
Factor XI/XIa Inhibition: The Arsenal in Development for a New Therapeutic Target in Cardio- and Cerebrovascular Disease

Abstract
:1. Introduction
2. Distinguishing Physiological Hemostasis from Pathological Thrombosis
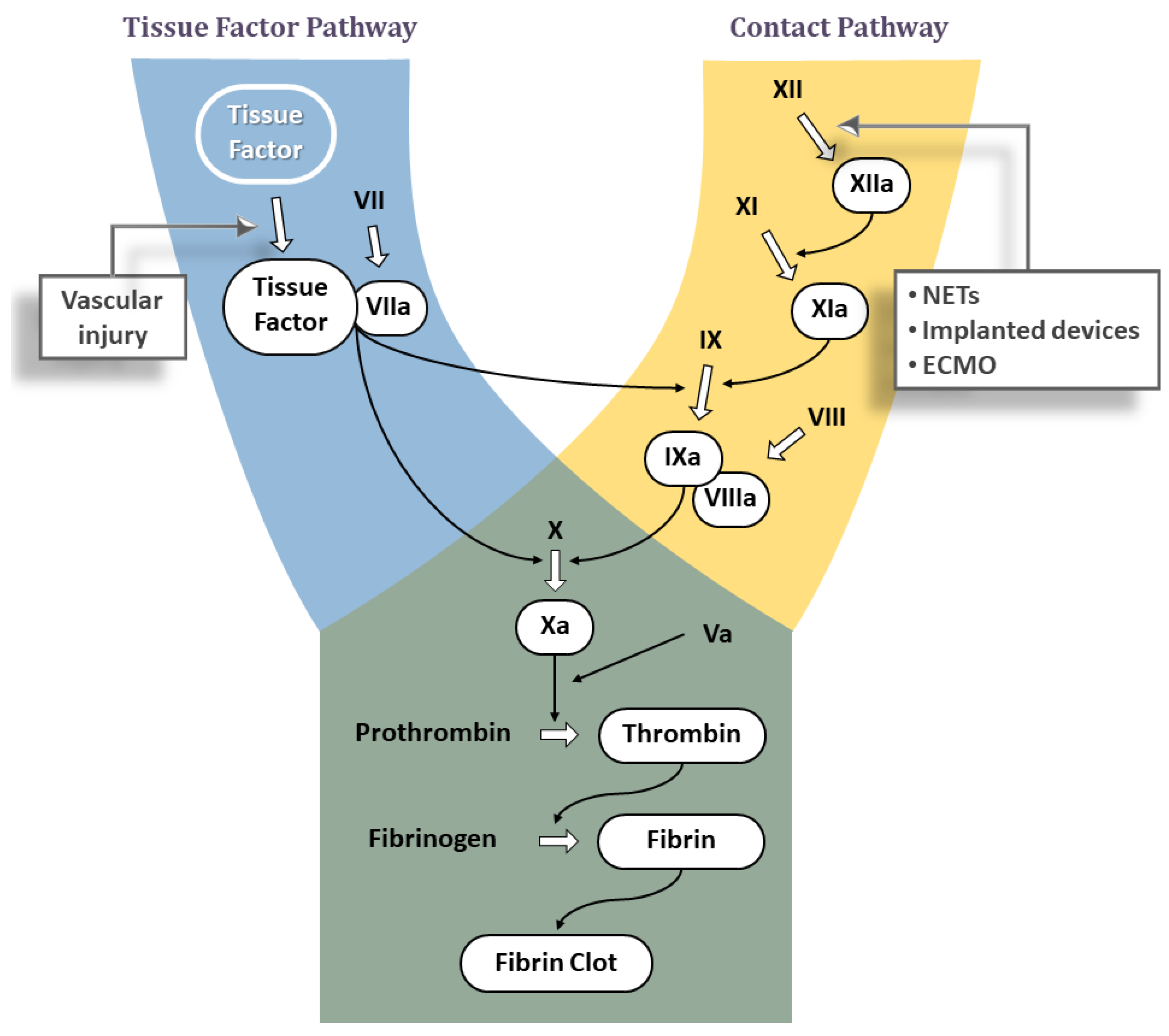
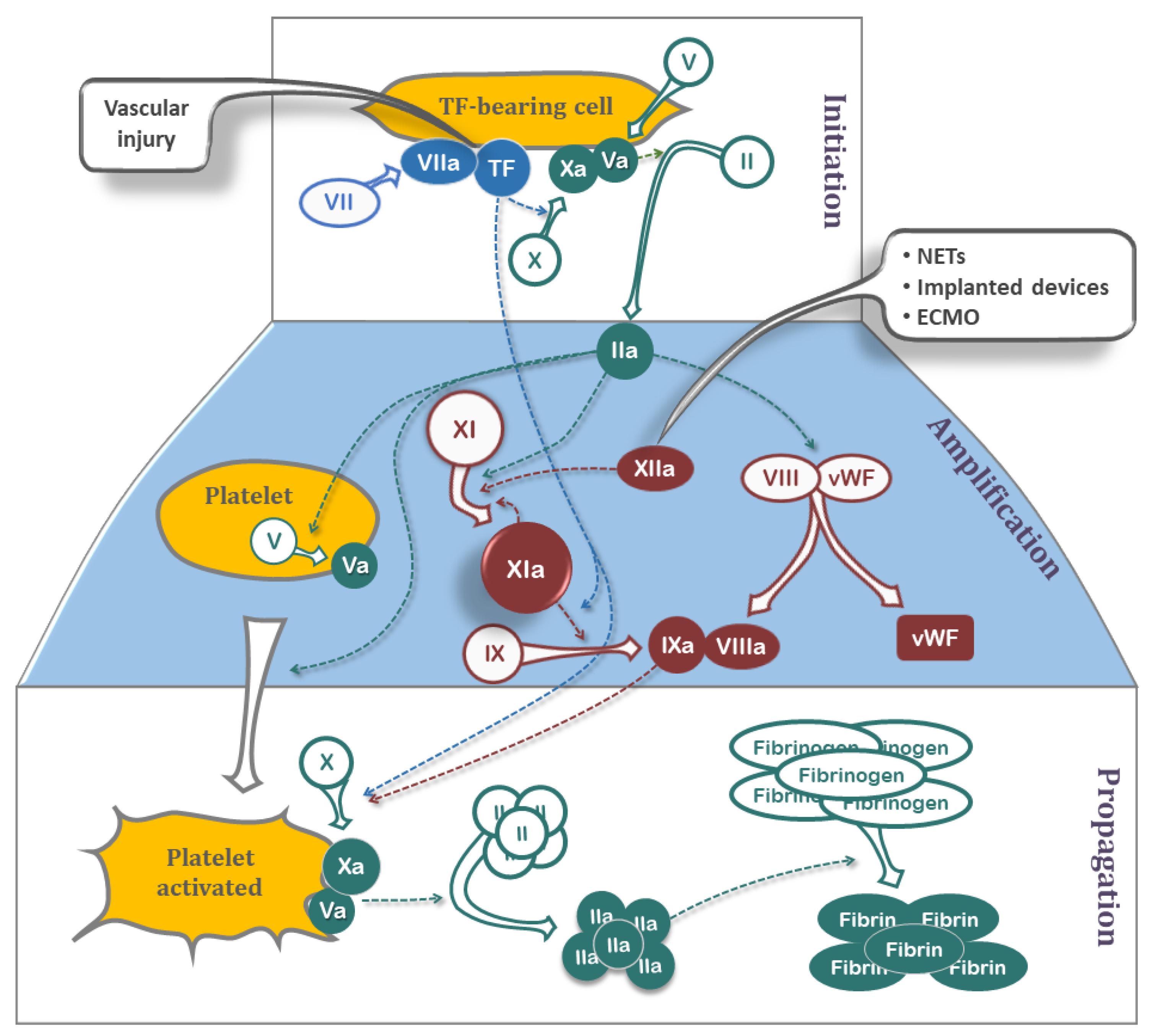
3. A Brief Review of the Classic Coagulation Cascade
3.1. Tissue Factor Pathway
3.2. Contact Pathway
Significance of Contact Pathway in Thrombosis
4. Factor XI as a Therapeutic Target
5. Pharmacologic Strategies for Factor XI Inhibition
5.1. Antisense Oligonucleotides (ASOs)
- IONIS-FXIRx.
- 2.
- IONIS-FXI-LRx.

{kind=link}
{kind=link}
{kind=link}
| Compounds | Route | Stage | Indication | N | Status |
|---|---|---|---|---|---|
| ASO 1 | |||||
| IONIS-FXIRx | S.C. 2 | Phase II | Total knee arthroplasty | 315 | Published [52] |
| Phase II | ESRD 4 | 49 | Published [53] | ||
| Phase II | ESRD 4 | 213 | Completed (NCT03358030) | ||
| IONIS-FXI-LRx | S.C. 2 | Phase II | ESRD 4 | 307 | Completed (NCT04534114) |
| Small molecule | |||||
| Asundexian | Oral | Phase II | Myocardial infarction | 1601 | Published [54] |
| Phase II | Ischemic stroke | 1808 | Published [55] | ||
| Phase II | AF 5 | 753 | Published [56] | ||
| Phase III | AF 5; Stroke and TIA 6 | 30,000 | Announced [57] | ||
