3 | 26.4, CH3 |
| -OCH3 | 52.0, CH3 | 52.1, CH3 | ||||
| -OCH2CH3 | 60.7, CH2 | 60.6, CH2 | 61.0, CH2 | 61.3, CH2 | ||
| -OCH2CH3 | 29.8, CH3 | 14.2, CH3 | 14.3, CH3 | 14.3, CH3 |
a Measured at 500 MHz; b Measured at 600 MHz; c Measured at 800 MHz; d Measured in CDCl3.
Table 5.
1H NMR Spectroscopic Data of Compounds 11−13 (CDCl3).
| No. | 11 bc | 12 bc | 13 ac |
|---|---|---|---|
| 1 | 2.96, overlapped | 2.98, overlapped | 2.87, overlapped |
| 1.76, ddd (17.3, 9.9, 3.2) | 1.70, overlapped | 1.68, overlapped | |
| 2 | 2.60, overlapped | 2.61, ddd (14.9, 9.0, 5.4) | 2.55, ddd (15.5, 9.6, 6.0) |
| 2.48, overlapped | 2.45, ddd (14.9, 8.6, 6.5) | 2.45, overlapped | |
| 5 | 2.26, dd (15.1, 2.6) | 2.09, dd (9.9, 5.4) | 2.19, dd (15.1, 2.7) |
| 6 | 2.60, overlapped | 1.70, overlapped | 2.48, dd (15.1, 14.4) |
| 2.52, overlapped | 1.24, m | 2.34, dd (14.4, 2.7) | |
| 7 | 4.47, br.s | ||
| 12 | 2.87, d (16.8) | 2.78, d (17.4) | 2.76, d (16.3) |
| 2.44, d (16.8) | 2.37, d (17.4) | 2.40, d (16.3) | |
| 15 | 4.39, dd (9.7, 5.7) | 2.05, m | 2.27, overlapped |
| 1.87, m | 1.77, overlapped | ||
| 16 | 1.82, ddd (15.1, 10.2, 5.5) | 2.00, overlapped | 1.89, overlapped |
| 2.49, overlapped | 1.96, overlapped | 1.68, overlapped | |
| 17 | 2.92, overlapped | 2.96, overlapped | 2.80, overlapped |
| 18 | 0.73, s | 0.68, s | 0.63, s |
| 19 | 1.27, s | 1.02, s | 1.21, s |
| 21 | 2.10, s | 2.22, s | 2.15, s |
| 22 | 6.08, m | 6.25, s | 6.16, s |
| 24 | 2.52, overlapped | 4.21, br.s | 4.62, dd (5.2, 2.9) |
| 2.95, overlapped | |||
| 25 | 2.94, overlapped | 2.96, overlapped | 2.78, overlapped |
| 27 | 1.18, d (6.7) | 1.30, d (7.3) | 0.92, d (7.1) |
| 28 | 1.14, s | 1.14, s | 1.07, s |
| 29 | 1.12, s | 1.07, s | 1.04, s |
| 30 | 1.26, s | 1.36, s | 1.25, s |
| -OCH3 | 3.68, s | ||
| -OCH2CH3 | 4.08, m, 2H | 4.14, q (7.1), 2H | |
| -OCH2CH3 | 1.19, t (7.3) | 1.24, t (7.1) | |
| 24-OH | 3.93, br.s |
a Measured at 500 MHz; b Measured at 600 MHz; c Measured in CDCl3.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Zhou, L.; Guo, L.-L.; Isaka, M.; Li, Z.-H.; Chen, H.-P. [20(22)E]-Lanostane Triterpenes from the Fungus Ganoderma australe. J. Fungi 2022, 8, 503. https://doi.org/10.3390/jof8050503
AMA Style
Zhou L, Guo L-L, Isaka M, Li Z-H, Chen H-P. [20(22)E]-Lanostane Triterpenes from the Fungus Ganoderma australe. Journal of Fungi. 2022; 8(5):503. https://doi.org/10.3390/jof8050503
Chicago/Turabian StyleZhou, Lin, Li-Li Guo, Masahiko Isaka, Zheng-Hui Li, and He-Ping Chen. 2022. "[20(22)E]-Lanostane Triterpenes from the Fungus Ganoderma australe" Journal of Fungi 8, no. 5: 503. https://doi.org/10.3390/jof8050503
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.