New Concepts in the Development and Malformation of the Arterial Valves


Biosciences Institute, Newcastle University, Newcastle upon Tyne NE1 3BZ, UK
*
Author to whom correspondence should be addressed.
J. Cardiovasc. Dev. Dis. 2020, 7(4), 38; https://doi.org/10.3390/jcdd7040038
Submission received: 28 August 2020
/
Revised: 21 September 2020
/
Accepted: 23 September 2020
/
Published: 24 September 2020
(This article belongs to the Special Issue Leaders in Cardiovascular Research: A special issue dedicated to Professor Robert Anderson)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Although in many ways the arterial and atrioventricular valves are similar, both being derived for the most part from endocardial cushions, we now know that the arterial valves and their surrounding structures are uniquely dependent on progenitors from both the second heart field (SHF) and neural crest cells (NCC). Here, we will review aspects of arterial valve development, highlighting how our appreciation of NCC and the discovery of the SHF have altered our developmental models. We will highlight areas of research that have been particularly instructive for understanding how the leaflets form and remodel, as well as those with limited or conflicting results. With this background, we will explore how this developmental knowledge can help us to understand human valve malformations, particularly those of the bicuspid aortic valve (BAV). Controversies and the current state of valve genomics will be indicated.
1. Anatomy, Histology and Nomenclature of the Mature Arterial (Semilunar) Valves
The aortic and pulmonary valves, which sit within the arterial roots between the myocardium of the left and right ventricular outflow tracts and the aorta and pulmonary trunk, respectively, are structurally similar to one another, and are closely comparable in humans and mice [1,2,3]. Thus, in the mature heart both the aortic and pulmonary valves have three superficially similar leaflets that form the moving parts of the valve. The right (R) and left (L) sinuses of the aortic valve each host the ostium of a coronary artery, whilst the third non coronary/non-facing/posterior (N) and the three pulmonary valve sinuses are almost never associated with a coronary artery—even in congenitally malformed hearts. However, we now recognise the valve complexes to include more than just the three moving leaflets, with additional components including: the hinges, the attachment points of the leaflets to the wall; the commissures, the points of apposition of the leaflets close to the wall; the sinuses, pockets that form between the leaflets and the wall; and the interleaflet triangles, the regions of the wall that lie upstream (on the ventricular side) of the hinges, but are distal to the base of the sinuses that are found on the arterial side (Figure 1). The nomenclature used to describe the arterial roots is controversial. For example, the outflow tract has been divided by some authors into a proximal conus and a distal truncus, although this terminology is falling out of practice and three rather than two components are increasingly recognised. In this nomenclature, the distal component represents the intra-pericardial arterial trunks and the proximal component comprises the ventricular outflow tracts, with the valve complex appearing in the intermediate part [2]. In other contexts, anatomically artificial “rings” are described for clinically important measurements, although some publications have highlighted the misuse of these descriptions for developmental and anatomical purposes [4,5]. Thus, the development of this highly complex area remains the topic of speculation and controversy. One longstanding conundrum is, what are the similarities and differences between arterial and atrioventricular valve development, particularly those that relate to their pathology? How does the valve achieve an undulating crown-like attachment, crossing the arterial–myocardial boundary? How do the outflow cushions remodel to form the sculpted valve leaflets? Overall, the question of how aortic valve malformations relate to other cardiovascular malformations, both congenital and apparently acquired, has not been answered. The answer to all these puzzles may lie with the progenitor cells that form the arterial roots. Thus, a re-evaluation of arterial valve development focusing on the second heart field (SHF) and neural crest cells (NCC) may explain the relationships between these different elements and make sense of abnormalities in transgenic animal models and human malformations.
Cre-lox-based lineage tracing technology in the developing mouse has indicated how cells originating in the SHF and NCC are involved in the development of the outflow wall, as well as septal and valve structures. It is clear that the hinges and the leaflets have contributions from both SHF-derived cells and NCC [3,6]. Similarly, above the undulating leaflet hinge, the sinus wall is made up of vascular smooth muscle cells (SMC) that also derive from NCC and SHF. Within the roots, SHF-derived cells predominate, whilst more distally the entire tunica media is of NCC lineage. In the transition, these progenitors maintain separation as a thinning inner layer of NCC-derived SMC, and a thickening outer layer of SHF-derived SMC [3,7,8]. Proximal to the root, the myocardium is also derived from the SHF. Thus, the supporting structures for the leaflets, the hinge attachments and the interleaflet triangles, the fibroblasts and SMC at the base of the sinus and the subaortic and sub pulmonary myocardium, all arise from SHF progenitors and NCC to a greater or lesser extent. The question of the undulating attachment of the leaflets must be expanded to ask, how do the SHF-derived cells on the arterial side of the hinge become SMC whilst those on the ventricular side become myocardial? Moreover, how do the interleaflet triangles that are found on the ventricular side of the hinge attachments become fibrous, whilst apparently originating from the same SHF progenitors as the SMC within the sinus? Although these questions remain unresolved, it seems likely that local signalling events, rather than patterning within primitive fields of progenitors, are responsible for defining the precise characteristics of specific structures and tissues within the arterial roots.
2. Positioning of the Arterial Valves
The left and right leaflets of the aortic and pulmonary valves, as well as the septal leaflets of the mitral and tricuspid valves, are known to be derived from endocardial cushions. The main aspects of initial formation and positioning are similar for all these cushions, particularly those relating to cellular invasion through the endocardial to mesenchymal transition (EndMT) [9,10,11]. These endocardial cushions initially appear as expansions of pre-existing extracellular matrix (ECM; cardiac jelly) between the outer myocardium and inner endocardial layer, at approximately embryonic day (E) 9.5 in the mouse embryo (Carnegie stage (CS) 10–11 in the human embryo). In both the outflow tract and atrioventricular canal, the initial positioning of the cushions is regulated by local Bmp signalling and a complex interaction between several T-box transcription factors and Notch signalling (reviewed recently [11]). The expanded cardiac jelly of the outflow cushions is also in continuity with the much thinner layer of ECM that extends proximally towards the ventricular chambers. This can, at least in theory, allow for the movement of the cellular component of the forming cushions relative to the overlying myocardial wall. This slippage may allow for repositioning of the three valve primordia in each arterial root relative to each other and could also provide an explanation of how the undulating line of hinge attachment can later span both myocardial and arterial aspects of the roots.
Whilst Bmp signalling determines the positioning of the forming cushions within the myocardial part of the outflow tract, little is known about how the original circumferential deposition of ECM becomes two longitudinal cushions in the superior–inferior opposition. Our previous studies have suggested that the aggregation of NCC within the outflow tract cushions affects where the discrete outflow cushions will form in the initially circumferential cardiac jelly [12]. It is possible that this localised NCC aggregation could be directed by signals from the surrounding myocardium or endothelium, however, it seems more likely that this is dictated by the physical properties of vorticial blood flow through the looping, but as yet unseptated, heart tube. Indeed, it has been shown that shear stress is asymmetrically localised in the outflow wall as the cushions are beginning to cellularise [13]; this could help to position and/or stabilise the cellularising cushions within the circumference of the outflow tract ([12]; Figure 2A). Later, the fusion of these spiralling cushions leads to placing of the root of the aorta to the left of the pulmonary trunk. The deletion of the heparan sulphate proteoglycan perlecan is associated with hyperplastic and malpositioned outflow cushions that ultimately result in transposition of the great arteries in a high proportion of mutant embryos [14]. In this model, the excessive numbers of mesenchymal cells (probably of NCC origin) do not form well defined “ridges” within the cardiac jelly. It can be imagined that this failure to form discrete aggregates of cells within the circumferential cardiac jelly will lead to aberrant positioning of the aorta and pulmonary trunk after septation.
3. The Transition Zone and the Arterial-Myocardial Boundary
Initially, the main outflow cushions are completely contained within the SHF-derived myocardial part of the outflow tract [15]. However, at E10.5, SHF cells continue to add to the outflow tract and, although they initially remain undifferentiated, will later form SMC [3,15,16]. This produces a histologically defined arterial–myocardial junction within the aortic root. Thus, the defining process in the formation and positioning of the aortic root may be the switch from differentiation of the SHF progenitors to cardiomyocytes proximal to the boundary, to SMC distal to the boundary (Figure 2B–D). This boundary between myocardial and non-myocardial fate is first apparent at E10.5 in mice, as a region we named the transition zone [17]. The transition zone does not have an anatomical identity but is identified as a region with cells that express the SHF-specific transcription factor Isl1, but at the same time co-express differentiated cardiomyocyte markers. Thus, cells in this region are transitioning from a progenitor to a cardiomyocyte fate. Above the transition zone, the SHF progenitors are in continuity with similar cells in the dorsal pericardial wall and will later downregulate Isl1 expression; the majority of these will go on to form the arterial SMC of the aorta and pulmonary trunk. However, not all the undifferentiated SHF cells within this boundary region become cardiomyocytes. At E10.5 (CS12 or around 28 dpc in human), two tongues of undifferentiated SHF cells can be seen to extend laterally into the otherwise myocardial outflow tract ([15,18]; Figure 2B). By E11.5, these non-myocardial extensions, surrounded laterally by cardiomyocytes, form the intercalated valve swellings (ICVS; sometimes erroneously called the intercalated cushions—see later) and are the primordia of the posterior (P) aortic (non-coronary) and anterior (A) pulmonary intercalated leaflets [1,19,20]. The origin and remodelling of these ICVS to valve leaflets will be discussed in more detail later, but it is important to emphasise that their localisation within the wall, at the boundary between the myocardial and presumptive-arterial parts of the outflow tract, is unlikely to be coincidental to the remodelling of the main outflow cushions and may play an important role in defining the position of the arterial valve complex.
4. EndMT and Cushion Formation
The process of endocardial cushion mesenchyme formation has been reviewed extensively [10,11,21,22,23]. There is evidence from numerous sources that the outflow endocardium is derived from the second heart field (SHF). Thus, in lineage tracing studies utilising the “SHF-specific” Isl1-Cre and Mef2c-AHF-Cre mouse lines, outflow cushion endocardium and mesenchyme is labelled [24,25]. This contrasts with the atrioventricular endocardium where only a few cells are labelled by these SHF-specific Cre lines [6]. It remains to be seen whether there are significant implications of this different origin or whether, in practice, origin is of less importance than specification to undergo EndMT. EndMT initially occurs in the proximal outflow cushions, with only limited EndMT-derived mesenchyme observed within the distal cushions until after E11.5 [26,27]. Thus, EndMT-derived cells are not initially found in the region of the outflow cushions where the valves will form. The endocardium overlying the valve primordia expresses Nfatc1, which is important for valve development [28,29,30]. There appears to be two roles for Nfatc1 in valve formation, playing an early role in initiating EndMT and then a later role in valve remodelling [30]. Cells labelled by Nfatc1-Cre (in which the Nfatc1 has ever been active) remain in the endocardium and do not undergo EndMT [27]. Targeted deletion of Nfatc1 in the endocardium results in an over-abundance of EndMT-derived cushion mesenchyme in the proximal outflow tract cushions at E10.5, and an extension of these cells into the distal region [27], perhaps indicating that EndMT continues unchecked in the absence of Nfatc1. The observation that the arterial valve leaflets later appear under-developed in the mutant embryos [28,29,30] seems counter-intuitive, but it may be that appropriate interactions between EndMT-derived cells and NCC are essential for leaflet remodelling, and that if this balance is disrupted, underdeveloped leaflets result. Many other signals that induce or repress EndMT of endocardial cells have now been described, including the Notch, BMP and TGFβ signalling pathways [31,32,33,34]. For example, the Nf1/Ras pathway has also been implicated in repressing EndMT in the outflow and atrioventricular cushions [35,36,37], with exacerbated Ras signalling leading to hyperplastic cushions.
Although EndMT-derived cells are not initially found in the distal outflow cushions, by E12.5 they are abundant in the forming leaflets [12,26,38] and are maintained in the postnatal valves (Figure 3A,B). These endocardial-derived cells (EDC) seem to play a crucial role in regulating the growth and remodelling of the cushion. There seems to be a functional difference between early endocardium and the Nfatc1-expressing late valve endocardium regarding Notch signalling. The loss of Notch signalling from the endocardium before EndMT results in grossly hypoplastic cushions and early death. However, if this stage is bypassed, perhaps by using Nfatc1-enCre (valve endocardium-specific [27]) rather than Tie2-Cre (all endocardium and endothelium [39]), or by deleting a Notch ligand that is only active in the endocardium after EndMT (e.g., [40]), then thickened arterial valve leaflets are the usual result. Recently, it has been suggested that some macrophages in the late foetal and postnatal valve mesenchyme are derivatives of the endocardium [41]. However, it remains unclear whether other EndMT-derived cells in the interstitium play specific roles within the adult valve.
5. Neural Crest Cells, Outflow Tract Septation and Valve Development
Outflow tract septation is intimately involved in arterial valve formation through shared cells, cushions and timing of events. Analysing human embryos, Kramer [19] showed that the fusion of the main outflow cushions (at around CS 12–13 or ~30 dpc in human embryos, corresponding to E11.5 in the mouse embryo) produces the precursors of the right and left leaflets of the forming arterial valves, with the parietal main outflow cushion contributing to the left leaflets of the aortic and pulmonary valve, and the septal leaflet contributing to the right leaflets (see Figure 3C). The fusion of the main cushions separates the channels of the aorta and pulmonary trunk and subsequent separation of the aorta from the pulmonary trunk in a plane perpendicular to the line of cushion fusion, results in the formation of two, free-standing arterial trunks with their distinct valves. This corresponds well with what is seen in the mouse embryo (Figure 3; [12]). This septation is dependent on NCC and cannot occur if there are significantly reduced numbers within the outflow cushions [42].
A great deal is known about NCC and their developmental roles outside the heart. NCC are a population of progenitor cells that arise in the dorsal neural tube and migrate throughout the body to contribute to a variety of cell types and organ systems, including the heart [43]. There is an extensive literature reviewing the processes of NCC induction, migration and differentiation (for example [44,45,46]) and these will not be considered further here. In contrast, although there have been many studies that have shown that NCC are critically required for outflow tract septation, their roles in heart development are relatively poorly understood and there have been only limited studies in recent years (reviewed in [47,48,49]). NCC migrate through the pharyngeal arches and first appear in the cardiac jelly of the distal outflow cushions early on E10 of mouse development (CS11 in human). At this stage, the distal outflow cushions are acellular, as although EndMT is initiating in the outflow cushions, this occurs in the proximal region. NCC that remain in the pharyngeal region rapidly differentiate into SMC and in doing so stabilise the nascent endocardial tubes that will become the pharyngeal arch arteries [50,51,52]. In contrast, the NCC entering the outflow cushions initially remain relatively undifferentiated (do not express for example αSMA or SM22α; [3]) and retain their expression of early NCC markers including PlexinA2 and Ap2α [53,54]. The main function of these NCC appears to be to provide bulk to the distal cushions, causing them to come into sustained contact, and permitting fusion, with each other and the dorsal wall of the aortic sac [15]. This fusion event initiates outflow tract septation and separates the systemic (aortic) and pulmonary circulations. Once septation is complete, the majority of the NCC in the proximal cushions die by apoptosis [55] and are replaced by ingrowth of myocardial cells to form the muscular subpulmonary infundibulum [56,57]. It has been suggested that the NCC might be involved in defining the position of the arterial valves, as at E11.5, NCC are restricted to the distal cushions and appear to form a boundary with the EndMT-derived mesenchymal population in the proximal region [27]. However, this is a transient boundary, as by E12.5, NCC are found throughout the outflow cushions, apart from their most proximal tips [15,52]. Whilst the role of NCC in the proximal outflow tract appears to be a transient one, the NCC make a permanent contribution to the arterial valve complex, contributing to the fibroblast-like cells found within the commissures, interleaflet triangles and the leaflets of the valves (Figure 3A,B; [3]). How the NCC-derived cells in these different components of the valves differ from one another remains unclear, and they may have more similarities than differences. Unfortunately, there is no mechanism currently to specifically disrupt the NCC that contribute to the valve leaflets, leaving those that are involved in other structures within the arterial roots, or those required for septation, intact. Thus, we do not know for certain if loss of these cells, in the setting of normal outflow septation, would disrupt valve formation and/or remodelling.