| Milvexian | Oral | Phase II | Total knee arthroplasty | 1242 | Published [58] |
| Phase II | Stroke and brain MRI 7 | NCT03766581 [59] | |||
| ONO-7684 | Oral | Phase I | PK 8 & PD 9 in healthy | 48 + 24 | Published [60] |
| EP-7041 | I.V. 3 | Phase II | Thrombocytopenia, COVID-19 | 90 | Not recruiting (NCT05040776) |
| BMS-962212 | I.V. 3 | Phase I | PK 8 & PD 9 in healthy | 691 | Completed (NCT03197779) |
| Antibodies | |||||
| Abelacimab | S.C. 2 | Phase II | Total knee arthroplasty | 412 | Published [61] |
| Phase II | AF 5 | 1200 | Not recruiting (NCT04755283) | ||
| Phase III | Cancer-associated VTE 10 | 1655 | Recruiting (NCT05171049) | ||
| Phase III | GI/GU-associated VTE 10 | 1020 | Recruiting (NCT05171075) | ||
| Osocimab | I.V. 3 | Phase II | Total knee arthroplasty | 813 | Published [62] |
| Xisomab 3G3 | I.V. 3 | Phase II | ESRD 4 | 27 | Published [63] |
| Phase II | Thrombosis in chemotherapy | 50 | Recruiting (NCT04465760) | ||
| MK-2060 | I.V. 3 | Phase II | ESRD 4 | 489 | Recruiting (NCT05027074) |
| REGN9933 | I.V. 3 | Phase I | PK 8 & PD 9 in healthy | 72 | Recruiting (NCT05102136) |
5.2. Small Molecules
5.2.1. Small Molecules Targeting the Active Site on FXIa
- Asundexian (BAY 2433334).
- 2.
- Milvexian (JNJ-70033093/BMS-986177).
- 3.