6. Direct Differentiation of SHF Progenitors into Valve Mesenchyme
The intercalated leaflet valve swellings (ICVS), the precursors of the anterior/posterior leaflets, were first noted by Kramer [19] and are seen clearly in cross sections through the outflow tract as lateral bulges within the outflow wall ([20]; Figure 3 and Figure 4). Importantly, although they are overlaid by a thin layer of ECM, the bulk of these structures is completely distinct from the main endocardial cushions and, at their first appearance at E10.5, they do not contain ECM or form by EndMT. Nor do they contain significant numbers of NCC [12,20,58,59]. Lineage tracing studies have shown that the majority of the cells contained within the ICVS are derived from the SHF [20,59], with much smaller contributions from EndMT or NCC than are seen for the main endocardial cushions [12,20,58,59]. Although in some ways they have similarities to the lateral cushions that form the mural leaflets of the mitral and tricuspid valve [20], they do not contain cells derived from the epicardium. Thus, they are distinct from other valve-forming structures within the developing heart. Although the ICVS are continuous with the outflow wall at E10.5–E11.5, they do not label with cardiomyocyte markers and are instead immuno-labelled with antibodies specific to Isl1. Whilst maintaining this progenitor-phenotype, they begin to express Sox9, a characteristic marker of cushion mesenchymal cells [60], and thus differentiate to this fate without passing through the endocardial lineage during EndMT. The conclusion from this is that the ICVS are not endocardial cushions and that the cells within them differentiate directly from the SHF [20]. The confusion arises because although these ICVS arise from the outflow wall and their early formation is entirely distinct from the main outflow cushions [20,58], they rapidly take on the superficial appearance of endocardial cushions by generating large amounts of ECM and expressing typical cushion markers such as Sox9 [20]. Hence, by E12.5 they are almost indistinguishable from other cellularised valve primordia originating from endocardial cushions. Thus, after septation it is reasonable to called them “intercalated cushions”. However, subtle differences remain, for example, the ICVS are more cell-dense and express tropoelastin which the primordia derived from the main cushions do not (Eley, Chaudhry, Henderson; unpublished data). As well as having distinct origins, the ICVS also appear more distally than the right and left cushions, although later remodelling places all the leaflets within each root on their own orthogonal planes. Despite these later similarities, the undifferentiated SHF cells that form the ICVS can still be recognised as they are labelled by the “myocardial-specific” Tnnt2-Cre transgenic marker ([20,58,61]; Figure 3A,B). Despite this they are not transdifferentiated cardiomyocytes (they never at any point express markers of differentiated cardiomyocytes) but at a very early stage activated the promoter used to produce the Tnnt2-Cre transgene. Whilst the Tnnt2-Cre line is therefore extremely useful for following the ICVS cells, it is important to recognise that if is used to drive Cre in the myocardium, there could also be direct effects on arterial valve development.
Although the mechanisms underpinning the formation of the ICVS have only recently been elucidated, we do have some ideas about the molecular mechanisms that are likely to be involved as there are several mouse mutants where the non-coronary leaflet is missing or hypoplastic. This includes mice null for Tbx1 [62], Alk2 [63], Robo1/2 [64] and when Vangl2 is knocked out specifically in the developing SHF [20]. Of particular interest, Notch1-ICD and Jag1/2 are localised within the ICVS during its formation at E10.5–E11.5 [20], in a pattern that suggests that Notch signalling may play an important role during their development. Although it is clear that Notch signalling is important in valve development in general, there is also specific evidence that it is important in the developing ICVS. Knockout of Jag1 and Jag2 in the developing ICVS using Tnnt2-Cre results in defects in the formation of the ICVS that result in a dysplastic or missing non-coronary leaflet of the aortic valve (and to a lesser extent the posterior leaflet of the pulmonary valve; [20]). Thus, there appears to be a specific signalling network acting within the ICVS to bring about direct differentiation of SHF cells to valve mesenchyme; this network utilises Notch signalling.
In summary, it seems that NCC and SHF-derived cells (both via direct differentiation and transitory passage though the endocardial state) are crucial to the formation of arterial valve primordia, although their roles in these processes may be complimentary rather than overlapping. Further work will be needed to better define these specific requirements and roles.
7. Cushion Expansion to Form Arterial Valve Primordia
The period between E10.5 and E12.5 is crucial for the appearance of discrete arterial valve primordia that remodel at the distal end of the outflow cushions, distinct from the septal components that form from the proximal cushions (Figure 4). Although the factors that regulate this process remain unclear, the presence of the transitionary tissues at the myocardial–arterial boundary, or indeed the formation of the ICVS adjacent to the main cushions, may be significant. Thus, it is possible that unique factors within the boundary region influence the development of the adjacent cushion tissue to become valve-like, whilst the more distant proximal cushion acquires a septal phenotype, becoming muscularised by cardiomyocytes migrating from the enveloping myocardial outflow wall [56,57]. The resolution of this point will require further investigation.
Once the relevant progenitor lineages have contributed cells to the mesenchyme by late on E10.5, they undergo a period of proliferation that results in rapid cushion growth. A number of molecules are implicated in this process, most of which affect both the atrioventricular and outflow regions. These include Vegf and FGFs (particularly FGF4) which are major drivers of cushion mesenchyme proliferation [65,66,67,68]. Other genes negatively regulate cushion mesenchyme proliferation, for example, EGF plays an important role in limiting proliferation in the cushion mesenchyme, with mice hypomorphic for the EGF receptor having increased mesenchymal cell numbers and enlarged arterial valve leaflets [69]. Similarly, Jag1, a Notch ligand, also appears to limit cushion mesenchyme proliferation [40]. Thus, too much [35,36,37,70], or too little (for example [71,72]) cell division is associated with defects in the leaflets as development proceeds.
As well as increasing cell numbers, the ECM matures and increases in complexity as the cushions expand. Knockout of the ECM components found in the remodelling cushions, such as versican, frequently result in acellular cushions (e.g., [73,74]) as these molecules are required for cushion formation as well as remodelling. Thus, it will take more nuanced approaches to reveal the specific roles for these molecules in the latter stages of cushion development and maturation. However, mouse embryos lacking Adamts5, which is required for versican cleavage, have grossly hyperplastic arterial valves (interestingly the pulmonary valves are most affected) due to the accumulation of uncleaved versican that, as well as increasing the ECM component, also results in increased mesenchymal cell numbers, with abnormalities seen as early as E12.5 [75]. Thus, along with its early role in supporting EndMT, versican also appears to be important for remodelling of the cushion/valve mesenchyme.
Recently, there has been much interest in the potential role of cilia in the development and maintenance of valve leaflets, largely because of the association between mutations in cilia-associated genes and valve defects (e.g., [76]). Primary cilia are small projections of membrane with a microtubule core that are implicated in cell signalling and mechano-sensation [77]. There is considerable evidence to show that the presence of cilia is temporally and spatially regulated during valve formation, and that although they are rarely found on the surface of endocardial cells, they are found on almost all cushion mesenchyme cells at mid gestation [78]. Cilia numbers decrease on the cushion mesenchyme from E11.5 onwards, declining to around 15% of adult VIC [78]. Although the reasons for this decline are not clear, it may be related to differentiation, as in a variety of cell types, cilia are required for this process [79]. The loss of cilia from the EndMT-derived component of the cushion mesenchyme (using Nfatc1-Cre to delete Ift88) resulted in dysplastic aortic valves, with an enlargement of the hinge region between the leaflets suggestive of BAV, and over-expression of ECM molecules including versican and collagen I [78]. Although it is unclear when or how the BAV arose, this suggests that cilia regulate ECM production or turnover in the developing valves and may play a similar role in the adult. This is supported by other studies that have shown that defects in cilia can lead to mitral valve prolapse, a disease where myxomatous degeneration is a characteristic feature [76]. It is not clear whether a specific subset of VIC retain a cilium in the adult valve, although it is interesting to speculate that these cells are the ones that are activated in response to stress. Absence from the valve endocardium suggests that their role is unlikely to be related to the detection of shear stress.
8. Valve Sculpting
Over the remaining days of foetal and early postnatal life, the bulky, primitive valve precursors are sculpted into thin fibrous leaflets, connected to the vessel wall by hinges that delineate the sinuses (Figure 1). These sculpting stages of arterial valve development remain the least well understood but may be the most relevant to human disease. Recent studies generally refer to a process of valve elongation or lengthening that is required to produce well-functioning and coapted leaflets [11,80,81,82]. However, by E11.5 in the mouse heart, before the outflow tract has septated and the cushions have remodelled to form distinct valve primordia, the outflow cushions cyclically occlude the outflow tract [83], allowing only unidirectional flow of blood. Thus, the valve leaflets do not need to lengthen in order to coapt (and thus function efficiently) when the valve is closed. This suggests that lengthening of valves may not be necessary, but that maintaining the overall proportions of valves during growth and enlargement of this area may be more important. The molecular mechanism(s) underpinning sculpting of the arterial valve leaflets have not been described in detail although there are some hypotheses, with varying amounts of supporting data. One hypothesis suggests that cushion mesenchyme is removed close to the wall, by a process of cell death. High levels of apoptotic cell death have been reported in the developing outflow cushions [84,85,86,87,88] and removal of cushion tissue by cell death in order to refine their shape continues to be an attractive model or leaflet sculpting (Figure 5). However, apoptosis has not been described in the sculpting arterial valve leaflets, although detailed studies are lacking. In contrast to cell death, phagocytosis (cell engulfment), carried out by macrophages derived from the endocardium, plays an essential role in remodelling the mature leaflets and in their absence, the leaflets become dysplastic [41]. Thus, the targeted removal of cushion mesenchyme and/or turnover of mature interstitial cells may be an important component of leaflet sculpting and homeostasis. Alternatives mechanisms of leaflet sculpting have been suggested. Early investigations, before the era of molecular biology, utilised electron microscopy to show that there is ingrowth of a thickened endocardium into the outflow cushion mesenchyme in both chicken and mouse embryos (from approximately E12.5; [89,90]). Furthermore, it was suggested that this ingrowth is associated with cell death of endocardial cells, although the data to support this was minimal. The valve endocardium expresses a number of signalling molecules including Fgfs, Wnts and Hh [68,91,92] and similarities have been drawn with the apical ectodermal ridge that acts as a signalling centre for limb bud outgrowth [22]. These signalling molecules may modify proliferation, differentiation and/or cell death within the underlying cushion mesenchyme, thereby regulating the process of valve leaflet formation and sculpting. Although there have been no detailed studies of how the valve sinuses form since those of Hurle, 40 years ago [89,90], it seems possible that their formation is intimately linked with leaflet sculpting, as the cavity of the sinus takes up much of the space previously occupied by cushion tissue (Figure 5). Whatever the cellular mechanism, high resolution episcopic microscopy (HREM) has revealed that the first indications of cushion excavation to form the sinuses are apparent late on E12.5 of mouse development [2], and that the part of the valve primordium closest to the muscular walls of the arterial root excavates, whilst the part closest to the lumen is retained as the valve leaflet. As the gradual excavation of the sinus simultaneously results in the free (lumenal) edge of the valve leaflet getting longer, this may explain why the valve has been interpreted as lengthening as it remodels (Figure 5).
Although the precise mechanisms are unclear, evidence from the zebrafish and chicken embryo suggest that haemodynamic forces play an important part in the morphogenesis [93] and remodelling of cushions to form sculpted valve leaflets [94,95,96,97]. In the absence of normal blood flow through the heart, either as a result of reduced contractility, disturbed flow resulting from anatomical alterations, or the absence of molecules that sense the flow and send signals that lead to a change in cell/tissue behaviour, the cushions remain bulky, are malpositoned and the leaflets do not remodel properly. Although a detailed discussion of this topic is beyond the scope of this manuscript, there have been several excellent reviews published recently (for example, [98,99]). These have highlighted the importance of shear stress in sculpting the leaflets and have shown that mechanosensors in the valve endocardium activate the transcription Klf2, which brings about the cellular changes involved in valve sculpting via a Wnt signalling cascade [94,100]. For example, Piezo1, a mechanosensor, has been implicated in valve sculpting [101]. In the absence of this mechanosensory signalling cascade, the cushions fail to remodel appropriately resulting in dysplastic leaflets. It should be noted that as the mechanisms underpinning valve sculpting remain unclear, the details of how shear stress brings about this remodelling are correspondingly poorly defined.
9. Maturation of the Arterial Valve Leaflets
Mature valve leaflets contain valvular interstitial cells (VIC) that are derivatives of the cushion mesenchyme. These VIC form three cellular layers, characterized by distinct ECM profiles: the fibrosa (on the arterial side of the leaflet) composed mostly of collagens; the spongiosa made up mostly of proteoglycans; and the ventricularis, which contain elastin fibres. The leaflets are also covered by a specialised endocardial layer (valve endocardial cells; VEC) [80]. This stratification of the valve into its three layers occurs relatively late, first becoming apparent at approximately E16.5 of mouse development [80], at the end of the first trimester in human embryos. Initially, collagens and proteoglycans, including versican, hyaluronan and perlecan [14,102,103] are widespread in the developing cushions. Elastin, in contrast, is found only at low levels in the valve leaflets before birth in the mouse heart (Henderson; unpublished data), although it has been shown to be present by Hamburger and Hamilton stage 30 in the chicken embryo ([104]; equivalent to approximately E13.5 in the mouse) and as early as 7 weeks of gestation in developing human arterial valves [105]. However, after birth, elastin is markedly upregulated in mammalian valves and it is well recognized that remodelling of the ECM continues into postnatal life [80]. Recently, the relationship between the three layers of extracellular proteins and subgroups of VIC has begun to be elucidated [106]. Thus, subpopulations of VIC that express high levels of fibrillar collagen, as well as other genes known to be involved in fibril organisation and ECM maturation have been identified at postnatal day 7 in the mouse heart. Similarly, VIC expressing high levels of glycosaminoglycan-containing proteins including versican, fibulin-2 and lumican, have been identified at the same stage [106]. Surprisingly, elastin was expressed by both subgroups, but a specific elastin-rich subgroup of VIC was not identified. Indeed, there is a disconnection between the initial lineage of VIC and those in mature valves. Whilst mouse studies have shown that the distinct embryonic lineages that give rise to VIC are differentially localised within the foetal and postnatal valves [3,12,107], this has not yet been mapped in the adult valve and it remains unknown how the subgroups of VIC identified on the basis of their transcriptome [106] correspond to the different lineages, or to different disease states. It is possible that distinct embryonic lineages may predispose to disease and this is supported by the observation that when only one of the three leaflets is thickened, it is usually the non-coronary [108], suggesting that its different origin may predispose it to disease in some scenarios. However, studies where the three valve leaflets are examined separately are rare. Current evidence leads us to believe that there are distinct groups of VIC (potentially with distinct origins) with differing roles and susceptibilities to disease, but there may be a significant degree of overlap between these factors. Whilst all three lineages (EndMT-derived, NCC and directly differentiated SHF-derived cells) are retained into postnatal life [12,20,39,43,109], cells derived from the bone marrow—largely macrophages and dendritic cells—are mostly found in the valve leaflets postnatally, expanding to make up almost 20% of cells in the valve mesenchyme by a few weeks after birth [110,111,112]. Other studies have suggested that some macrophages in the valve interstitium are derivatives of the endocardium and that these cells appear to be more phagocytic than those derived from the bone marrow [41]. In healthy, mature valve leaflets, the VIC are generally quiescent with only a low level of cell turnover. These cells maintain normal valve structure and function and express a variety of markers that place them within the phenotypic spectrum of fibroblasts (although they are distinct from fibroblasts in other tissues). Similarly, the endocardial and immune cells found in the arterial valves appear to change little in the first few weeks of postnatal life. However, in response to disease or injury, a subpopulation of these apparently quiescent VIC can be activated to take on features of myofibroblasts. This activation occurs in response to signalling factors such as TGFβ which rapidly results in the formation of stress fibres and alignment, leading to increased contractility and increased mechanical stress. This may be the first step in a pathway that ultimately leads to gross remodelling of the valve matrix and valve disease. In diseased valves, some VIC begin to differentiate into osteoblasts to promote calcification of the leaflets. The transcriptional and signalling factors underlying this transition have been reviewed in detail [80,113]. However, the lineage of these VIC has not yet been determined. It is equally possible that different lineages have specific functions, or conversely, that there are no functional differences between them, with each are as likely as any other to remain in the mature valves.