- Other Small-Molecules in Early Development
5.2.2. Small Molecules Targeting Heparin Allosteric Site on FXIa
5.3. Monoclonal Antibodies
- Abelacimab (MAA868).
- 2.
- Osocimab (BAY 1213790).
- 3.
- Xisomab 3G3 (AB023).
- 4.
- Other Antibodies in Clinical Testing.
5.4. Aptamers
6. Fields for Therapeutic Investigation
6.1. Active Areas of Investigations
6.1.1. Atrial Fibrillation
6.1.2. Venous Thromboembolism
6.2. Potential Areas for Therapeutic Investigations
6.2.1. Antiphospholipid Syndrome
6.2.2. Sickle Cell Disease (SCD)
6.2.3. Implantable Devices/Blood Contact with Artificial Surfaces
6.2.4. Myocardial Infarction
7. Conclusions
8. Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Lozano, R.; Naghavi, M.; Foreman, K.; Lim, S.; Shibuya, K.; Aboyans, V.; Abraham, J.; Adair, T.; Aggarwal, R.; Ahn, S.Y.; et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: A systematic analysis for the Global Burden of Disease Study 2010. Lancet 2012, 380, 2095–2128. [Google Scholar] [CrossRef] [PubMed]
- Day, I.S.C.f.W.T. Thrombosis: A major contributor to the global disease burden. J. Thromb. Haemost. 2014, 12, 1580–1590. [Google Scholar] [CrossRef]
- O’Brien, M.P.; Zafar, M.U.; Rodriguez, J.C.; Okoroafor, I.; Heyison, A.; Cavanagh, K.; Rodriguez-Caprio, G.; Weinberg, A.; Escolar, G.; Aberg, J.A.; et al. Targeting thrombogenicity and inflammation in chronic HIV infection. Sci. Adv. 2019, 5, eaav5463. [Google Scholar] [CrossRef] [Green Version]
- Hemkens, L.G.; Bucher, H.C. HIV infection and cardiovascular disease. Eur. Heart J. 2014, 35, 1373–1381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alonso, A.; Barnes, A.E.; Guest, J.L.; Shah, A.; Shao, I.Y.; Marconi, V. HIV Infection and Incidence of Cardiovascular Diseases: An Analysis of a Large Healthcare Database. J. Am. Heart Assoc. 2019, 8, e012241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia, D.A.; Baglin, T.P.; Weitz, J.I.; Samama, M.M. Parenteral anticoagulants: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest 2012, 141, e24S–e43S. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ageno, W.; Gallus, A.S.; Wittkowsky, A.; Crowther, M.; Hylek, E.M.; Palareti, G. Oral anticoagulant therapy: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest 2012, 141, e44S–e88S. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zafar, M.U.; Vorchheimer, D.A.; Gaztanaga, J.; Velez, M.; Yadegar, D.; Moreno, P.R.; Kunitada, S.; Pagan, J.; Fuster, V.; Badimon, J.J. Antithrombotic effects of factor Xa inhibition with DU-176b: Phase-I study of an oral, direct factor Xa inhibitor using an ex-vivo flow chamber. Thromb. Haemost. 2007, 98, 883–888. [Google Scholar] [CrossRef]
- Zafar, M.U.; Farkouh, M.E.; Osende, J.; Shimbo, D.; Palencia, S.; Crook, J.; Leadley, R.; Fuster, V.; Chesebro, J.H. Potent arterial antithrombotic effect of direct factor-Xa inhibition with ZK-807834 administered to coronary artery disease patients. Thromb. Haemost. 2007, 97, 487–492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viles-Gonzalez, J.F.; Gaztanaga, J.; Zafar, U.M.; Fuster, V.; Badimon, J.J. Clinical and experimental experience with factor Xa inhibitors. Am. J. Cardiovasc. Drugs 2004, 4, 379–384. [Google Scholar] [CrossRef] [PubMed]
- Sardar, P.; Chatterjee, S.; Lavie, C.J.; Giri, J.S.; Ghosh, J.; Mukherjee, D.; Lip, G.Y. Risk of major bleeding in different indications for new oral anticoagulants: Insights from a meta-analysis of approved dosages from 50 randomized trials. Int. J. Cardiol. 2015, 179, 279–287. [Google Scholar] [CrossRef] [Green Version]
- Hellenbart, E.L.; Faulkenberg, K.D.; Finks, S.W. Evaluation of bleeding in patients receiving direct oral anticoagulants. Vasc. Health Risk Manag. 2017, 13, 325–342. [Google Scholar] [CrossRef] [Green Version]
- Ruff, C.T.; Giugliano, R.P.; Braunwald, E.; Hoffman, E.B.; Deenadayalu, N.; Ezekowitz, M.D.; Camm, A.J.; Weitz, J.I.; Lewis, B.S.; Parkhomenko, A.; et al. Comparison of the efficacy and safety of new oral anticoagulants with warfarin in patients with atrial fibrillation: A meta-analysis of randomised trials. Lancet 2014, 383, 955–962. [Google Scholar] [CrossRef]
- Alamneh, E.A.; Chalmers, L.; Bereznicki, L.R. Suboptimal Use of Oral Anticoagulants in Atrial Fibrillation: Has the Introduction of Direct Oral Anticoagulants Improved Prescribing Practices? Am. J. Cardiovasc. Drugs 2016, 16, 183–200. [Google Scholar] [CrossRef]