10. Mechanisms Underpinning BAV
Congenital malformations of arterial valves include valve leaflet dysplasia, in which the leaflets are typically thickened and/or shortened, and valves with abnormal numbers of leaflets, including unicuspid, bicuspid and quadricuspid variants [114,115]. In all cases, the valves are predisposed to stenosis and/or regurgitation and degeneration in later life. Clinical series from the U.S. and Europe have suggested a common left-right leaflet (LR) occurs in about 85% of non-syndromic BAV cases [116,117]. However, studies from South Korea and Japan suggest that the LR pattern may be found in less than 60% of Asian patients [118,119]. Common left/non-coronary (LN) leaflets are rare in any population. Recent studies have suggested that genetic syndromes can be associated with different patterns of leaflet fusion. For example, Down’s syndrome is associated with a common RN leaflet, whilst Turner and DiGeorge syndrome with a common LR leaflet [120]. Together, all this evidence suggests that there is likely to be a strong genetic element to leaflet patterning. A well-recognised feature of bicuspid valves is the presence or absence of a raphe, presumed to represent a fusion seam, which in some cases is associated with a notch or an asymmetry suggesting a third leaflet formed during development, but fused with one of the others. Although it is commonly stated that 85–90% of BAV have a raphe [116,117], recent studies suggest as many as 25% may have no raphe [121].
Numerous mechanisms to explain development of BAV have been proposed (Figure 6A) over the last century [122,123,124,125,126,127]. These include excessive cushion/leaflet fusion, failure to form the main cushions, abnormal outflow tract septation, and the absence of one of the leaflet primordia, primarily the ICVS that will form the non-coronary leaflet. Although many of these suggestions were made decades before developmental molecular genetics existed, these potential mechanisms remain and there is evidence for each of them as potential mechanisms for BAV in the literature today.
11. Hyperplastic Cushions/Leaflets and Excessive Fusion
Whilst we now know that severe disruption of EndMT results in acellular or severely hypoplastic cushions and embryonic death in mid development (e.g., [128]), it remains possible that more subtle defects, for instance those that prevent appropriate resolution of EndMT, might disrupt cushion positioning or lead to hyperplastic cushions and leaflets that are forced into apposition and thus are more likely to fuse. There is accumulating evidence to support the idea that cushion or leaflet fusion may be a cause of BAV. BAV resulting from excessive fusion of the main outflow cushions has been described in the Syrian hamster, leading to LR fusions. This includes BAV and more rarely bicuspid pulmonary valves (BPV; [126,129,130]) as well as quadricuspid leaflets, all in the same colony of animals [130,131]. An investigation of the developmental defects underlying the abnormal leaflet fusion suggest that hyperplasia of the main cushions shortly after outflow septation pushes the cushions into apposition and this leads to fusion; a fusion seam can sometimes but not always be seen later in gestation in these animals [126,132]. Interestingly, in an inbred colony of the hamsters, even those animals determined to have a tricuspid valve frequently had excessive fusion of the left and right leaflets [132] suggesting that the affected gene (currently unknown) shows variable expressivity, or that other non-genetic factors affect the phenotype. Quadricuspid aortic valves in the outbred colony appear to result from splitting of one of the cushions (usually the right) into two relatively undeveloped cushions [133]; how this relates to the BAV remains unclear.
Recently, another well-described example of leaflet fusion, but this time later in gestation, has been described. In this case Krox20, a transcriptional regulator, was analysed during heart development revealing that the loss of Krox20 in NCC (and more rarely when removed from EDC) can lead to BAV [134,135]. Interestingly, the loss of Krox20 increased the numbers of NCC whilst the numbers of the EDC population remained normal. Excessive numbers of NCC, particularly in the non-coronary leaflet, led to malpositioned and hyperplastic leaflets that fused by late gestation [135]; a raphe could clearly be seen in these cases of BAV. These studies thus confirm that fusions between the cushions or leaflets, at different stages of development, can lead to BAV.
12. Displaced Cushions
A novel mechanism for the development of BAV has been suggested based on anatomical examination of a series of human embryos [136]. These authors observed that in some embryos the aortic ICVS was displaced proximally, with the developing valve tissue first appearing external to the myocardium. The valve then appeared to develop without this displaced ICVS, giving rise to a biscuspid phenotype. Interestingly, this did not appear to occur in the developing pulmonary valve, suggesting that there may be some subtle differences between the formation of the aortic and pulmonary valves [20,136]. The abnormal positioning of the outflow cushions in early development has been suggested to cause BAV in eNOS mutants [137]. In this case, the endocardial cushions were found to be misplaced and the main cushions were fused with the ICVS. More recently, this has been suggested to result from relatively small changes in the contributions of SHF and NCC to the forming cushions [59]. These authors also suggested that the BAV in the eNOS null mice might arise from a distinct mechanism, where the precursor of the anterior/posterior leaflet fails to separate from the main cushions. However, this implies that the ICVS does not exist as a separate entity, and thus is at odds to what we have described earlier, and what seems to be the generally accepted view [2,12,18,19,20,58]. Interestingly, it has been shown that Krox20 can regulate eNOS expression, and that mice heterozygous for mutations in Krox20 and eNOS develop BAV, whereas single heterozygous mutants do not [138], supporting the genetic interaction between these two genes. Similarly, abnormal cushion positioning in mice expressing a dominant-negative form of Rho kinase (ROCK; a modifier of the cytoskeleton) in the NCC lineage results in their fusion. In this case, the abnormal positioning of the cushions appears to result from disrupted aggregation of NCC within the forming cushions [12].
13. Bicuspid Valve without Raphe and the Absent Leaflet
Although the majority of human BAV present with a raphe [115,116] some studies suggest that BAV without raphe is more common than generally reported. For example, Koenraadt et al. [120] suggested that absence of raphe was represented in 25% of Dutch patients with BAV. Moreover, in cases of BPV, the majority also had BAV without raphe, suggesting that whatever process causes bicuspid valves without raphe, it affects both valves at the same time [139]. However, it should be remembered that a raphe might be present initially and then disappear later, particularly if the fusion event occurred very early in development [125]. When the complete absence of a leaflet is considered, the non-coronary leaflet is usually suggested to be the missing one [20,62,63,140]. As the non-coronary leaflet has a distinct origin from the main cushions (although the SHF is crucial to all; [20,59]) it can readily be seen how formation of this leaflet could be disrupted whilst leaving the right and left leaflets intact and with no evidence of raphe. Interestingly, there appears to be an over-representation of this type of BAV (absent non-coronary leaflet) within the mouse literature, suggesting that this mechanism may be common, at least in this species.
14. Abnormal Leaflet Numbers in the Setting of Defects in Outflow Tract Septation
Abnormalities in outflow tract septation, for example, common arterial trunk, are accompanied by defects in the arterial valves, with dysplasia and abnormal numbers of leaflets seen in the common truncal valve. However, the number of leaflets observed varies, with 3 or 4 leaflets being most common. It seems probable that the number of leaflets varies according to the cause of the common trunk, with abnormalities in cushion fusion, as is seen in the setting of NCC deficiency, usually resulting in four leaflets, (for example the Splotch (Pax3) mutant; [141]), as might be expected if the two main cushions do not fuse and separate (Figure 6B). In contrast, three leaflets are more commonly seen in the setting of single trunk associated with pulmonary atresia, as has been suggested in the Tbx1 mouse mutant [61], and are likely to result from loss of the SHF-derived tissue that would normally form the pulmonary trunk. However, there is considerable variation in the numbers of leaflets found in the setting of common arterial trunk, even those where the lineage origin of the defect is clear, showing that there is not a straightforward relationship between abnormalities of particular cell types and valve phenotype.
15. Bicuspid Valves and Congenital Heart Malformations
BAV is frequently found in combination with other left ventricular outflow tract malformations, including coarctation of the aorta (which is particularly common in Turner syndrome) ventricular septal defects and hypoplastic left heart syndrome (reviewed in [116]). It is reasonable to suggest that when malformations frequently occur together, they share a common aetiology. This could be due to the disruption of specific genes or signalling pathways, or to disruption of related developmental processes. For example, it has been speculated that defects in NCC could lead to a range of defects that include BAV, coarctation of the aorta, common arterial trunk and VSD [142,143,144,145]. As all the affected areas have major contributions from NCC, this seems a reasonable conclusion.
Alternatively, some groupings of malformations could be sequences rather than syndromes, meaning that a primary abnormality leads to secondary consequences that are not directly linked to the primary insult. An example of this might be some sub-types of HLHS, where it is speculated that the primary defect specifically affects the mitral valve, with all of the other defects (ventricular hypoplasia, aortic stenosis/atresia and aortic hypoplasia) being secondary consequences of this that result from disrupted blood flow (the “no flow no grow” hypothesis (reviewed in [6]). It remains to be seen whether the association of left-sided ventricular outflow tract (LVOT) anomalies, that includes BAV, aortic stenosis, VSD and coarctation of the aorta and HLHS, is linked to a genetic defect that disrupts all of the affected areas of the heart, with variable expressivity. BAV has also been linked to atherosclerotic vascular disease as well as aortopathy and linked syndromes, e.g., Marfan, (reviewed in [116]). In all these cases it will be important to determine whether there is a common genetic pathway or progenitor cell linking the conditions or whether they are linked by physiological disturbance.
In contrast to BAV, isolated bicuspid pulmonary valve (BPV) is extremely rare, except in patients with tetralogy of Fallot, where it affects half to 2/3 of patients [146]. Surprisingly, the morphology of these valves has not been systematically described, probably because it has no bearing on the immediate surgical issues. However, as during surgical repair the transannular patch is placed on the accessible anterior aspect of the pulmonary trunk, it is possible that the anterior intercalated leaflet is missing in the majority of cases. Anecdotally, it has been suggested that raphes are not seen in these BPV. Clearly, an analysis of the morphology of BPV is needed. If the anterior intercalated leaflet is missing, this would support the notion that tetralogy of Fallot is due to an abnormality of the SHF. This is important since it was assumed for many years that the outflow defects seen in DiGeorge syndrome—most commonly tetralogy of Fallot or common arterial trunk—were due to a defect in NCC [147,148]. However, since the identification of TBX1 as the main cardiovascular gene linked to DiGeorge syndrome, it has become clear that this is an indirect effect on NCC, as TBX1 is not expressed in NCC, but it influences their migration (reviewed in [149,150,151]). Furthermore, three of the characteristic malformations found in tetralogy of Fallot (pulmonary atresia, deviation of the outlet septum and ventricular septal defect) can be linked to defects in the SHF [61,151,152], whilst the fourth, right ventricular hypertrophy does not manifest until after birth and may be secondary to the other defects.
16. The Genomics of BAV
Genomic analyses of congenital heart disease have been successful in discovering a small number of variants in the small percentage of patients with syndromic conditions [153,154,155,156]. However, the genetic basis of common conditions such as BAV, even when appearing to affect families, has remained elusive. A number of genes including NOTCH1, GATA 4, 5 and 6, and have been previously implicated in CHD from mouse models [157,158,159,160,161,162,163,164,165,166]. Thus, there is obviously good reason to consider these genes as candidates for human BAV. However, knockout of most of these genes results in severe, complex cardiovascular defects, rather than simply BAV, and modelling of human variants of these genes is limited and often disappointing. This is particularly so with NOTCH1. Variants are readily reported in genomic studies, but mouse knock out studies do not recapitulate the BAV (see [127]). Some of this may relate to incomplete penetrance where the mice carry a variant but are not subject to malformation (as is commonly seen in BAV families) or variable expressivity where variations of phenotype are seen. If so, this questions the utility of functional assays in preclinical models, and also preventative treatment or preconceptual counselling for families. Alternatively, some variants may not be in themselves pathological in isolation but may interact with the individual’s developmental genetic landscape to make abnormalities more likely. If there is a specific modifier gene, we might call this oligogenic inheritance, but a range of modifiers may equally contribute. Again, in this scenario, it is very difficult to perform functional assays. There is also a problem regarding what functional assays to perform. The most popular functional assay for BAV variants is an EndMT assay. However, the failure of EndMT results in hypoplasia of the cushions (e.g., [167]) and this causes severe septal as well as valve defects (e.g., [168]). Hence, there is no experimental evidence to indicate a failure of EndMT could cause isolated BAV. Furthermore, EndMT is only one discrete process in valve development—much of which we still do not mechanistically understand. Another example of a suggested BAV-causing gene is GATA4 (e.g., [163,169,170]). Patient variants in GATA4 have been shown to disrupt EndMT in induced pluripotent stem cells [163] and Gata4 has been shown to be required for EndMT to form the endocardial cushions in the mouse heart [26]; indeed, mice lacking functional Gata4 have severe heart defects. Unfortunately, to our knowledge, BAV has not been reported in any mouse models of Gata4 insufficiency (e.g., [171,172,173]). In contrast, GATA6 seems more likely to be relevant to BAV. GATA6 variants have been reported in patients with BAV [161,174,175] and these have been at least partially validated, in some cases by in vitro assays (showing that patient variants had reduced transcriptional activity; [174]) and importantly by knocking out Gata6 in mice and showing that BAV results [161]. Of relevance to this review, it was also shown that knockout of Gata6 solely in SHF progenitors was enough to recapitulate the BAV seen in total knockouts [161], highlighting the importance of this lineage in the development of the arterial valve leaflets. Studies using zebrafish have also been able to validate the importance of BAV variants for cardiovascular development (e.g., [100]), although these have the disadvantage of not being able to recapitulate the BAV phenotype itself, as zebrafish ordinarily have a bicuspid aortic valve. The best validated BAV gene currently is ROBO4 [166]. A potential disease-causing variant in this gene was originally identified by whole exome sequencing in a family with BAV, although in the presence of thoracic aortic aneurysm. Further variants were then identified in another small family and in sporadic cases with a mixture of RL and RN fusions. The relevance of ROBO4 for BAV was confirmed in a variety of ways. This included in vitro studies that showed that ROBO4 variant alleles disrupt endothelial barrier function, a mouse knockout for Robo4 that developed BAV and aortic aneurysm, and most impressively, a mouse knock-in of the specific variant that was identified in the original family, which also developed BAV and aortic aneurysm [166]. This comprehensively validates the ROBO4 gene as a cause of BAV in the setting of aortopathy. Although this sets a “gold standard” for such studies it is again remarkable that a gene has been fully validated for a syndromic/familial condition rather than in sporadic cases. Finally, it may be time to re-evaluate the mechanisms by which sporadic developmental defects occur as arguably, the sum of experience to date suggests that the simple concept of the disease-causing gene variant is not valid. Other modes of disturbing genetic cascades such as alternative splicing or stochastic mechanisms may need to be explored.