- Sorvillo, N.; Cherpokova, D.; Martinod, K.; Wagner, D.D. Extracellular DNA NET-Works With Dire Consequences for Health. Circ. Res. 2019, 125, 470–488. [Google Scholar] [CrossRef]
- Baker, C.J.; Smith, S.A.; Morrissey, J.H. Polyphosphate in thrombosis, hemostasis, and inflammation. Res. Pract. Thromb. Haemost. 2019, 3, 18–25. [Google Scholar] [CrossRef] [Green Version]
- Nossel, H.L. Differential consumption of coagulation factors resulting from activation of the extrinsic (tissue thromboplastin) or the intrinsic (foreign surface contact) pathways. Blood 1967, 29, 331–340. [Google Scholar] [CrossRef]
- Silverberg, M.; Dunn, J.T.; Garen, L.; Kaplan, A.P. Autoactivation of human Hageman factor. Demonstration utilizing a synthetic substrate. J. Biol. Chem. 1980, 255, 7281–7286. [Google Scholar] [CrossRef] [PubMed]
- Tankersley, D.L.; Finlayson, J.S. Kinetics of activation and autoactivation of human factor XII. Biochemistry 1984, 23, 273–279. [Google Scholar] [CrossRef] [PubMed]
- Franke, L.; Schewe, H.J.; Muller, B.; Campman, V.; Kitzrow, W.; Uebelhack, R.; Berghofer, A.; Muller-Oerlinghausen, B. Serotonergic platelet variables in unmedicated patients suffering from major depression and healthy subjects: Relationship between 5HT content and 5HT uptake. Life Sci. 2000, 67, 301–315. [Google Scholar] [CrossRef]
- Wood, J.P.; Ellery, P.E.; Maroney, S.A.; Mast, A.E. Biology of tissue factor pathway inhibitor. Blood 2014, 123, 2934–2943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griffin, J.H.; Fernandez, J.A.; Gale, A.J.; Mosnier, L.O. Activated protein C. J. Thromb. Haemost. 2007, 5 (Suppl. S1), 73–80. [Google Scholar] [CrossRef] [PubMed]
- Olson, S.T.; Swanson, R.; Raub-Segall, E.; Bedsted, T.; Sadri, M.; Petitou, M.; Herault, J.P.; Herbert, J.M.; Bjork, I. Accelerating ability of synthetic oligosaccharides on antithrombin inhibition of proteinases of the clotting and fibrinolytic systems. Comparison with heparin and low-molecular-weight heparin. Thromb. Haemost. 2004, 92, 929–939. [Google Scholar] [CrossRef]
- Larsen, K.S.; Ostergaard, H.; Bjelke, J.R.; Olsen, O.H.; Rasmussen, H.B.; Christensen, L.; Kragelund, B.B.; Stennicke, H.R. Engineering the substrate and inhibitor specificities of human coagulation Factor VIIa. Biochem. J. 2007, 405, 429–438. [Google Scholar] [CrossRef] [Green Version]
- Zhao, M.; Abdel-Razek, T.; Sun, M.F.; Gailani, D. Characterization of a heparin binding site on the heavy chain of factor XI. J. Biol. Chem. 1998, 273, 31153–31159. [Google Scholar] [CrossRef] [Green Version]
- Renne, T.; Schmaier, A.H.; Nickel, K.F.; Blomback, M.; Maas, C. In vivo roles of factor XII. Blood 2012, 120, 4296–4303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colhoun, H.M.; Zito, F.; Norman Chan, N.; Rubens, M.B.; Fuller, J.H.; Humphries, S.E. Activated factor XII levels and factor XII 46C>T genotype in relation to coronary artery calcification in patients with type 1 diabetes and healthy subjects. Atherosclerosis 2002, 163, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Grundt, H.; Nilsen, D.W.; Hetland, O.; Valente, E.; Fagertun, H.E. Activated factor 12 (FXIIa) predicts recurrent coronary events after an acute myocardial infarction. Am. Heart J. 2004, 147, 260–266. [Google Scholar] [CrossRef]
- Doggen, C.J.; Rosendaal, F.R.; Meijers, J.C. Levels of intrinsic coagulation factors and the risk of myocardial infarction among men: Opposite and synergistic effects of factors XI and XII. Blood 2006, 108, 4045–4051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salomon, O.; Steinberg, D.M.; Koren-Morag, N.; Tanne, D.; Seligsohn, U. Reduced incidence of ischemic stroke in patients with severe factor XI deficiency. Blood 2008, 111, 4113–4117. [Google Scholar] [CrossRef] [PubMed]
- Salomon, O.; Steinberg, D.M.; Zucker, M.; Varon, D.; Zivelin, A.; Seligsohn, U. Patients with severe factor XI deficiency have a reduced incidence of deep-vein thrombosis. Thromb. Haemost. 2011, 105, 269–273. [Google Scholar] [CrossRef] [PubMed]
- Renne, T.; Pozgajova, M.; Gruner, S.; Schuh, K.; Pauer, H.U.; Burfeind, P.; Gailani, D.; Nieswandt, B. Defective thrombus formation in mice lacking coagulation factor XII. J. Exp. Med. 2005, 202, 271–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kleinschnitz, C.; Stoll, G.; Bendszus, M.; Schuh, K.; Pauer, H.U.; Burfeind, P.; Renne, C.; Gailani, D.; Nieswandt, B.; Renne, T. Targeting coagulation factor XII provides protection from pathological thrombosis in cerebral ischemia without interfering with hemostasis. J. Exp. Med. 2006, 203, 513–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gailani, D.; Bane, C.E.; Gruber, A. Factor XI and contact activation as targets for antithrombotic therapy. J. Thromb. Haemost. 2015, 13, 1383–1395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muller, F.; Gailani, D.; Renne, T. Factor XI and XII as antithrombotic targets. Curr. Opin. Hematol. 2011, 18, 349–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schumacher, W.A.; Luettgen, J.M.; Quan, M.L.; Seiffert, D.A. Inhibition of factor XIa as a new approach to anticoagulation. Arter. Thromb. Vasc. Biol. 2010, 30, 388–392. [Google Scholar] [CrossRef]
- Key, N.S. Epidemiologic and clinical data linking factors XI and XII to thrombosis. Hematol. Am. Soc. Hematol. Educ. Prog. 2014, 2014, 66–70. [Google Scholar] [CrossRef] [Green Version]
- Davie, E.W.; Fujikawa, K.; Kisiel, W. The coagulation cascade: Initiation, maintenance, and regulation. Biochemistry 1991, 30, 10363–10370. [Google Scholar] [CrossRef]
- Vondemborne, P.A.K.; Meijers, J.C.M.; Bouma, B.N. Feedback Activation of Factor-Xi by Thrombin in Plasma Results in Additional Formation of Thrombin That Protects Fibrin Clots from Fibrinolysis. Blood 1995, 86, 3035–3042. [Google Scholar] [CrossRef] [Green Version]
- Meijers, J.C.; Tekelenburg, W.L.; Bouma, B.N.; Bertina, R.M.; Rosendaal, F.R. High levels of coagulation factor XI as a risk factor for venous thrombosis. N. Engl. J. Med. 2000, 342, 696–701. [Google Scholar] [CrossRef] [PubMed]
- Gill, D.; Georgakis, M.K.; Laffan, M.; Sabater-Lleal, M.; Malik, R.; Tzoulaki, I.; Veltkamp, R.; Dehghan, A. Genetically Determined FXI (Factor XI) Levels and Risk of Stroke. Stroke J. Cereb. Circ. 2018, 49, 2761–2763. [Google Scholar] [CrossRef] [PubMed]
- Salomon, O.; Steinberg, D.M.; Seligshon, U. Variable bleeding manifestations characterize different types of surgery in patients with severe factor XI deficiency enabling parsimonious use of replacement therapy. Haemophilia 2006, 12, 490–493. [Google Scholar] [CrossRef] [PubMed]
- Preis, M.; Hirsch, J.; Kotler, A.; Zoabi, A.; Stein, N.; Rennert, G.; Saliba, W. Factor XI deficiency is associated with lower risk for cardiovascular and venous thromboembolism events. Blood 2017, 129, 1210–1215. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Li, Q.; Du, F.; Caruso, J.; Nawrocki, A.; Chintala, M. Reversal of Antithrombotic Effects of FXIa Inhibitor Milvexian (BMS-986177/JNJ-70033093) by Non-specific Agents in a Rabbit AV-shunt Model of Thrombosis. In Proceedings of the ISTH 2021 Congress, Philadelphia, PA, USA, 17–21 July 2021. [Google Scholar]
- David, T.; Kim, Y.C.; Ely, L.K.; Rondon, I.; Gao, H.; O’Brien, P.; Bolt, M.W.; Coyle, A.J.; Garcia, J.L.; Flounders, E.A.; et al. Factor XIa-specific IgG and a reversal agent to probe factor XI function in thrombosis and hemostasis. Sci. Transl. Med. 2016, 8, 353ra112. [Google Scholar] [CrossRef]
- Vilahur, G.; Choi, B.G.; Zafar, M.U.; Viles-Gonzalez, J.F.; Vorchheimer, D.A.; Fuster, V.; Badimon, J.J. Normalization of platelet reactivity in clopidogrel-treated subjects. J. Thromb. Haemost. 2007, 5, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Zafar, M.U.; Santos-Gallego, C.; Vorchheimer, D.A.; Viles-Gonzalez, J.F.; Elmariah, S.; Giannarelli, C.; Sartori, S.; Small, D.S.; Jakubowski, J.A.; Fuster, V.; et al. Platelet function normalization after a prasugrel loading-dose: Time-dependent effect of platelet supplementation. J. Thromb. Haemost. 2013, 11, 100–106. [Google Scholar] [CrossRef] [Green Version]
- Zafar, M.U.; Smith, D.A.; Baber, U.; Sartori, S.; Chen, K.; Lam, D.W.; Linares-Koloffon, C.A.; Rey-Mendoza, J.; Jimenez Britez, G.; Escolar, G.; et al. Impact of Timing on the Functional Recovery Achieved With Platelet Supplementation after Treatment With Ticagrelor. Circ. Cardiovasc. Interv. 2017, 10, 8. [Google Scholar] [CrossRef]
- Kole, R.; Krainer, A.R.; Altman, S. RNA therapeutics: Beyond RNA interference and antisense oligonucleotides. Nat. Rev. Drug Discov. 2012, 11, 125–140. [Google Scholar] [CrossRef] [Green Version]
- Shen, X.; Corey, D.R. Chemistry, mechanism and clinical status of antisense oligonucleotides and duplex RNAs. Nucleic Acids Res. 2018, 46, 1584–1600. [Google Scholar] [CrossRef]
- Younis, H.S.; Crosby, J.; Huh, J.I.; Lee, H.S.; Rime, S.; Monia, B.; Henry, S.P. Antisense inhibition of coagulation factor XI prolongs APTT without increased bleeding risk in cynomolgus monkeys. Blood 2012, 119, 2401–2408. [Google Scholar] [CrossRef]