17. Final Conclusions
In this review, we have indicated the major roles performed by NCC and the SHF in forming the arterial valves elucidated using animal models. In contrast the recent attempts to interpret the causes of human malformations through genomic approaches have been limited. It is clear that objective and detailed analysis of the arterial valve phenotypes in human aortic and pulmonary valve malformations has not been historically performed and this is especially relevant in bicuspid aortic and pulmonary valves. Similarly, in developmental studies there are areas of valve development, such as sculpting and sinus formation, that have received little attention so far, but are essential to understand if variants invoked in human disease conditions are to be correctly interpreted. Finally, the homeostatic biology of the valves is of great relevance for the aging human heart and in particular it will be essential to know whether developmental populations such as SHF and NCC, or indeed later additions from bone marrow-derived cells, have specific roles, or alternatively, the differentiated identity, regardless of origin, is what matters.
Funding
The authors were funded by British Heart Foundation Programme Grant RG/19/2/34256.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
- Webb, S.; Qayyum, S.R.; Anderson, R.H.; Lamers, W.H.; Richardson, M.K. Septation and separation within the outflow tract of the developing heart. J. Anat. 2003, 202, 327–342. [Google Scholar] [CrossRef]
- Anderson, R.H.; Mori, S.; Spicer, D.E.; Brown, N.A.; Mohun, T.J. Development and Morphology of the Ventricular Outflow Tracts. World J. Pediatr. Congenit. Hear. Surg. 2016, 7, 561–577. [Google Scholar] [CrossRef] [Green Version]
- Richardson, R.; Eley, L.; Donald-Wilson, C.; Davis, J.; Curley, N.; Alqahtani, A.; Murphy, L.; Anderson, R.H.; Henderson, D.J.; Chaudhry, B. Development and maturation of the fibrous components of the arterial roots in the mouse heart. J. Anat. 2017, 232, 554–567. [Google Scholar] [CrossRef] [Green Version]
- Sievers, H.H.; Hemmer, W.; Beyersdorf, F.; Moritz, A.; Moosdorf, R.; Lichtenberg, A.; Misfeld, M.; Charitos, E.I.; Working Group for Aortic Valve Surgery of German Society of Thoracic and Cardiovascular Surgery. The everyday used nomenclature of the aortic root components: The tower of Babel? Eur. J. Cardiothorac. Surg. 2012, 41, 478–482. [Google Scholar] [CrossRef] [Green Version]
- Anderson, R.H.; Devine, W.A.; Ho, S.Y.; Smith, A.; McKay, R. The myth of the aortic annulus: The anatomy of the subaortic outflow tract. Ann. Thorac. Surg. 1991, 52, 640–646. [Google Scholar] [CrossRef]
- Crucean, A.; Alqahtani, A.; Barron, D.J.; Brawn, W.J.; Richardson, R.V.; O’Sullivan, J.; Anderson, R.H.; Henderson, D.J.; Chaudhry, B. Re-evaluation of hypoplastic left heart syndrome from a developmental and morphological perspective. Orphanet J. Rare Dis. 2017, 12, 1–10. [Google Scholar] [CrossRef]
- Choudhary, B.; Zhou, J.; Li, P.; Thomas, S.; Kaartinen, V.; Sucov, H.M. Absence of TGFβ signaling in embryonic vascular smooth muscle leads to reduced lysyl oxidase expression, impaired elastogenesis, and aneurysm. Genesis 2009, 47, 115–121. [Google Scholar] [CrossRef]
- Harmon, A.; Nakano, A. Nkx2-5 lineage tracing visualizes the distribution of second heart field-derived aortic smooth muscle. Genesis 2013, 51, 862–869. [Google Scholar] [CrossRef] [Green Version]
- Von Gise, A.; Pu, W.T. Endocardial and epicardial epithelial to mesenchymal transitions in heart development and disease. Circ. Res. 2012, 110, 1628–1645. [Google Scholar] [CrossRef]
- MacGrogan, D.; Luxán, G.; Driessen-Mol, A.; Bouten, C.V.; Baaijens, F.; De La Pompa, J.L. How to Make a Heart Valve: From Embryonic Development to Bioengineering of Living Valve Substitutes. Cold Spring Harb. Perspect. Med. 2014, 4, a013912. [Google Scholar] [CrossRef]
- O’Donnell, A.; Yutzey, K.E. Mechanisms of heart valve development and disease. Development 2020, 147, dev183020. [Google Scholar] [CrossRef]
- Phillips, H.M.; Mahendran, P.; Singh, E.; Anderson, R.H.; Chaudhry, B.; Henderson, D.J. Neural crest cells are required for correct positioning of the developing outflow cushions and pattern the arterial valve leaflets. Cardiovasc. Res. 2013, 99, 452–460. [Google Scholar] [CrossRef] [Green Version]
- Courchaine, K.; Gray, M.J.; Beel, K.; Thornburg, K.L.; Rugonyi, S. 4-D Computational Modeling of Cardiac Outflow Tract Hemodynamics over Looping Developmental Stages in Chicken Embryos. J. Cardiovasc. Dev. Dis. 2019, 6, 11. [Google Scholar] [CrossRef] [Green Version]
- Costell, M.; Carmona, R.; Gustafsson, E.; González-Iriarte, M.; Faessler, R.; Muñoz-Chápuli, R. Hyperplastic conotruncal endocardial cushions and transposition of great arteries in perlecan-null mice. Circ. Res. 2002, 91, 158–164. [Google Scholar] [CrossRef] [Green Version]
- Anderson, R.H.; Chaudhry, B.; Mohun, T.J.; Bamforth, S.D.; Hoyland, D.; Phillips, H.M.; Webb, S.; Moorman, A.F.; Brown, N.A.; Henderson, D.J. Normal and abnormal development of the intrapericardial arterial trunks in humans and mice. Cardiovasc. Res. 2012, 95, 108–115. [Google Scholar] [CrossRef] [Green Version]
- Waldo, K.L.; Hutson, M.R.; Ward, C.C.; Zdanowicz, M.; Stadt, H.A.; Kumiski, D.; Abu-Issa, R.; Kirby, M.L. Secondary heart field contributes myocardium and smooth muscle to the arterial pole of the developing heart. Dev. Biol. 2005, 281, 78–90. [Google Scholar] [CrossRef] [Green Version]
- Ramsbottom, S.A.; Sharma, V.; Rhee, H.J.; Eley, L.; Phillips, H.M.; Rigby, H.F.; Dean, C.; Chaudhry, B.; Henderson, D.J. Vangl2-Regulated Polarisation of Second Heart Field-Derived Cells is Required for Outflow Tract Lengthening during Cardiac Development. PLoS Genet. 2014, 10, e1004871. [Google Scholar] [CrossRef] [Green Version]
- Sizarov, A.; Lamers, W.H.; Mohun, T.J.; Brown, N.A.; Anderson, R.H.; Moorman, A.F. Three-dimensional and molecular analysis of the arterial pole of the developing human heart. J. Anat. 2012, 220, 336–349. [Google Scholar] [CrossRef]
- Kramer, T.C. The partitioning of the truncus and conus and the formation of the membranous portion of the interventricular septum in the human heart. Am. J. Anat. 1942, 71, 343–370. [Google Scholar] [CrossRef] [Green Version]
- Eley, L.; Alqahtani, A.; MacGrogan, D.; Richardson, R.V.; Murphy, L.; Salguero-Jiménez, A.; Pedro, M.S.R.S.; Tiurma, S.; McCutcheon, L.; Gilmore, A.; et al. A novel source of arterial valve cells linked to bicuspid aortic valve without raphe in mice. eLife 2018, 7, e34110. [Google Scholar] [CrossRef]
- Combs, M.D.; Yutzey, K.E. Heart Valve Development. Circ. Res. 2009, 105, 408–421. [Google Scholar] [CrossRef] [Green Version]
- De Vlaming, A.; Sauls, K.; Hajdu, Z.; Visconti, R.P.; Mehesz, A.N.; Levine, R.A.; Slaugenhaupt, S.A.; Hagege, A.; Chester, A.H.; Markwald, R.R.; et al. Atrioventricular valve development: New perspectives on an old theme. Differentiation 2012, 84, 103–116. [Google Scholar] [CrossRef] [Green Version]
- Puceat, M. Embryological origin of the endocardium and derived valve progenitor cells: From developmental biology to stem cell-based valve repair. Biochim. Biophys. Acta 2013, 1833, 917–922. [Google Scholar] [CrossRef] [Green Version]
- Cai, C.-L.; Liang, X.; Shi, Y.; Chu, P.-H.; Pfaff, S.L.; Chen, J.; Evans, S. Isl1 Identifies a Cardiac Progenitor Population that Proliferates Prior to Differentiation and Contributes a Majority of Cells to the Heart. Dev. Cell 2003, 5, 877–889. [Google Scholar] [CrossRef] [Green Version]
- Verzi, M.P.; McCulley, D.J.; De Val, S.; Dodou, E.; Black, B.L. The right ventricle, outflow tract, and ventricular septum comprise a restricted expression domain within the secondary/anterior heart field. Dev. Biol. 2005, 287, 134–145. [Google Scholar] [CrossRef] [Green Version]
- Rivera-Feliciano, J.; Lee, K.-H.; Kong, S.W.; Rajagopal, S.; Ma, Q.; Springer, Z.; Izumo, S.; Tabin, C.J.; Pu, W.T. Development of heart valves requires Gata4 expression in endothelial-derived cells. Development 2006, 133, 3607–3618. [Google Scholar] [CrossRef] [Green Version]
- Wu, B.; Wang, Y.; Lui, W.; Langworthy, M.; Tompkins, K.L.; Hatzopoulos, A.K.; Baldwin, H.S.; Zhou, B. Nfatc1 Coordinates Valve Endocardial Cell Lineage Development Required for Heart Valve Formation. Circ. Res. 2011, 109, 183–192. [Google Scholar] [CrossRef] [Green Version]
- De La Pompa, J.L.; Timmerman, L.A.; Takimoto, H.; Yoshida, H.; Elia, A.J.; Samper, E.; Potter, J.; Wakeham, A.; Marengere, L.; Langille, B.L.; et al. Role of the NF-ATc transcription factor in morphogenesis of cardiac valves and septum. Nature 1998, 392, 182–186. [Google Scholar] [CrossRef]
- Ranger, A.M.; Grusby, M.J.; Hodge, M.R.; Gravallese, E.M.; De La Brousse, F.C.; Hoey, T.; Mickanin, C.; Baldwin, H.S.; Glimcher, L.H. The transcription factor NF-ATc is essential for cardiac valve formation. Nature 1998, 392, 186–190. [Google Scholar] [CrossRef]
- Chang, C.-P.; Neilson, J.R.; Bayle, J.; Gestwicki, J.E.; Kuo, A.; Stankunas, K.; Graef, I.A.; Crabtree, G.R. A Field of Myocardial-Endocardial NFAT Signaling Underlies Heart Valve Morphogenesis. Cell 2004, 118, 649–663. [Google Scholar] [CrossRef] [Green Version]
- Timmerman, L.A.; Grego-Bessa, J.; Raya, A.; Bertran, E.; Pérez-Pomares, J.; Díez, J.; Aranda, S.; Palomo-Ponce, S.; McCormick, F.; Izpisúa-Belmonte, J.C.; et al. Notch promotes epithelial-mesenchymal transition during cardiac development and oncogenic transformation. Genes Dev. 2004, 18, 99–115. [Google Scholar] [CrossRef] [Green Version]
- Luna-Zurita, L.; Prados, B.; Grego-Bessa, J.; Luxán, G.; Del Monte, G.; Benguria, A.; Adams, R.H.; Pérez-Pomares, J.; De La Pompa, J.L. Integration of a Notch-dependent mesenchymal gene program and Bmp2-driven cell invasiveness regulates murine cardiac valve formation. J. Clin. Investig. 2010, 120, 3493–3507. [Google Scholar] [CrossRef] [Green Version]
- Ma, L.; Lu, M.-F.; Schwartz, R.J.; Martin, J.F. Bmp2 is essential for cardiac cushion epithelial-mesenchymal transition and myocardial patterning. Development 2005, 132, 5601–5611. [Google Scholar] [CrossRef] [Green Version]
- Kokudo, T.; Suzuki, Y.; Yoshimatsu, Y.; Yamazaki, T.; Watabe, T.; Miyazono, K. Snail is required for TGFbeta-induced endothelial-mesenchymal transition of embryonic stem cell-derived endothelial cells. J. Cell Sci. 2008, 121 Pt 20, 3317–3324. [Google Scholar] [CrossRef] [Green Version]
- Lakkis, M.M.; Epstein, J.A. Neurofibromin modulation of ras activity is required for normal endocardial-mesenchymal transformation in the developing heart. Development 1998, 125, 4359–4367. [Google Scholar]
- Gitler, A.D.; Zhu, Y.; Ismat, F.A.; Lu, M.M.; Yamauchi, Y.; Parada, L.F.; Epstein, J.A. Nf1 has an essential role in endothelial cells. Nat. Genet. 2002, 33, 75–79. [Google Scholar] [CrossRef] [Green Version]
- Araki, T.; Chan, G.; Newbigging, S.; Morikawa, L.; Bronson, R.T.; Neel, B.G. Noonan syndrome cardiac defects are caused by PTPN11 acting in endocardium to enhance endocardial-mesenchymal transformation. Proc. Natl. Acad. Sci. USA 2009, 106, 4736–4741. [Google Scholar] [CrossRef] [Green Version]
- Lincoln, J.; Alfieri, C.M.; Yutzey, K.E. Development of heart valve leaflets and supporting apparatus in chicken and mouse embryos. Dev. Dyn. 2004, 230, 239–250. [Google Scholar] [CrossRef]
- Kisanuki, Y.Y.; Ehammerbc, R.; Miyazaki, J.; Williamsba, S.C.; Arichardsonef, J.; Yanagisawabea, M. Tie2-Cre Transgenic Mice: A New Model for Endothelial Cell-Lineage Analysis In Vivo. Dev. Biol. 2001, 230, 230–242. [Google Scholar] [CrossRef] [Green Version]
- MacGrogan, D.; D’Amato, G.; Travisano, S.; Martínez-Poveda, B.; Luxán, G.; Del Monte-Nieto, G.; Papoutsi, T.; Sbroggiò, M.; Bou, V.; Arco, P.G.-D.; et al. Sequential Ligand-Dependent Notch Signaling Activation Regulates Valve Primordium Formation and Morphogenesis. Circ. Res. 2016, 118, 1480–1497. [Google Scholar] [CrossRef]
- Shigeta, A.; Huang, V.; Zuo, J.; Besada, R.; Nakashima, Y.; Lu, Y.; Ding, Y.; Pellegrini, M.; Kulkarni, R.P.; Hsiai, T.; et al. Endocardially Derived Macrophages are Essential for Valvular Remodeling. Dev. Cell 2019, 48, 617–630. [Google Scholar] [CrossRef] [Green Version]
- Kirby, M.L.; Gale, T.F.; Stewart, D.E. Neural crest cells contribute to normal aorticopulmonary septation. Science 1983, 220, 1059–1061. [Google Scholar] [CrossRef]
- Jiang, X.; Rowitch, D.H.; Soriano, P.; McMahon, A.P.; Sucov, H.M. Fate of the mammalian cardiac neural crest. Development 2000, 127, 1607–1616. [Google Scholar]
- Simões-Costa, M.; Bronner, M.E. Establishing neural crest identity: A gene regulatory recipe. Development 2015, 142, 242–257. [Google Scholar] [CrossRef] [Green Version]
- Leonard, C.E.; Taneyhill, L.A. The road best traveled: Neural crest migration upon the extracellular matrix. Semin. Cell Dev. Biol. 2020, 100, 177–185. [Google Scholar] [CrossRef]
- Dupin, E.; Calloni, G.W.; Coelho-Aguiar, J.D.M.; Le Douarin, N.M. The issue of the multipotency of the neural crest cells. Dev. Biol. 2018, 444, S47–S59. [Google Scholar] [CrossRef]