- Buller, H.R.; Bethune, C.; Bhanot, S.; Gailani, D.; Monia, B.P.; Raskob, G.E.; Segers, A.; Verhamme, P.; Weitz, J.I.; Investigators, F.-A.T. Factor XI antisense oligonucleotide for prevention of venous thrombosis. N. Engl. J. Med. 2015, 372, 232–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walsh, M.; Bethune, C.; Smyth, A.; Tyrwhitt, J.; Jung, S.W.; Yu, R.Z.; Wang, Y.; Geary, R.S.; Weitz, J.; Bhanot, S.; et al. Phase 2 Study of the Factor XI Antisense Inhibitor IONIS-FXIRx in Patients With ESRD. Kidney Int. Rep. 2022, 7, 200–209. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.V.; Kirsch, B.; Bhatt, D.L.; Budaj, A.; Coppolecchia, R.; Eikelboom, J.; James, S.K.; Jones, W.S.; Merkely, B.; Keller, L.; et al. A Multicenter, Phase 2, Randomized, Placebo-Controlled, Double-Blind, Parallel-Group, Dose-Finding Trial of the Oral Factor XIa Inhibitor Asundexian to Prevent Adverse Cardiovascular Outcomes Following Acute Myocardial Infarction. Circulation 2022, 146, 1196–1206. [Google Scholar] [CrossRef] [PubMed]
- Shoamanesh, A.; Mundl, H.; Smith, E.E.; Masjuan, J.; Milanov, I.; Hirano, T.; Agafina, A.; Campbell, B.; Caso, V.; Mas, J.L.; et al. Factor XIa inhibition with asundexian after acute non-cardioembolic ischaemic stroke (PACIFIC-Stroke): An international, randomised, double-blind, placebo-controlled, phase 2b trial. Lancet 2022, 400, 997–1007. [Google Scholar] [CrossRef] [PubMed]
- Piccini, J.P.; Caso, V.; Connolly, S.J.; Fox, K.A.A.; Oldgren, J.; Jones, W.S.; Gorog, D.A.; Durdil, V.; Viethen, T.; Neumann, C.; et al. Safety of the oral factor XIa inhibitor asundexian compared with apixaban in patients with atrial fibrillation (PACIFIC-AF): A multicentre, randomised, double-blind, double-dummy, dose-finding phase 2 study. Lancet 2022, 399, 1383–1390. [Google Scholar] [CrossRef]
- BusinessWire. Bayer Initiates Phase III Study Program for Investigational Oral FXIa Inhibitor Asundexian; BusinessWire: San Francisco, CA, USA, 2022. [Google Scholar]
- Weitz, J.I.; Strony, J.; Ageno, W.; Gailani, D.; Hylek, E.M.; Lassen, M.R.; Mahaffey, K.W.; Notani, R.S.; Roberts, R.; Segers, A.; et al. Milvexian for the Prevention of Venous Thromboembolism. N. Engl. J. Med. 2021, 385, 2161–2172. [Google Scholar] [CrossRef]
- ESCPressOffice. Milvexian Shows Potential to Reduce the Risk of Ischaemic Stroke; ESCPressOffice: Barcelona, Spain, 2022. [Google Scholar]
- Beale, D.; Dennison, J.; Boyce, M.; Mazzo, F.; Honda, N.; Smith, P.; Bruce, M. ONO-7684 a novel oral FXIa inhibitor: Safety, tolerability, pharmacokinetics and pharmacodynamics in a first-in-human study. Br. J. Clin. Pharm. 2021, 87, 3177–3189. [Google Scholar] [CrossRef]
- Verhamme, P.; Yi, B.A.; Segers, A.; Salter, J.; Bloomfield, D.; Buller, H.R.; Raskob, G.E.; Weitz, J.I.; Investigators, A.-T. Abelacimab for Prevention of Venous Thromboembolism. N. Engl. J. Med. 2021, 385, 609–617. [Google Scholar] [CrossRef]
- Weitz, J.I.; Bauersachs, R.; Becker, B.; Berkowitz, S.D.; Freitas, M.C.S.; Lassen, M.R.; Metzig, C.; Raskob, G.E. Effect of Osocimab in Preventing Venous Thromboembolism Among Patients Undergoing Knee Arthroplasty: The FOXTROT Randomized Clinical Trial. JAMA 2020, 323, 130–139. [Google Scholar] [CrossRef]
- Lorentz, C.U.; Tucker, E.I.; Verbout, N.G.; Shatzel, J.J.; Olson, S.R.; Markway, B.D.; Wallisch, M.; Ralle, M.; Hinds, M.T.; McCarty, O.J.T.; et al. The contact activation inhibitor AB023 in heparin-free hemodialysis: Results of a randomized phase 2 clinical trial. Blood 2021, 138, 2173–2184. [Google Scholar] [CrossRef]
- Al-Horani, R.A.; Desai, U.R. Factor XIa inhibitors: A review of the patent literature. Expert Opin. Ther. Pat. 2016, 26, 323–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayward, N.J.; Goldberg, D.I.; Morrel, E.M.; Friden, P.M.; Bokesch, P.M. Phase 1a/1b Study of EP-7041: A Novel, Potent, Selective, Small Molecule FXIa Inhibitor. Circulation 2017, 136, 13747. [Google Scholar]
- Perera, V.; Luettgen, J.M.; Wang, Z.; Frost, C.E.; Yones, C.; Russo, C.; Lee, J.; Zhao, Y.; LaCreta, F.P.; Ma, X.; et al. First-in-human study to assess the safety, pharmacokinetics and pharmacodynamics of BMS-962212, a direct, reversible, small molecule factor XIa inhibitor in non-Japanese and Japanese healthy subjects. Br. J. Clin. Pharm. 2018, 84, 876–887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Horani, R.A.; Gailani, D.; Desai, U.R. Allosteric inhibition of factor XIa. Sulfated non-saccharide glycosaminoglycan mimetics as promising anticoagulants. Thromb. Res. 2015, 136, 379–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koch, A.W.; Schiering, N.; Melkko, S.; Ewert, S.; Salter, J.; Zhang, Y.; McCormack, P.; Yu, J.; Huang, X.; Chiu, Y.H.; et al. MAA868, a novel FXI antibody with a unique binding mode, shows durable effects on markers of anticoagulation in humans. Blood 2019, 133, 1507–1516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, R.R.; Sullenger, B.A.; Rusconi, C.P. Developing aptamers into therapeutics. J. Clin. Investig. 2000, 106, 929–934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donkor, D.A.; Bhakta, V.; Eltringham-Smith, L.J.; Stafford, A.R.; Weitz, J.I.; Sheffield, W.P. Selection and characterization of a DNA aptamer inhibiting coagulation factor XIa. Sci. Rep. 2017, 7, 2102. [Google Scholar] [CrossRef] [Green Version]
- Woodruff, R.S.; Ivanov, I.; Verhamme, I.M.; Sun, M.F.; Gailani, D.; Sullenger, B.A. Generation and characterization of aptamers targeting factor XIa. Thromb. Res. 2017, 156, 134–141. [Google Scholar] [CrossRef]
- Rusconi, C.P.; Roberts, J.D.; Pitoc, G.A.; Nimjee, S.M.; White, R.R.; Quick, G., Jr.; Scardino, E.; Fay, W.P.; Sullenger, B.A. Antidote-mediated control of an anticoagulant aptamer in vivo. Nat. Biotechnol. 2004, 22, 1423–1428. [Google Scholar] [CrossRef]
- Oney, S.; Lam, R.T.; Bompiani, K.M.; Blake, C.M.; Quick, G.; Heidel, J.D.; Liu, J.Y.; Mack, B.C.; Davis, M.E.; Leong, K.W.; et al. Development of universal antidotes to control aptamer activity. Nat. Med. 2009, 15, 1224–1228. [Google Scholar] [CrossRef]
- Go, A.S.; Hylek, E.M.; Phillips, K.A.; Chang, Y.; Henault, L.E.; Selby, J.V.; Singer, D.E. Prevalence of diagnosed atrial fibrillation in adults: National implications for rhythm management and stroke prevention: The AnTicoagulation and Risk Factors in Atrial Fibrillation (ATRIA) Study. JAMA 2001, 285, 2370–2375. [Google Scholar] [CrossRef]
- Wolf, P.A.; Abbott, R.D.; Kannel, W.B. Atrial fibrillation as an independent risk factor for stroke: The Framingham Study. Stroke J. Cereb. Circ. 1991, 22, 983–988. [Google Scholar] [CrossRef] [Green Version]
- Kannel, W.B.; Benjamin, E.J. Status of the epidemiology of atrial fibrillation. Med. Clin. N. Am. 2008, 92, 17–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez-Lopez, J.A.; Sterne, J.A.C.; Thom, H.H.Z.; Higgins, J.P.T.; Hingorani, A.D.; Okoli, G.N.; Davies, P.A.; Bodalia, P.N.; Bryden, P.A.; Welton, N.J.; et al. Oral anticoagulants for prevention of stroke in atrial fibrillation: Systematic review, network meta-analysis, and cost effectiveness analysis. BMJ Clin. Res. Ed 2017, 359, j5058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Es, N.; Coppens, M.; Schulman, S.; Middeldorp, S.; Buller, H.R. Direct oral anticoagulants compared with vitamin K antagonists for acute venous thromboembolism: Evidence from phase 3 trials. Blood 2014, 124, 1968–1975. [Google Scholar] [CrossRef] [PubMed]
- Heit, J.A.; Petterson, T.M.; Farmer, S.A.; Bailey, K.R.; Joseph Melton, L. Trends in the incidence of deep vein thrombosis and pulmonary embolism: A 35-year population-based study. Blood 2006, 108, 1488. [Google Scholar] [CrossRef]
- van der Hulle, T.; Kooiman, J.; den Exter, P.L.; Dekkers, O.M.; Klok, F.A.; Huisman, M.V. Effectiveness and safety of novel oral anticoagulants as compared with vitamin K antagonists in the treatment of acute symptomatic venous thromboembolism: A systematic review and meta-analysis. J. Thromb. Haemost. 2014, 12, 320–328. [Google Scholar] [CrossRef]
- Cohen, H.; Hunt, B.J.; Efthymiou, M.; Arachchillage, D.R.; Mackie, I.J.; Clawson, S.; Sylvestre, Y.; Machin, S.J.; Bertolaccini, M.L.; Ruiz-Castellano, M.; et al. Rivaroxaban versus warfarin to treat patients with thrombotic antiphospholipid syndrome, with or without systemic lupus erythematosus (RAPS): A randomised, controlled, open-label, phase 2/3, non-inferiority trial. Lancet. Haematol. 2016, 3, e426–e436. [Google Scholar] [CrossRef] [Green Version]
- Woller, S.C.; Stevens, S.M.; Kaplan, D.A.; Branch, D.W.; Aston, V.T.; Wilson, E.L.; Gallo, H.M.; Johnson, E.G.; Rondina, M.T.; Lloyd, J.F.; et al. Apixaban for the Secondary Prevention of Thrombosis Among Patients With Antiphospholipid Syndrome: Study Rationale and Design (ASTRO-APS). Clin. Appl. Thromb./Hemost. Off. J. Int. Acad. Clin. Appl. Thromb./Hemost. 2016, 22, 239–247. [Google Scholar] [CrossRef]