- Scholl, A.M.; Kirby, M.L.; Reinhold, A.M. Signals controlling neural crest contributions to the heart. Wiley Interdiscip. Rev. Syst. Biol. Med. 2009, 1, 220–227. [Google Scholar] [CrossRef] [Green Version]
- Keyte, A.L.; Alonzo-Johnsen, M.; Hutson, M.R. Evolutionary and developmental origins of the cardiac neural crest: Building a divided outflow tract. Birth Defects Res. C Embryo Today 2014, 102, 309–323. [Google Scholar] [CrossRef] [Green Version]
- Plein, A.; Fantin, A.; Ruhrberg, C. Neural Crest Cells in Cardiovascular Development. Mech. Regen. 2015, 111, 183–200. [Google Scholar] [CrossRef]
- Bockman, D.E.; Redmond, M.E.; Kirby, M.L. Alteration of early vascular development after ablation of cranial neural crest. Anat. Rec. Adv. Integr. Anat. Evol. Biol. 1989, 225, 209–217. [Google Scholar] [CrossRef]
- Bockman, D.E.; Redmond, M.E.; Kirby, M.L. Altered Development of Pharyngeal Arch Vessels after Neural Crest Ablation. Ann. N. Y. Acad. Sci. 1990, 588, 296–304. [Google Scholar] [CrossRef]
- Bradshaw, L.; Chaudhry, B.; Hildreth, V.; Webb, S.; Henderson, D.J. Dual role for neural crest cells during outflow tract septation in the neural crest-deficient mutant Splotch2H. J. Anat. 2009, 214, 245–257. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.B.; Feiner, L.; Lu, M.M.; Li, J.; Ma, X.; Webber, A.L.; Jia, L.; Raper, J.A.; Epstein, J.A. PlexinA2 and semaphorin signaling during cardiac neural crest development. Development 2001, 128, 3071–3080. [Google Scholar] [PubMed]
- Luo, T.; Lee, Y.-H.; Saint-Jeannet, J.-P.; Sargent, T.D. Induction of neural crest in Xenopus by transcription factor AP2. Proc. Natl. Acad. Sci. USA 2003, 100, 532–537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poelmann, R.; Groot, A.G.-D. A Subpopulation of Apoptosis-Prone Cardiac Neural Crest Cells Targets to the Venous Pole: Multiple Functions in Heart Development? Dev. Biol. 1999, 207, 271–286. [Google Scholar] [CrossRef] [PubMed]
- Hoff, M.J.V.D.; Moorman, A.F.; Ruijter, J.M.; Lamers, W.H.; Bennington, R.W.; Markwald, R.R.; Wessels, A. Myocardialization of the Cardiac Outflow Tract. Dev. Biol. 1999, 212, 477–490. [Google Scholar] [CrossRef] [Green Version]
- Hoff, M.J.V.D.; Kruithof, B.P.; Moorman, A.F.; Markwald, R.R.; Wessels, A. Formation of Myocardium after the Initial Development of the Linear Heart Tube. Dev. Biol. 2001, 240, 61–76. [Google Scholar] [CrossRef] [Green Version]
- Mifflin, J.J.; Dupuis, L.E.; Alcala, N.E.; Russell, L.G.; Kern, C.B. Intercalated cushion cells within the cardiac outflow tract are derived from the myocardial troponin T type 2 (Tnnt2) Cre lineage. Dev. Dyn. 2018, 247, 1005–1017. [Google Scholar] [CrossRef]
- Peterson, J.C.; Chughtai, M.; Wisse, L.J.; Groot, A.C.G.-D.; Feng, Q.; Goumans, M.-J.; VanMunsteren, J.C.; Jongbloed, M.R.M.; DeRuiter, M.C. Bicuspid aortic valve formation: Nos3 mutation leads to abnormal lineage patterning of neural crest cells and the second heart field. Dis. Model. Mech. 2018, 11, dmm034637. [Google Scholar] [CrossRef] [Green Version]
- Akiyama, H.; Chaboissier, M.-C.; Behringer, R.R.; Rowitch, D.H.; Schedl, A.; Epstein, J.A.; De Crombrugghe, B. Essential role of Sox9 in the pathway that controls formation of cardiac valves and septa. Proc. Natl. Acad. Sci. USA 2004, 101, 6502–6507. [Google Scholar] [CrossRef] [Green Version]
- Jiao, K.; Kulessa, H.; Tompkins, K.; Zhou, Y.; Batts, L.; Baldwin, H.S.; Hogan, B. An essential role of Bmp4 in the atrioventricular septation of the mouse heart. Genes Dev. 2003, 17, 2362–2367. [Google Scholar] [CrossRef] [Green Version]
- Théveniau-Ruissy, M.; Dandonneau, M.; Mesbah, K.; Ghez, O.; Mattei, M.; Miquerol, L.; Kelly, R.G. The del22q11.2 Candidate GeneTbx1Controls Regional Outflow Tract Identity and Coronary Artery Patterning. Circ. Res. 2008, 103, 142–148. [Google Scholar] [CrossRef] [Green Version]
- Thomas, P.S.; Sridurongrit, S.; Ruiz-Lozano, P.; Kaartinen, V. Deficient Signaling via Alk2 (Acvr1) Leads to Bicuspid Aortic Valve Development. PLoS ONE 2012, 7, e35539. [Google Scholar] [CrossRef] [Green Version]
- Mommersteeg, M.T.; Yeh, M.L.; Parnavelas, J.G.; Andrews, W.D. Disrupted Slit-Robo signalling results in membranous ventricular septum defects and bicuspid aortic valves. Cardiovasc. Res. 2015, 106, 55–66. [Google Scholar] [CrossRef] [Green Version]
- Miquerol, L.; Langille, B.L.; Nagy, A. Embryonic development is disrupted by modest increases in vascularendothelial growth factor gene expression. Development 2000, 127, 3941–3946. [Google Scholar]
- Dor, Y.; Camenisch, T.D.; Itin, A.I.; Fishman, G.; McDonald, J.A.; Carmeliet, P.; Keshet, E. A novel role for VEGF in endocardial cushion formation and its potential contribution to congenital heart defects. Development 2001, 128, 1531–1538. [Google Scholar]
- Lee, Y.M.; Cope, J.J.; Ackermann, G.E.; Goishi, K.; Armstrong, E.J.; Paw, B.H.; Bischoff, J. Vascular endothelial growth factor receptor signaling is required for cardiac valve formation in zebrafish. Dev. Dyn. 2006, 235, 29–37. [Google Scholar] [CrossRef] [Green Version]
- Sugi, Y.; Ito, N.; Szebenyi, G.; Myers, K.; Fallon, J.F.; Mikawa, T.; Markwald, R.R. Fibroblast growth factor (FGF)-4 can induce proliferation of cardiac cushion mesenchymal cells during early valve leaflet formation. Dev. Biol. 2003, 258, 252–263. [Google Scholar] [CrossRef] [Green Version]
- Chen, B.; Bronson, R.T.; Klaman, L.D.; Hampton, T.G.; Wang, J.-F.; Green, P.J.; Magnuson, T.; Douglas, P.S.; Morgan, J.P.; Neel, B.G. Mice mutant for Egfr and Shp2 have defective cardiac semilunar valvulogenesis. Nat. Genet. 2000, 24, 296–299. [Google Scholar] [CrossRef]
- Chen, Y.-H.; Ishii, M.; Sun, J.; Sucov, H.M.; Maxson, R.E.; Maxson, R.E. Msx1 and Msx2 regulate survival of secondary heart field precursors and post-migratory proliferation of cardiac neural crest in the outflow tract. Dev. Biol. 2007, 308, 421–437. [Google Scholar] [CrossRef]
- Srinivasan, D.K.; Dheen, S.T.; Tay, S.S.-W. Maternal diabetes induces congenital heart defects in mice by altering the expression of genes involved in cardiovascular development. Cardiovasc. Diabetol. 2007, 6, 34. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Von Gise, A.; Liu, Q.; Hu, T.; Tian, X.; He, L.; Pu, W.; Huang, X.; He, L.; Cai, C.-L.; et al. Yap1 is Required for Endothelial to Mesenchymal Transition of the Atrioventricular Cushion. J. Biol. Chem. 2014, 289, 18681–18692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mjaatvedt, C.; Yamamura, H.; Capehart, A.; Turner, D.; Markwald, R. The Cspg2 Gene, Disrupted in thehdfMutant, is Required for Right Cardiac Chamber and Endocardial Cushion Formation. Dev. Biol. 1998, 202, 56–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camenisch, T.D.; Spicer, A.P.; Brehm-Gibson, T.; Biesterfeldt, J.; Augustine, M.L.; Calabro, A.; Kubalak, S.; Klewer, S.E.; McDonald, J.A. Disruption of hyaluronan synthase-2 abrogates normal cardiac morphogenesis and hyaluronan-mediated transformation of epithelium to mesenchyme. J. Clin. Investig. 2000, 106, 349–360. [Google Scholar] [CrossRef] [Green Version]
- Dupuis, L.E.; McCulloch, D.R.; McGarity, J.D.; Bahan, A.; Wessels, A.; Weber, D.; Diminich, A.M.; Nelson, C.M.; Apte, S.S.; Kern, C.B. Altered versican cleavage in ADAMTS5 deficient mice; a novel etiology of myxomatous valve disease. Dev. Biol. 2011, 357, 152–164. [Google Scholar] [CrossRef] [Green Version]
- Durst, R.; Sauls, K.; Peal, D.S.; DeVlaming, A.; Toomer, K.; Leyne, M.; Salani, M.; Talkowski, M.E.; Brand, H.; Perrocheau, M.; et al. Mutations in DCHS1 cause mitral valve prolapse. Nature 2015, 525, 109–113. [Google Scholar] [CrossRef]
- Anvarian, Z.; Mykytyn, K.; Mukhopadhyay, S.; Pedersen, L.B.; Christensen, S.T. Cellular signalling by primary cilia in development, organ function and disease. Nat. Rev. Nephrol. 2019, 15, 199–219. [Google Scholar] [CrossRef]
- Toomer, K.A.; Fulmer, D.; Guo, L.; Drohan, A.; Peterson, N.; Swanson, P.; Brooks, B.; Mukherjee, R.; Body, S.; Lipschutz, J.H.; et al. A role for primary cilia in aortic valve development and disease. Dev. Dyn. 2017, 246, 625–634. [Google Scholar] [CrossRef] [Green Version]
- Irigoín, F.; Badano, J.L. Keeping the Balance Between Proliferation and Differentiation: The Primary Cilium. Curr. Genom. 2011, 12, 285–297. [Google Scholar] [CrossRef] [Green Version]
- Hinton, R.B.; Lincoln, J.; Deutsch, G.H.; Osinska, H.; Manning, P.B.; Benson, D.W.; Yutzey, K.E. Extracellular Matrix Remodeling and Organization in Developing and Diseased Aortic Valves. Circ. Res. 2006, 98, 1431–1438. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.-J.; Chen, C.-H.; Zhou, B.; Chang, C.-P. Partitioning the heart: Mechanisms of cardiac septation and valve development. Development 2012, 139, 3277–3299. [Google Scholar] [CrossRef] [Green Version]
- Wu, B.; Baldwin, H.S.; Zhou, B. Nfatc1 directs the endocardial progenitor cells to make heart valve primordium. Trends Cardiovasc. Med. 2013, 23, 294–300. [Google Scholar] [CrossRef] [Green Version]
- Nomura-Kitabayashi, A.; Phoon, C.K.; Kishigami, S.; Rosenthal, J.; Yamauchi, Y.; Abe, K.; Yamamura, K.-I.; Samtani, R.; Lo, C.W.; Mishina, Y. Outflow tract cushions perform a critical valve-like function in the early embryonic heart requiring BMPRIA-mediated signaling in cardiac neural crest. Am. J. Physiol. Circ. Physiol. 2009, 297, H1617–H1628. [Google Scholar] [CrossRef] [Green Version]
- Keyes, W.M.; Sanders, E.J. Regulation of apoptosis in the endocardial cushions of the developing chick heart. Am. J. Physiol. Physiol. 2002, 282, C1348–C1360. [Google Scholar] [CrossRef] [Green Version]
- Cheng, G.; Wessels, A.; Gourdie, R.G.; Thompson, R.P. Spatiotemporal and tissue specific distribution of apoptosis in the developing chick heart. Dev. Dyn. 2002, 223, 119–133. [Google Scholar] [CrossRef]
- Abdelwahid, E.; Rice, D.P.; Pelliniemi, L.J.; Jokinen, E. Overlapping and differential localization of Bmp-2, Bmp-4, Msx-2 and apoptosis in the endocardial cushion and adjacent tissues of the developing mouse heart. Cell Tissue Res. 2001, 305, 67–78. [Google Scholar] [CrossRef]
- Zhao, Z.; Rivkees, S.A. Programmed cell death in the developing heart: Regulation by BMP4 and FGF2. Dev. Dyn. 2000, 217, 388–400. [Google Scholar] [CrossRef]
- Sharma, P.R.; Anderson, R.H.; Copp, A.J.; Henderson, D.J. Spatiotemporal analysis of programmed cell death during mouse cardiac septation. Anat. Rec. Adv. Integr. Anat. Evol. Biol. 2004, 277, 355–369. [Google Scholar] [CrossRef]
- Hurle, J.; Blanco, A.M. Development of mouse semilunar valves. Brain Struct. Funct. 1980, 160, 83–91. [Google Scholar] [CrossRef]
- Hurle, J. Scanning and light microscope studies of the development of the chick embryo semilunar heart valves. Brain Struct. Funct. 1979, 157, 69–80. [Google Scholar] [CrossRef]
- Alfieri, C.M.; Cheek, J.; Chakraborty, S.; Yutzey, K.E. Wnt signaling in heart valve development and osteogenic gene induction. Dev. Biol. 2010, 338, 127–135. [Google Scholar] [CrossRef] [Green Version]
- Bitgood, M.J.; McMahon, A.P. Hedgehog and Bmp genes are coexpressed at many diverse sites of cell-cell interaction in the mouse embryo. Dev. Biol 1995, 172, 126–138. [Google Scholar] [CrossRef] [Green Version]
- Biechler, S.V.; Junor, L.; Evans, A.N.; Eberth, J.F.; Price, R.L.; Potts, J.D.; Yost, M.J.; Goodwin, R.L. The impact of flow-induced forces on the morphogenesis of the outflow tract. Front. Physiol. 2014, 5. [Google Scholar] [CrossRef]
- Goddard, L.M.; Duchemin, A.-L.; Ramalingan, H.; Wu, B.; Chen, M.; Bamezai, S.; Yang, J.; Li, L.; Morley, M.P.; Wang, T.; et al. Hemodynamic Forces Sculpt Developing Heart Valves through a KLF2-WNT9B Paracrine Signaling Axis. Dev. Cell 2017, 43, 274–289. [Google Scholar] [CrossRef]
- Hsu, J.J.; Vedula, V.; Baek, K.I.; Chen, C.; Chen, J.; Chou, M.I.; Lam, J.; Subhedar, S.; Wang, J.; Ding, Y.; et al. Contractile and hemodynamic forces coordinate Notch1b-mediated outflow tract valve formation. JCI Insight 2019, 5. [Google Scholar] [CrossRef] [Green Version]
- Menon, V.; Eberth, J.F.; Goodwin, R.L.; Potts, J.D. Altered Hemodynamics in the Embryonic Heart Affects Outflow Valve Development. J. Cardiovasc. Dev. Dis. 2015, 2, 108–124. [Google Scholar] [CrossRef]
- Menon, V.; Eberth, J.F.; Junor, L.; Potts, A.J.; Belhaj, M.; DiPette, N.J.; Jenkins, M.W.; Potts, J.D. Removing vessel constriction on the embryonic heart results in changes in valve gene expression, morphology, and hemodynamics. Dev. Dyn. 2017, 247, 531–541. [Google Scholar] [CrossRef] [Green Version]
- Steed, E.; Boselli, F.; Vermot, J. Hemodynamics driven cardiac valve morphogenesis. Biochim. Biophys. Acta 2016, 1863, 1760–1766. [Google Scholar] [CrossRef] [PubMed]
- Poelmann, R.E.; Groot, A.C.G.-D. Hemodynamics in Cardiac Development. J. Cardiovasc. Dev. Dis. 2018, 5, 54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heckel, E.; Boselli, F.; Roth, S.; Krudewig, A.; Belting, H.-G.; Charvin, G.; Vermot, J. Oscillatory Flow Modulates Mechanosensitive klf2a Expression through trpv4 and trpp2 during Heart Valve Development. Curr. Biol. 2015, 25, 1354–1361. [Google Scholar] [CrossRef] [Green Version]
- Faucherre, A.; Maati, H.M.O.; Nasr, N.; Pinard, A.; Theron, A.; Odelin, G.; Desvignes, J.-P.; Salgado, D.; Collod-Béroud, G.; Avierinos, J.-F.; et al. Piezo1 is required for outflow tract and aortic valve development. J. Mol. Cell. Cardiol. 2020, 143, 51–62. [Google Scholar] [CrossRef]
- Henderson, D.J.; Copp, A.J. Versican expression is associated with chamber specification, septation, and valvulogenesis in the developing mouse heart. Circ. Res. 1998, 83, 523–532. [Google Scholar] [CrossRef] [Green Version]
- Fenderson, B.A.; Stamenkovic, I.; Aruffo, A. Localization of hyaluronan in mouse embryos during implantation, gastrulation and organogenesis. Differentiation 1993, 54, 85–98. [Google Scholar] [CrossRef]
- Hurle, J.; Kitten, G.; Sakai, L.Y.; Volpin, D.; Solursh, M. Elastic extracellular matrix of the embryonic chick heart: An immunohistological study using laser confocal microscopy. Dev. Dyn. 1994, 200, 321–332. [Google Scholar] [CrossRef] [PubMed]
- Votteler, M.; Berrio, D.A.C.; Horke, A.; Sabatier, L.; Reinhardt, D.P.; Nsair, A.; Aikawa, E.; Schenke-Layland, K. Elastogenesis at the onset of human cardiac valve development. Development 2013, 140, 2345–2353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hulin, A.; Hortells, L.; Gomez-Stallons, M.V.; O’Donnell, A.; Chetal, K.; Adam, M.; Lancellotti, P.; Oury, C.; Potter, S.S.; Salomonis, N.; et al. Maturation of heart valve cell populations during postnatal remodeling. Development 2019, 146, dev173047. [Google Scholar] [CrossRef] [Green Version]
- Wu, B.; Wang, Y.; Xiao, F.; Butcher, J.T.; Yutzey, K.E.; Zhou, B. Developmental Mechanisms of Aortic Valve Malformation and Disease. Annu. Rev. Physiol. 2017, 79, 21–41. [Google Scholar] [CrossRef]
- Cujec, B.; Pollick, C. Isolated Thickening of One Aortic Cusp: Preferential Thickening of the Noncoronary Cusp. J. Am. Soc. Echocardiogr. 1988, 1, 430–432. [Google Scholar] [CrossRef]
- Nakamura, T.; Colbert, M.C.; Robbins, J. Neural Crest Cells Retain Multipotential Characteristics in the Developing Valves and Label the Cardiac Conduction System. Circ. Res. 2006, 98, 1547–1554. [Google Scholar] [CrossRef] [Green Version]
- Anstine, L.J.; Horne, T.E.; Horwitz, E.M.; Lincoln, J. Contribution of Extra-Cardiac Cells in Murine Heart Valves is Age-Dependent. J. Am. Hear. Assoc. 2017, 6. [Google Scholar] [CrossRef] [Green Version]
- Hulin, A.; Anstine, L.J.; Kim, A.J.; Potter, S.J.; DeFalco, T.J.; Lincoln, J.; Yutzey, K.E. Macrophage Transitions in Heart Valve Development and Myxomatous Valve Disease. Arter. Thromb. Vasc. Biol. 2018, 38, 636–644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deb, A.; Wang, S.H.; Skelding, K.; Miller, D.; Simper, D.; Caplice, N. Bone marrow-derived myofibroblasts are present in adult human heart valves. J. Hear. Valve Dis. 2005, 14, 674–678. [Google Scholar]
- Tao, G.; Kotick, J.D.; Lincoln, J. Heart Valve Development, Maintenance, and Disease. Mech. Regen. 2012, 100, 203–232. [Google Scholar] [CrossRef]
- Krepp, J.M.; Roman, M.J.; Devereux, R.B.; Bruce, A.; Prakash, S.K.; Morris, S.A.; Milewicz, D.M.; Holmes, K.W.; Ravekes, W.; Shohet, R.V.; et al. Bicuspid and unicuspid aortic valves: Different phenotypes of the same disease? Insight from the GenTAC Registry. Congenit. Hear. Dis. 2017, 12, 740–745. [Google Scholar] [CrossRef]
- Lopez, A.; Fernández, M.C.; Durán, A.C.; Sans-Coma, V.; Fernández, B. Quadricuspid aortic valves in Syrian hamsters and their formation according to current knowledge on valvulogenesis. Jpn. J. Veter. Res. 2015, 63, 37–43. [Google Scholar]
- Sievers, H.-H.; Schmidtke, C. A classification system for the bicuspid aortic valve from 304 surgical specimens. J. Thorac. Cardiovasc. Surg. 2007, 133, 1226–1233. [Google Scholar] [CrossRef] [Green Version]
- Urena, M.; Doyle, D.; Dumont, É.; Ribeiro, H.B.; Bilodeau, S.; Rodés-Cabau, J. Transcatheter Aortic Valve Replacement with a Balloon-expandable Valve for the Treatment of Noncalcified Bicuspid Aortic Valve Disease. Rev. Española Cardiol. 2014, 67, 327–329. [Google Scholar] [CrossRef]
- Kinoshita, T.; Naito, S.; Tomoaki, S.; Asai, T. Valve Phenotype and Risk Factors of Aortic Dilatation After Aortic Valve Replacement in Japanese Patients With Bicuspid Aortic Valve. Circ. J. 2016, 80, 1356–1361. [Google Scholar] [CrossRef] [Green Version]
- Sun, B.J.; Jin, X.; Song, J.-K.; Lee, S.; Lee, J.H.; Park, J.-B.; Lee, S.-P.; Kim, D.-H.; Park, S.-J.; Kim, Y.-J.; et al. Clinical Characteristics of Korean Patients with Bicuspid Aortic Valve Who Underwent Aortic Valve Surgery. Korean Circ. J. 2018, 48, 48. [Google Scholar] [CrossRef] [Green Version]
- Niaz, T.; Poterucha, J.T.; Olson, T.M.; Johnson, J.N.; Craviari, C.; Nienaber, T.; Palfreeman, J.; Cetta, F.; Hagler, D.J. Characteristic Morphologies of the Bicuspid Aortic Valve in Patients with Genetic Syndromes. J. Am. Soc. Echocardiogr. 2017, 31, 194–200. [Google Scholar] [CrossRef]
- Koenraadt, W.M.C.; Grewal, N.; Gaidoukevitch, O.Y.; DeRuiter, M.C.; Groot, A.C.G.-D.; Bartelings, M.M.; Holman, E.R.; Klautz, R.J.M.; Schalij, M.J.; Jongbloed, M.R. The extent of the raphe in bicuspid aortic valves is associated with aortic regurgitation and aortic root dilatation. Neth. Hear. J. 2016, 24, 127–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Vries, W.M. Ueber Abweichungen in der Zahl der Semilunar-klappen. Beitr. Pathol. Anat. 1918, 64, 39–54. [Google Scholar]
- Simonds, J.P. Congenital Malformations of the Aortic and Pulmonary Valves. Am. J. Med Sci. 1923, 166, 584–595. [Google Scholar] [CrossRef]
- Koletsky, S. Congenital bicuspid pulmonary valves. Arch. Pathol. 1941, 31, 338–353. [Google Scholar]
- Shaner, R.F. Abnormal pulmonary and aortic semilunar valves in embryos. Anat. Rec. Adv. Integr. Anat. Evol. Biol. 1963, 147, 5–13. [Google Scholar] [CrossRef]
- Sans-Coma, V.; Fernández, B.; Durán, A.C.; Thiene, G.; Arqué, J.M.; Muñoz-Chápuli, R.; Cardo, M. Fusion of valve cushions as a key factor in the formation of congenital bicuspid aortic valves in Syrian hamsters. Anat. Rec. 1996, 244, 490–498. [Google Scholar] [CrossRef]
- Soto-Navarrete, M.T.; López-Unzu, M.Á.; Durán, A.C.; Fernandez, B. Embryonic development of bicuspid aortic valves. Prog. Cardiovasc. Dis. 2020, 25. [Google Scholar] [CrossRef]
- Luxán, G.; D’Amato, G.; MacGrogan, D.; De La Pompa, J.L. Endocardial Notch Signaling in Cardiac Development and Disease. Circ. Res. 2016, 118. [Google Scholar] [CrossRef]
- Sans-Coma, V.; Cardo, M.; Thiene, G.; Fernández, B.; Arqué, J.M.; Durán, A.C. Bicuspid aortic and pulmonary valves in the Syrian hamster. Int. J. Cardiol. 1992, 34, 249–254. [Google Scholar] [CrossRef]
- Fernández, B.; Fernandez, M.C.; Durán, A.C.; Lopez, D.; Martire, A.; Sans-Coma, V. Anatomy and formation of congenital bicuspid and quadricuspid pulmonary valves in Syrian hamsters. Anat. Rec. Adv. Integr. Anat. Evol. Biol. 1998, 250, 70–79. [Google Scholar] [CrossRef]
- Fernández, B.; Durán, A.C.; Thiene, G.; Cardo, M.; Arqué, J.M.; Sans-Coma, V. Embryological evidence for the formation of a quadricuspid aortic valve in the Syrian hamster. Cardiovasc. Pathol. 1994, 3, 287–291. [Google Scholar] [CrossRef]
- Sans-Coma, V.; Fernandez, M.C.; Fernández, B.; Durán, A.C.; Anderson, R.H.; Arqué, J.M. Genetically alike Syrian hamsters display both bifoliate and trifoliate aortic valves. J. Anat. 2011, 220, 92–101. [Google Scholar] [CrossRef]
- Fernandez, B.; Durán, A.; Martire, A.; López, D.; Sans-Coma, V. New Embryological Evidence for the Formation of Quadricuspid Aortic Valves in the Syrian Hamster (Mesocricetus auratus). J. Comp. Pathol. 1999, 121, 89–94. [Google Scholar] [CrossRef]
- Odelin, G.; Faure, E.; Kober, F.; Maurel-Zaffran, C.; Theron, A.; Coulpier, F.; Guillet, B.; Bernard, M.; Avierinos, J.-F.; Charnay, P.; et al. Loss of Krox20 results in aortic valve regurgitation and impaired transcriptional activation of fibrillar collagen genes. Cardiovasc. Res. 2014, 104, 443–455. [Google Scholar] [CrossRef] [Green Version]
- Odelin, G.; Faure, E.; Coulpier, F.; Di Bonito, M.; Bajolle, F.; Studer, M.; Avierinos, J.-F.; Charnay, P.; Topilko, P.; Zaffran, S. Krox20 defines a subpopulation of cardiac neural crest cells contributing to arterial valves and bicuspid aortic valve. Development 2017, 145, dev151944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milos, N.C.; Nordstrom, D.B.; Ongaro, I.; Chow, A.K. Variations in structure of the outflow tract of the human embryonic heart: A new hypothesis for generating bicuspid aortic semilunar valves. Ann. Anat. Anat. Anz. 2017, 211, 88–103. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, B.; Durán, A.C.; Fernández-Gallego, T.; Fernandez, M.C.; Such, M.; Arqué, J.M.; Sans-Coma, V. Bicuspid Aortic Valves With Different Spatial Orientations of the Leaflets Are Distinct Etiological Entities. J. Am. Coll. Cardiol. 2009, 54, 2312–2318. [Google Scholar] [CrossRef] [Green Version]
- Odelin, G.; Faure, E.; Maurel-Zaffran, C.; Zaffran, S. Krox20 Regulates Endothelial Nitric Oxide Signaling in Aortic Valve Development and Disease. J. Cardiovasc. Dev. Dis. 2019, 6, 39. [Google Scholar] [CrossRef] [Green Version]
- Koenraadt, W.M.C.; Bartelings, M.M.; Groot, A.C.G.-D.; Bökenkamp, R.; DeRuiter, M.C.; Schalij, M.J.; Jongbloed, M.R. Pulmonary Valve Morphology in Patients with Bicuspid Aortic Valves. Pediatr. Cardiol. 2018, 39, 690–694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laforest, B.; Andelfinger, G.; Nemer, M. Loss of Gata5 in mice leads to bicuspid aortic valve. J. Clin. Investig. 2011, 121, 2876–2887. [Google Scholar] [CrossRef] [Green Version]
- Franz, T. Persistent truncus arteriosus in the Splotch mutant mouse. Brain Struct. Funct. 1989, 180, 457–464. [Google Scholar] [CrossRef] [PubMed]
- Sundeen, J.T.; Bloom, S. Sinus of valsalva aneurysm associated with multiple conotruncal congenital malformations. Hum. Pathol. 1987, 18, 96–99. [Google Scholar] [CrossRef]
- Kappetein, A.; Groot, A.G.-D.; Zwinderman, A.; Rohmer, J.; Poelmann, R.; Huysmans, H. The neural crest as a possible pathogenetic factor in coarctation of the aorta and bicuspid aortic valve. J. Thorac. Cardiovasc. Surg. 1991, 102, 830–836. [Google Scholar] [CrossRef]
- Jain, R.; Engleka, K.A.; Rentschler, S.L.; Manderfield, L.J.; Li, L.; Yuan, L.; Epstein, J.A. Cardiac neural crest orchestrates remodeling and functional maturation of mouse semilunar valves. J. Clin. Investig. 2011, 121, 422–430. [Google Scholar] [CrossRef] [Green Version]
- Komiyama, M. Cardio-cephalic neural crest syndrome: A novel hypothesis of vascular neurocristopathy. Interv. Neuroradiol. 2017, 23, 572–576. [Google Scholar] [CrossRef]
- Quintessenza, J.A. Tetralogy of Fallot: Management of the Pulmonary Valve. In Surgery of Conotruncal Anomalies; Lacour-Gayet, F., Bove, E.L., Hraska, V., Morell, V.O., Spray, T.L., Eds.; Springer International Publishing: Cham, Switzerland, 2016; pp. 139–147. [Google Scholar]
- Lammer, E.J.; Opitz, J.M.; Reynolds, J.F. The DiGeorge anomaly as a developmental field defect. Am. J. Med. Genet. 1986, 25, 113–127. [Google Scholar] [CrossRef]
- Van Mierop, L.H.; Kutsche, L.M. Cardiovascular anomalies in digeorge syndrome and importance of neural crest as a possible pathogenetic factor. Am. J. Cardiol. 1986, 58, 133–137. [Google Scholar] [CrossRef]
- Keyte, A.; Hutson, M.R. The neural crest in cardiac congenital anomalies. Differentiation 2012, 84, 25–40. [Google Scholar] [CrossRef] [Green Version]
- Parisot, P.; Mesbah, K.; Théveniau-Ruissy, M.; Kelly, R.G. Tbx1, subpulmonary myocardium and conotruncal congenital heart defects. Birth Defects Res. Part A Clin. Mol. Teratol. 2011, 91, 477–484. [Google Scholar] [CrossRef]
- Hasten, E.; McDonald-McGinn, D.M.; Crowley, T.B.; Zackai, E.; Emanuel, B.S.; Morrow, B.E.; Racedo, S.E. Dysregulation of TBX1 dosage in the anterior heart field results in congenital heart disease resembling the 22q11.2 duplication syndrome. Hum. Mol. Genet. 2018, 27, 1847–1857. [Google Scholar] [CrossRef]
- Zhou, Z.; Wang, J.; Guo, C.; Chang, W.; Zhuang, J.; Zhu, P.; Li, X. Temporally Distinct Six2-Positive Second Heart Field Progenitors Regulate Mammalian Heart Development and Disease. Cell Rep. 2017, 18, 1019–1032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaidi, S.; Choi, M.; Wakimoto, H.; Ma, L.; Jiang, J.; Overton, J.D.; Romano-Adesman, A.; Bjornson, R.D.; Breitbart, R.E.; Brown, K.K.; et al. De novo mutations in histone-modifying genes in congenital heart disease. Nature 2013, 498, 220–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Homsy, J.; Zaidi, S.; Shen, Y.; Ware, J.S.; Samocha, K.E.; Karczewski, J.; DePalma, S.R.; Mckean, D.; Wakimoto, H.; Gorham, J.; et al. De novo mutations in congenital heart disease with neurodevelopmental and other congenital anomalies. Science 2015, 350, 1262–1266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glessner, J.T.; Bick, A.G.; Ito, K.; Homsy, J.G.; Rodriguez-Murillo, L.; Fromer, M.; Mazaika, E.; Vardarajan, B.; Italia, M.; Leipzig, J.; et al. Increased Frequency of De Novo Copy Number Variants in Congenital Heart Disease by Integrative Analysis of Single Nucleotide Polymorphism Array and Exome Sequence Data. Circ. Res. 2014, 115, 884–896. [Google Scholar] [CrossRef] [PubMed]