- Ohene-Frempong, K.; Weiner, S.J.; Sleeper, L.A.; Miller, S.T.; Embury, S.; Moohr, J.W.; Wethers, D.L.; Pegelow, C.H.; Gill, F.M. Cerebrovascular accidents in sickle cell disease: Rates and risk factors. Blood 1998, 91, 288–294. [Google Scholar]
- Adams, R.; McKie, V.; Nichols, F.; Carl, E.; Zhang, D.L.; McKie, K.; Figueroa, R.; Litaker, M.; Thompson, W.; Hess, D. The use of transcranial ultrasonography to predict stroke in sickle cell disease. N. Engl. J. Med. 1992, 326, 605–610. [Google Scholar] [CrossRef] [PubMed]
- Adams, R.J.; McKie, V.C.; Carl, E.M.; Nichols, F.T.; Perry, R.; Brock, K.; McKie, K.; Figueroa, R.; Litaker, M.; Weiner, S.; et al. Long-term stroke risk in children with sickle cell disease screened with transcranial Doppler. Ann. Neurol. 1997, 42, 699–704. [Google Scholar] [CrossRef] [PubMed]
- Switzer, J.A.; Hess, D.C.; Nichols, F.T.; Adams, R.J. Pathophysiology and treatment of stroke in sickle-cell disease: Present and future. Lancet. Neurol. 2006, 5, 501–512. [Google Scholar] [CrossRef]
- Hillery, C.A.; Panepinto, J.A. Pathophysiology of stroke in sickle cell disease. Microcirculation 2004, 11, 195–208. [Google Scholar] [CrossRef]
- Shet, A.S.; Aras, O.; Gupta, K.; Hass, M.J.; Rausch, D.J.; Saba, N.; Koopmeiners, L.; Key, N.S.; Hebbel, R.P. Sickle blood contains tissue factor-positive microparticles derived from endothelial cells and monocytes. Blood 2003, 102, 2678–2683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glassberg, J.; Rahman, A.H.; Zafar, M.; Cromwell, C.; Punzalan, A.; Badimon, J.J.; Aledort, L. Application of phospho-CyTOF to characterize immune activation in patients with sickle cell disease in an ex vivo model of thrombosis. J. Immunol. Methods 2018, 453, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Adams, R.J.; McKie, V.C.; Hsu, L.; Files, B.; Vichinsky, E.; Pegelow, C.; Abboud, M.; Gallagher, D.; Kutlar, A.; Nichols, F.T.; et al. Prevention of a first stroke by transfusions in children with sickle cell anemia and abnormal results on transcranial Doppler ultrasonography. N. Engl. J. Med. 1998, 339, 5–11. [Google Scholar] [CrossRef]
- Sy, E.; Sklar, M.C.; Lequier, L.; Fan, E.; Kanji, H.D. Anticoagulation practices and the prevalence of major bleeding, thromboembolic events, and mortality in venoarterial extracorporeal membrane oxygenation: A systematic review and meta-analysis. J. Crit. Care 2017, 39, 87–96. [Google Scholar] [CrossRef]
- McMichael, A.B.V.; Ryerson, L.M.; Ratano, D.; Fan, E.; Faraoni, D.; Annich, G.M. 2021 ELSO Adult and Pediatric Anticoagulation Guidelines. ASAIO J. 2022, 68, 303–310. [Google Scholar] [CrossRef]
- Eikelboom, J.W.; Connolly, S.J.; Brueckmann, M.; Granger, C.B.; Kappetein, A.P.; Mack, M.J.; Blatchford, J.; Devenny, K.; Friedman, J.; Guiver, K.; et al. Dabigatran versus warfarin in patients with mechanical heart valves. N. Engl. J. Med. 2013, 369, 1206–1214. [Google Scholar] [CrossRef] [Green Version]
- Jaffer, I.H.; Fredenburgh, J.C.; Hirsh, J.; Weitz, J.I. Medical device-induced thrombosis: What causes it and how can we prevent it? J. Thromb. Haemost. 2015, 13 (Suppl. S1), S72–S81. [Google Scholar] [CrossRef] [PubMed]
- Jaffer, I.H.; Stafford, A.R.; Fredenburgh, J.C.; Whitlock, R.P.; Chan, N.C.; Weitz, J.I. Dabigatran is Less Effective Than Warfarin at Attenuating Mechanical Heart Valve-Induced Thrombin Generation. J. Am. Heart Assoc. 2015, 4, e002322. [Google Scholar] [CrossRef] [PubMed]
- Mega, J.L.; Braunwald, E.; Wiviott, S.D.; Bassand, J.P.; Bhatt, D.L.; Bode, C.; Burton, P.; Cohen, M.; Cook-Bruns, N.; Fox, K.A.; et al. Rivaroxaban in patients with a recent acute coronary syndrome. N. Engl. J. Med. 2012, 366, 9–19. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Badimon, J.J.; Escolar, G.; Zafar, M.U. Factor XI/XIa Inhibition: The Arsenal in Development for a New Therapeutic Target in Cardio- and Cerebrovascular Disease. J. Cardiovasc. Dev. Dis. 2022, 9, 437. https://doi.org/10.3390/jcdd9120437
Badimon JJ, Escolar G, Zafar MU. Factor XI/XIa Inhibition: The Arsenal in Development for a New Therapeutic Target in Cardio- and Cerebrovascular Disease. Journal of Cardiovascular Development and Disease. 2022; 9(12):437. https://doi.org/10.3390/jcdd9120437
Chicago/Turabian StyleBadimon, Juan J., Gines Escolar, and M. Urooj Zafar. 2022. "Factor XI/XIa Inhibition: The Arsenal in Development for a New Therapeutic Target in Cardio- and Cerebrovascular Disease" Journal of Cardiovascular Development and Disease 9, no. 12: 437. https://doi.org/10.3390/jcdd9120437