- Sifrim, A.; Hitz, M.-P.; Wilsdon, A.; Breckpot, J.; Al Turki, S.H.; Thienpont, B.; McRae, J.; Fitzgerald, T.W.; Singh, T.; Swaminathan, G.J.; et al. Distinct genetic architectures for syndromic and nonsyndromic congenital heart defects identified by exome sequencing. Nat. Genet. 2016, 48, 1060–1065. [Google Scholar] [CrossRef] [PubMed]
- Wooten, E.C.; Iyer, L.K.; Montefusco, M.C.; Hedgepeth, A.K.; Payne, D.D.; Kapur, N.K.; Housman, D.E.; Mendelsohn, M.E.; Huggins, G.S. Application of gene network analysis techniques identifies AXIN1/PDIA2 and endoglin haplotypes associated with bicuspid aortic valve. PLoS ONE 2010, 5, e8830. [Google Scholar] [CrossRef] [PubMed]
- Martin, L.J.; Pilipenko, V.; Kaufman, K.M.; Cripe, L.H.; Kottyan, L.; Keddache, M.; Dexheimer, P.; Weirauch, M.T.; Benson, D.W. Whole Exome Sequencing for Familial Bicuspid Aortic Valve Identifies Putative Variants. Circ. Cardiovasc. Genet. 2014, 7, 677–683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonachea, E.M.; Zender, G.; White, P.; Corsmeier, D.J.; Newsom, D.; Fitzgerald-Butt, S.; Garg, V.; McBride, K.L. Use of a targeted, combinatorial next-generation sequencing approach for the study of bicuspid aortic valve. BMC Med. Genom. 2014, 7, 56. [Google Scholar] [CrossRef] [Green Version]
- Dargis, N.; Lamontagne, M.; Gaudreault, N.; Sbarra, L.; Henry, C.; Pibarot, P.; Mathieu, P.; Bossé, Y. Identification of Gender-Specific Genetic Variants in Patients With Bicuspid Aortic Valve. Am. J. Cardiol. 2016, 117, 420–426. [Google Scholar] [CrossRef]
- Gharibeh, L.; Komati, H.; Bossé, Y.; Boodhwani, M.; Heydarpour, M.; Fortier, M.; Hassanzadeh, R.; Ngu, J.; Mathieu, P.; Body, S.; et al. GATA6 Regulates Aortic Valve Remodeling, and Its Haploinsufficiency Leads to Right-Left Type Bicuspid Aortic Valve. Circulation 2018, 138, 1025–1038. [Google Scholar] [CrossRef] [PubMed]
- Giusti, B.; Sticchi, E.; De Cario, R.; Magi, A.; Nistri, S.; Pepe, G. Genetic Bases of Bicuspid Aortic Valve: The Contribution of Traditional and High-Throughput Sequencing Approaches on Research and Diagnosis. Front. Physiol. 2017, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, B.; Zhou, W.; Jiao, J.; Nielsen, J.B.; Mathis, M.R.; Heydarpour, M.; Lettre, G.; Folkersen, L.; Prakash, S.; Schurmann, C.; et al. Protein-altering and regulatory genetic variants near GATA4 implicated in bicuspid aortic valve. Nat. Commun. 2017, 8, 15481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKellar, S.H.; Tester, D.J.; Yagubyan, M.; Majumdar, R.; Ackerman, M.J.; Sundt, T.M. Novel NOTCH1 mutations in patients with bicuspid aortic valve disease and thoracic aortic aneurysms. J. Thorac. Cardiovasc. Surg. 2007, 134, 290–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohamed, S.A.; Aherrahrou, Z.; Liptau, H.; Erasmi, A.W.; Hagemann, C.; Wrobel, S.; Borzym, K.; Schunkert, H.; Sievers, H.-H.; Erdmann, J. Novel missense mutations (p.T596M and p.P1797H) in NOTCH1 in patients with bicuspid aortic valve. Biochem. Biophys. Res. Commun. 2006, 345, 1460–1465. [Google Scholar] [CrossRef]
- Gould, R.A.; Genomics, B.-H.C.F.M.; Aziz, H.; Woods, C.E.; Seman-Senderos, M.A.; Sparks, E.; Preuss, C.; Wünnemann, F.; Bedja, D.; Moats, C.R.; et al. ROBO4 variants predispose individuals to bicuspid aortic valve and thoracic aortic aneurysm. Nat. Genet. 2018, 51, 42–50. [Google Scholar] [CrossRef]
- Wang, Y.; Wu, B.; Chamberlain, A.A.; Lui, W.; Koirala, P.; Susztak, K.; Klein, D.; Taylor, V.; Zhou, B. Endocardial to Myocardial Notch-Wnt-Bmp Axis Regulates Early Heart Valve Development. PLoS ONE 2013, 8, e60244. [Google Scholar] [CrossRef] [Green Version]
- Koenig, S.N.; Bosse, K.; Majumdar, U.; Bonachea, E.M.; Radtke, F.; Garg, V. Endothelial Notch1 is Required for Proper Development of the Semilunar Valves and Cardiac Outflow Tract. J. Am. Hear. Assoc. 2016, 5. [Google Scholar] [CrossRef] [Green Version]
- Li, R.-G.; Xu, Y.; Wang, J.; Liu, X.-Y.; Yuan, F.; Huang, R.-T.; Xue, S.; Li, L.; Liu, H.; Li, Y.-J.; et al. GATA4 Loss-of-Function Mutation and the Congenitally Bicuspid Aortic Valve. Am. J. Cardiol. 2018, 121, 469–474. [Google Scholar] [CrossRef]
- Musfee, F.I.; Guo, D.; Pinard, A.C.; Hostetler, E.M.; Blue, E.E.; Nickerson, D.A.; Bamshad, M.J.; Milewicz, D.M.; Prakash, S.K. Rare deleterious variants of NOTCH1, GATA4, SMAD6, and ROBO4 are enriched in BAV with early onset complications but not in BAV with heritable thoracic aortic disease. Mol. Genet. Genom. Med. 2020, e1406. [Google Scholar] [CrossRef]
- Moskowitz, I.P.; Wang, J.; Peterson, M.A.; Pu, W.T.; MacKinnon, A.C.; Oxburgh, L.; Chu, G.C.; Sarkar, M.; Berul, C.; Smoot, L.; et al. Transcription factor genes Smad4 and Gata4 cooperatively regulate cardiac valve development. Proc. Natl. Acad. Sci. USA 2011, 108, 4006–4011. [Google Scholar] [CrossRef] [Green Version]
- Misra, C.; Chang, S.-W.; Basu, M.; Huang, N.; Garg, V. Disruption of myocardial Gata4 and Tbx5 results in defects in cardiomyocyte proliferation and atrioventricular septation. Hum. Mol. Genet. 2014, 23, 5025–5035. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Liu, J.; Xiang, M.; Olson, P.; Guzzetta, A.; Zhang, K.; Moskowitz, I.P.; Xie, L. Gata4 potentiates second heart field proliferation and Hedgehog signaling for cardiac septation. Proc. Natl. Acad. Sci. USA 2017, 114, E1422–E1431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.; Di, R.-M.; Qiao, Q.; Li, X.-M.; Huang, R.-T.; Xue, S.; Liu, X.-Y.; Wang, J.; Yang, Y.-Q. GATA6 loss-of-function mutation contributes to congenital bicuspid aortic valve. Gene 2018, 663, 115–120. [Google Scholar] [CrossRef] [PubMed]
- Montes, C.A.; Martín, M.; Arias, L.M.; Coto, E.; Naves-Díaz, M.; Moris, C.; Andía, J.B.C.; Rodríguez, I. Variants in cardiac GATA genes associated with bicuspid aortic valve. Eur. J. Clin. Investig. 2018, 48, e13027. [Google Scholar] [CrossRef]
Figure 1.
Anatomy of the arterial roots. (A) The arterial valve complex is made up of: three moving leaflets; the hinges, the attachment points of the leaflets to the wall; the commissures, the points of apposition of the leaflets close to the wall; the sinuses, pockets that form between the leaflets and the wall; and the interleaflet triangles, the regions of the wall that lie upstream (on the ventricular side) of the hinges, but are distal to the base of the sinuses that are found on the arterial side. Green = cardiomyocytes, blue = smooth muscle cells, yellow = fibrous tissue, purple = valve leaflet. The dotted line represents the position of the cross section through the valve complex. (B) Wholemount view of the adult mouse aortic root. The leaflets have been removed to allow the view of the other structures of the valve complex. (C,D) Masson’s trichrome images of the mouse aortic root at P21 in longitudinal and transverse planes. Red staining is muscle tissue, blue staining is fibrous tissue. c = commissure, h = hinge, ilt = interleaflet triangle, l = leaflet, s = sinus, sw = sinus wall.
Figure 1.
Anatomy of the arterial roots. (A) The arterial valve complex is made up of: three moving leaflets; the hinges, the attachment points of the leaflets to the wall; the commissures, the points of apposition of the leaflets close to the wall; the sinuses, pockets that form between the leaflets and the wall; and the interleaflet triangles, the regions of the wall that lie upstream (on the ventricular side) of the hinges, but are distal to the base of the sinuses that are found on the arterial side. Green = cardiomyocytes, blue = smooth muscle cells, yellow = fibrous tissue, purple = valve leaflet. The dotted line represents the position of the cross section through the valve complex. (B) Wholemount view of the adult mouse aortic root. The leaflets have been removed to allow the view of the other structures of the valve complex. (C,D) Masson’s trichrome images of the mouse aortic root at P21 in longitudinal and transverse planes. Red staining is muscle tissue, blue staining is fibrous tissue. c = commissure, h = hinge, ilt = interleaflet triangle, l = leaflet, s = sinus, sw = sinus wall.

Figure 2.
Early outflow tract development. (A) Model representing how flow of blood through the unseptated outflow tract may lead to formation of the spiralling outflow septum. Vorticial flow of blood through the outflow tract as the cushions are beginning to cellularise leads to aggregation of these cells (purple dots) in columns that prefigure the spiralling cushions (c). This positions and/or stabilises the cellularising cushions within the circumference of the outflow tract. The ensuing fusion of these cushions leads to formation of a spiralling septum (s) that places the root of the aorta to the left of the pulmonary trunk. (B) The boundary between myocardial and non-myocardial fate is first apparent at 10.5 in the mouse as the transition zone. The cells in this region are transitioning from a progenitor (grey) to a cardiomyocyte (green) fate. However, not all the undifferentiated SHF cells within this boundary region become cardiomyocytes. Two tongues of undifferentiated SHF cells can be seen to extend laterally into the myocardial outflow tract and these form the intercalated valve swellings (ICVS) that are the primordia of the posterior (non-coronary) and anterior intercalated leaflets of the aortic and pulmonary valves. (C,D) The myocardial-non-myocardial boundary is labelled by Isl1 (pink) and cTnI (green) at E10.5, with the cells in the transition zone labelled by both markers (arrow). By E11.5 the ICVS can clearly be seen in the outflow wall, as it continues to express Isl1 (arrows). By E15.5 the myocardial (green)-SMC (red) boundary at the level of the valve complex is well defined.
Figure 2.
Early outflow tract development. (A) Model representing how flow of blood through the unseptated outflow tract may lead to formation of the spiralling outflow septum. Vorticial flow of blood through the outflow tract as the cushions are beginning to cellularise leads to aggregation of these cells (purple dots) in columns that prefigure the spiralling cushions (c). This positions and/or stabilises the cellularising cushions within the circumference of the outflow tract. The ensuing fusion of these cushions leads to formation of a spiralling septum (s) that places the root of the aorta to the left of the pulmonary trunk. (B) The boundary between myocardial and non-myocardial fate is first apparent at 10.5 in the mouse as the transition zone. The cells in this region are transitioning from a progenitor (grey) to a cardiomyocyte (green) fate. However, not all the undifferentiated SHF cells within this boundary region become cardiomyocytes. Two tongues of undifferentiated SHF cells can be seen to extend laterally into the myocardial outflow tract and these form the intercalated valve swellings (ICVS) that are the primordia of the posterior (non-coronary) and anterior intercalated leaflets of the aortic and pulmonary valves. (C,D) The myocardial-non-myocardial boundary is labelled by Isl1 (pink) and cTnI (green) at E10.5, with the cells in the transition zone labelled by both markers (arrow). By E11.5 the ICVS can clearly be seen in the outflow wall, as it continues to express Isl1 (arrows). By E15.5 the myocardial (green)-SMC (red) boundary at the level of the valve complex is well defined.

Figure 3.
Lineage tracing of cells in the foetal and neonatal arterial valves. (A) At E12.5, Tie2-Cre labelled endocardial-derived cells, Wnt1-Cre-labelled NCC and Tnnt2-Cre labelled cells derived directly from the SHF, via the outflow wall, are found within the pulmonary valve leaflet primordia. Whilst Tie2-Cre and Wnt1-Cre-labelled cells are most abundant in the left (L) and right (R) leaflets, the Tnnt2-Cre labelled cells are the most abundant in the anterior (A) leaflet derived from the ICVS. Similar observations are made for the aortic valve (not shown). (B) At P2, each of the three lineages of cells are maintained within the leaflets of the aortic valve. As at E12.5, Wnt1-Cre and Tie2-Cre labelled cells are most abundant in the L and R leaflets, whilst Tnnt2-Cre labelled cells are most abundant in the posterior (P) leaflet derived from the ICVS. Similar observations are made for the pulmonary valve (not shown). (C) Cartoon illustrating how arterial valve leaflet formation is intimately linked to outflow tract septation. At E10.5, the forming endocardial cushions (buff coloured) are circumferential within the outflow vessel and the ICVS (grey) can first be seen within the outflow wall (green). By E11.5 the endocardial cushions (buff) have expanded and are coming together to fuse in the midline. Fusion of these cushions separates the outflow tract and gives rise to the L and R valve primordia. The ICVS give rise to the A and P valve primordia. By E13.5 the aortic and pulmonary valve are distinct, and the sinuses are beginning to appear. At E15.5, valve sculpting is almost complete, and the aorta and pulmonary valves are offset. Green = myocardium, yellow = SMC, red = endocardium.
Figure 3.
Lineage tracing of cells in the foetal and neonatal arterial valves. (A) At E12.5, Tie2-Cre labelled endocardial-derived cells, Wnt1-Cre-labelled NCC and Tnnt2-Cre labelled cells derived directly from the SHF, via the outflow wall, are found within the pulmonary valve leaflet primordia. Whilst Tie2-Cre and Wnt1-Cre-labelled cells are most abundant in the left (L) and right (R) leaflets, the Tnnt2-Cre labelled cells are the most abundant in the anterior (A) leaflet derived from the ICVS. Similar observations are made for the aortic valve (not shown). (B) At P2, each of the three lineages of cells are maintained within the leaflets of the aortic valve. As at E12.5, Wnt1-Cre and Tie2-Cre labelled cells are most abundant in the L and R leaflets, whilst Tnnt2-Cre labelled cells are most abundant in the posterior (P) leaflet derived from the ICVS. Similar observations are made for the pulmonary valve (not shown). (C) Cartoon illustrating how arterial valve leaflet formation is intimately linked to outflow tract septation. At E10.5, the forming endocardial cushions (buff coloured) are circumferential within the outflow vessel and the ICVS (grey) can first be seen within the outflow wall (green). By E11.5 the endocardial cushions (buff) have expanded and are coming together to fuse in the midline. Fusion of these cushions separates the outflow tract and gives rise to the L and R valve primordia. The ICVS give rise to the A and P valve primordia. By E13.5 the aortic and pulmonary valve are distinct, and the sinuses are beginning to appear. At E15.5, valve sculpting is almost complete, and the aorta and pulmonary valves are offset. Green = myocardium, yellow = SMC, red = endocardium.
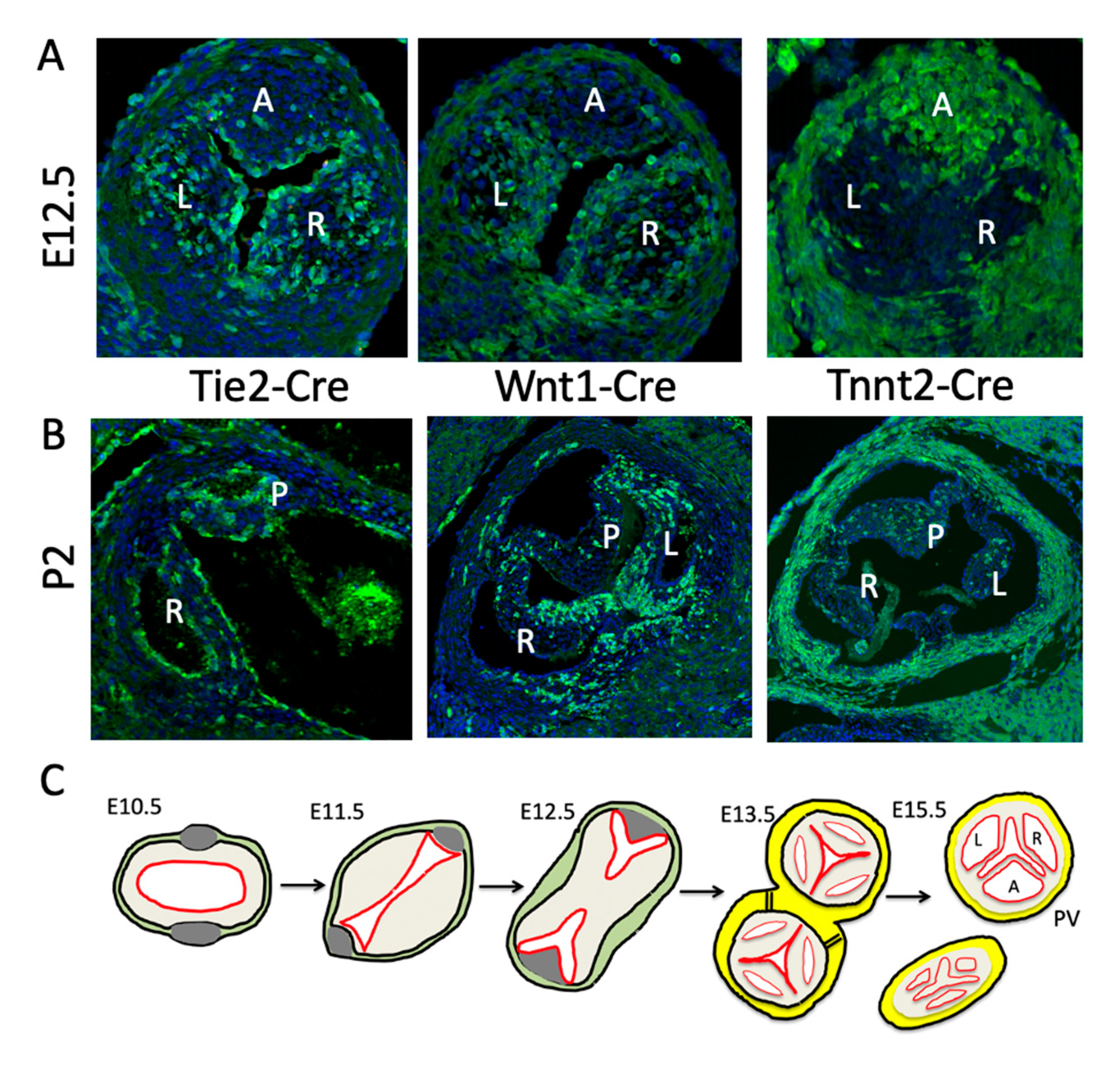
Figure 4.
Contributions of the cushions and ICVS to the arterial valve primordia. Histological sections of mouse hearts stained with H&E. (A) At E10.5 the outflow is unseptated. The cushions (arrows) are forming and can be seen to extend along the length of the outflow tract in transverse view and to be circumferential in both the distal and proximal regions, in frontal view. The ICVS cannot be seen in transverse sections at this stage, but in frontal sections can be seen distally as cell dense regions within the outflow wall (arrowheads). (B) By E11.5, the cushions are more cell dense and in transverse sections the valve forming part (red bar) can be seen to be distinct from the proximal part of the cushions that will form the sub-pulmonary infundibulum in transverse sections. The ICVS (arrowhead) can now be seen adjacent to the valve-forming parts of the cushions. In frontal sections the main cushions (arrows) are coming together towards the midline of the outflow tract in the distal region. The ICVS (arrowheads) have expanded but are still continuous with the outflow wall. More proximally, the cushions (arrows) are still widely separated. (C) By E12.5 the outflow tract has septated and the primordia of all three leaflets are apparent in transverse and frontal views. The proximal region of the outflow tract remains unseptated at this time point, although the cushions (arrows) are beginning to come together. A = anterior leaflet; L=left leaflet; P = posterior leaflet; R = right leaflet.
Figure 4.
Contributions of the cushions and ICVS to the arterial valve primordia. Histological sections of mouse hearts stained with H&E. (A) At E10.5 the outflow is unseptated. The cushions (arrows) are forming and can be seen to extend along the length of the outflow tract in transverse view and to be circumferential in both the distal and proximal regions, in frontal view. The ICVS cannot be seen in transverse sections at this stage, but in frontal sections can be seen distally as cell dense regions within the outflow wall (arrowheads). (B) By E11.5, the cushions are more cell dense and in transverse sections the valve forming part (red bar) can be seen to be distinct from the proximal part of the cushions that will form the sub-pulmonary infundibulum in transverse sections. The ICVS (arrowhead) can now be seen adjacent to the valve-forming parts of the cushions. In frontal sections the main cushions (arrows) are coming together towards the midline of the outflow tract in the distal region. The ICVS (arrowheads) have expanded but are still continuous with the outflow wall. More proximally, the cushions (arrows) are still widely separated. (C) By E12.5 the outflow tract has septated and the primordia of all three leaflets are apparent in transverse and frontal views. The proximal region of the outflow tract remains unseptated at this time point, although the cushions (arrows) are beginning to come together. A = anterior leaflet; L=left leaflet; P = posterior leaflet; R = right leaflet.
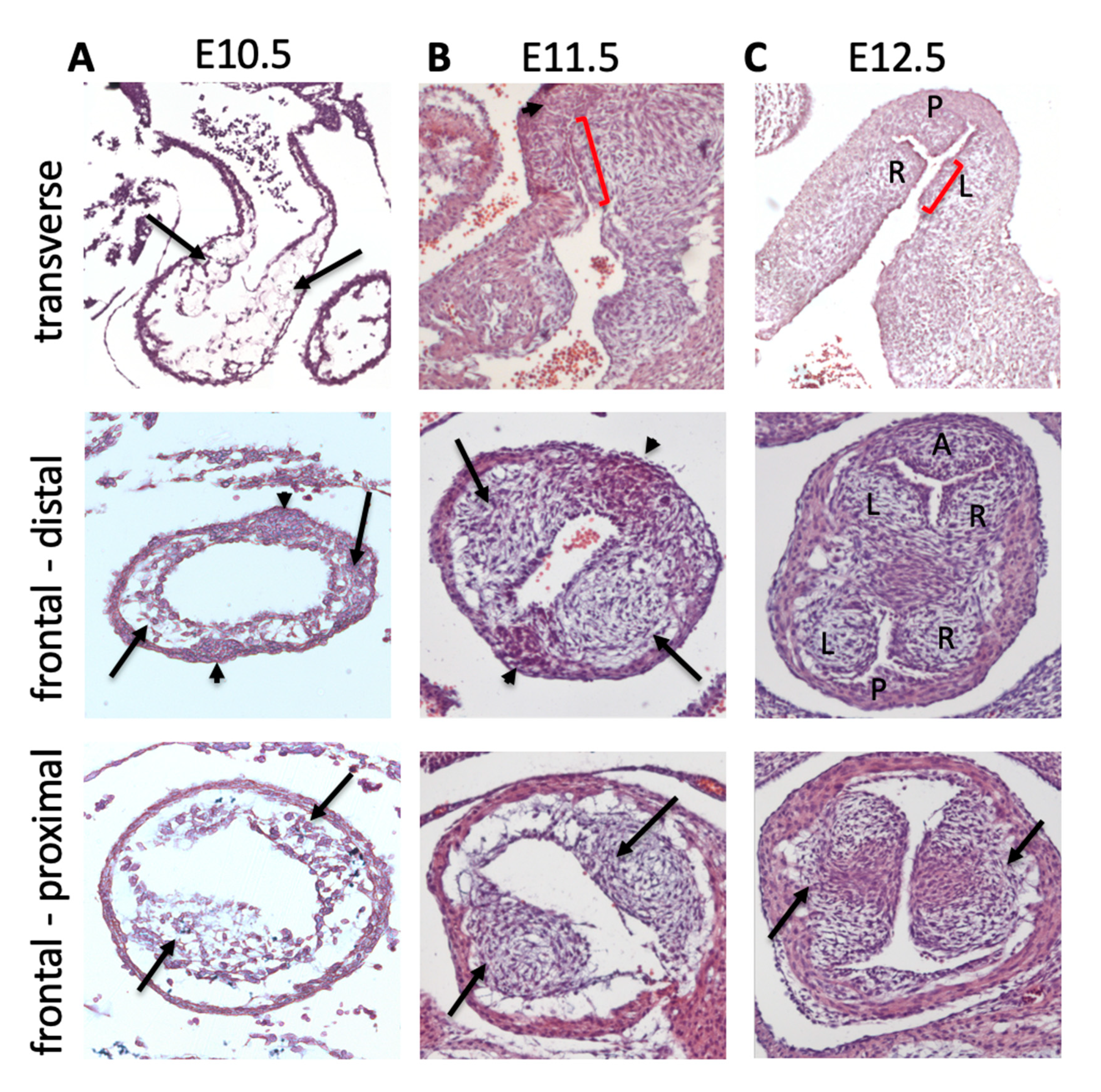
Figure 5.
Models of valve sculpting. (A) Histological images showing how the leaflet sculpts from a bulky cushion at E11.5, to a well-defined leaflet by E14.5. (B) The leaflet elongation model proposes that under the influence of factors secreted by the thickened valve endocardium, the leaflet actively elongates to acquire its sculpted form. (C) In the second model a process of cell death removes cushion tissue at the boundary with the outflow wall and in this way excavates the sinus, leading to freeing of the leaflet. (D) In the final model, the endocardium bends into the cushion tissue and in so doing creates the sinus, again freeing the leaflet. Experimental evidence is minimal for each of the three models and further work is needed to determine which, if any, are accurate.
Figure 5.
Models of valve sculpting. (A) Histological images showing how the leaflet sculpts from a bulky cushion at E11.5, to a well-defined leaflet by E14.5. (B) The leaflet elongation model proposes that under the influence of factors secreted by the thickened valve endocardium, the leaflet actively elongates to acquire its sculpted form. (C) In the second model a process of cell death removes cushion tissue at the boundary with the outflow wall and in this way excavates the sinus, leading to freeing of the leaflet. (D) In the final model, the endocardium bends into the cushion tissue and in so doing creates the sinus, again freeing the leaflet. Experimental evidence is minimal for each of the three models and further work is needed to determine which, if any, are accurate.

Figure 6.
Possible mechanisms underlying THE bicuspid aortic valve BAV. (A) Cartoon illustrating several possible mechanisms underlying BAV in the situation of outflow tract septation. These can result in different numbers of sinuses and raphes and can occur early in development (as shown) or later in development as a consequence of pathological processes. Valvar stenosis could also be caused by over fusion of the cushions, at the commissures. (B) Cartoon illustrating possible valve outcomes when outflow septation fails. In the case of the proposed situation where the SHF is deficient, the numbers of leaflets can be highly variable, possibly depending on the extent of deficiency.
Figure 6.
Possible mechanisms underlying THE bicuspid aortic valve BAV. (A) Cartoon illustrating several possible mechanisms underlying BAV in the situation of outflow tract septation. These can result in different numbers of sinuses and raphes and can occur early in development (as shown) or later in development as a consequence of pathological processes. Valvar stenosis could also be caused by over fusion of the cushions, at the commissures. (B) Cartoon illustrating possible valve outcomes when outflow septation fails. In the case of the proposed situation where the SHF is deficient, the numbers of leaflets can be highly variable, possibly depending on the extent of deficiency.

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Henderson, D.J.; Eley, L.; Chaudhry, B. New Concepts in the Development and Malformation of the Arterial Valves. J. Cardiovasc. Dev. Dis. 2020, 7, 38. https://doi.org/10.3390/jcdd7040038
AMA Style
Henderson DJ, Eley L, Chaudhry B. New Concepts in the Development and Malformation of the Arterial Valves. Journal of Cardiovascular Development and Disease. 2020; 7(4):38. https://doi.org/10.3390/jcdd7040038
Chicago/Turabian StyleHenderson, Deborah J., Lorraine Eley, and Bill Chaudhry. 2020. "New Concepts in the Development and Malformation of the Arterial Valves" Journal of Cardiovascular Development and Disease 7, no. 4: 38. https://doi.org/10.3390/jcdd7040038
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.