
Preliminary Evidence for Aortopathy and an X-Linked Parent-of-Origin Effect on Aortic Valve Malformation in a Mouse Model of Turner Syndrome

Abstract
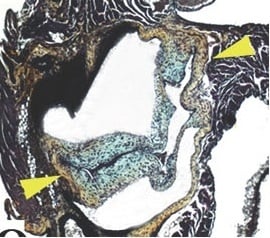
:
1. Introduction
2. Methods and Materials
3. Results and Discussion
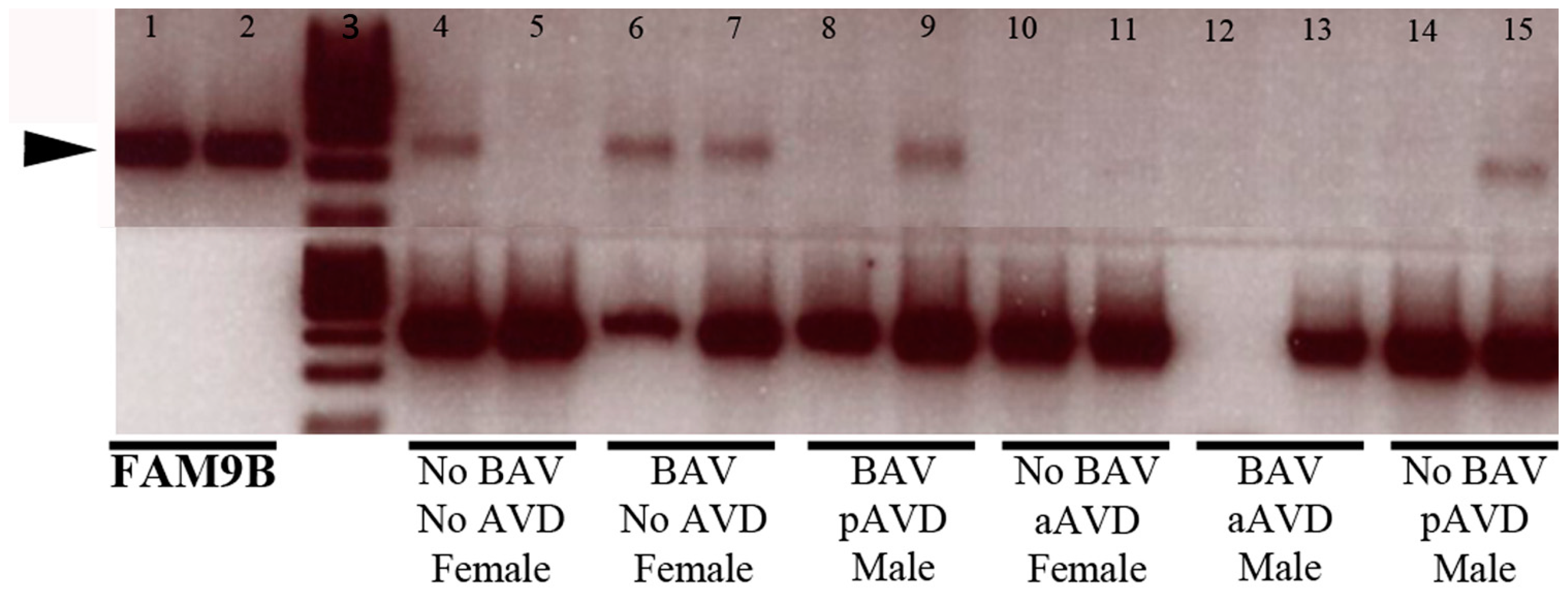
3.1. Aortic Valve
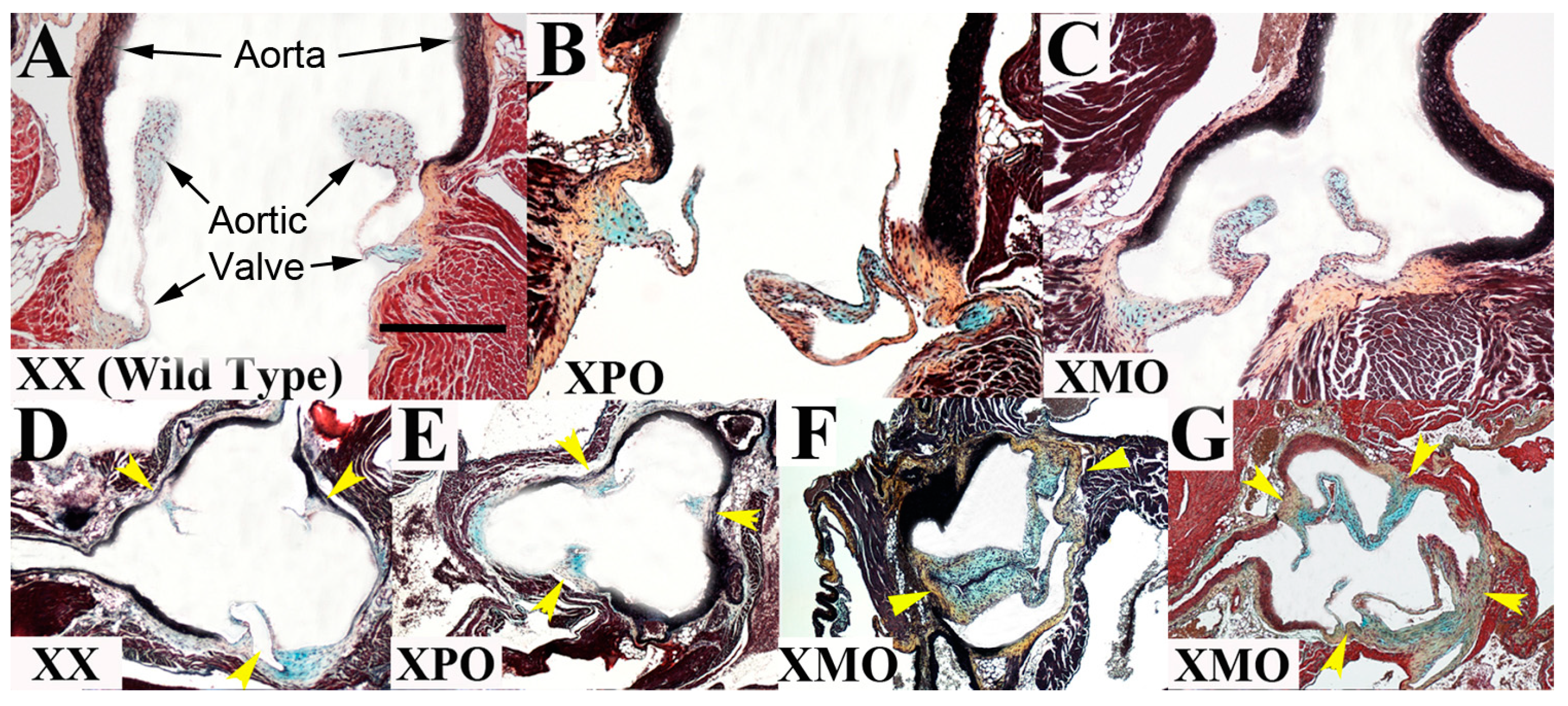

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Valve Annulus Area (μm2) | Valve Hinge Thickness (μm) | Valve Cusp Length (μm) | Aortic Root Thickness (μm) | Ascending Aorta Thickness (μm) | |
|---|---|---|---|---|---|
| XX ( n = 8) | 5563 ± 1628 | 47 ± 14 | 279 ± 57 | 58 ± 8 | 111 ± 16 |
| XPO (n = 6) | 4738 ± 1639 | 50 ± 6 | 325 ± 101 | 63 ± 4 | 103 ± 5 |
| XMO (n = 12) | 7180 ± 1328 | 59 ± 19 | 318 ± 112 | 57 ± 7 | 104 ± 14 |
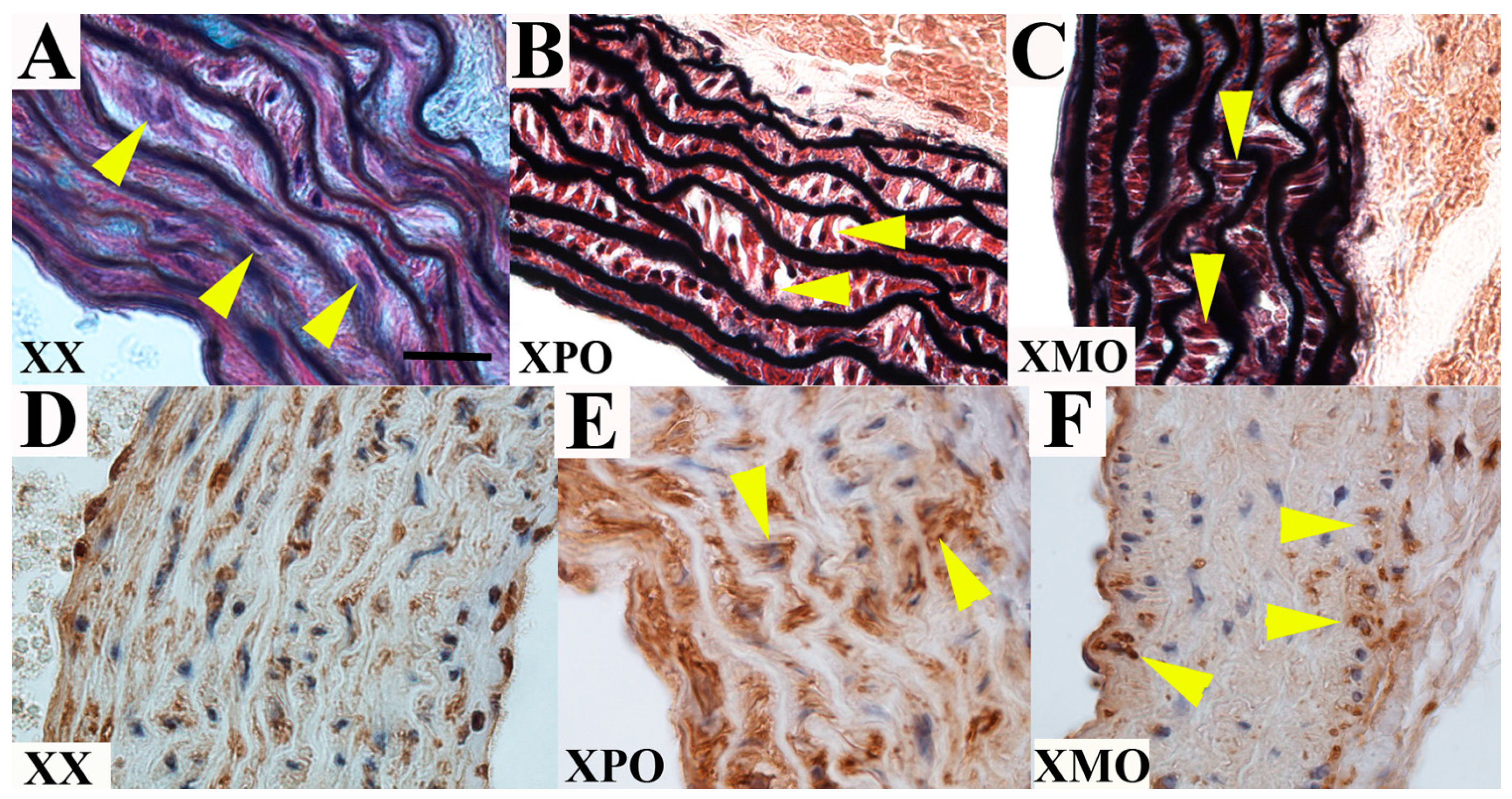
3.2. Ascending Aorta
| Valve Annulus Dimension (μm) | Aortic Root Dimension (μm) | Sinotubular Junction Dimension (μm) | Ascending Aorta Dimension (μm) | |
|---|---|---|---|---|
| XX (n = 5) | 486 ± 40 | 735 ± 52 | 502 ± 54 | 517 ± 41 |
| XPO (n = 5) | 500 ± 36 | 778 ± 65 | 531 ± 62 | 559 ± 25 |
| (0/5, 0%) | (1/5, 20%) | (1/5, 20%) | (1/5, 20%) | |
| XMO (n = 9) | 537 ± 50 | 826 ± 63 | 540 ± 59 | 566 ± 44 |
| (3/9, 33%) | (5/9, 56%) | (3/9, 33%) | (2/7, 29%) |
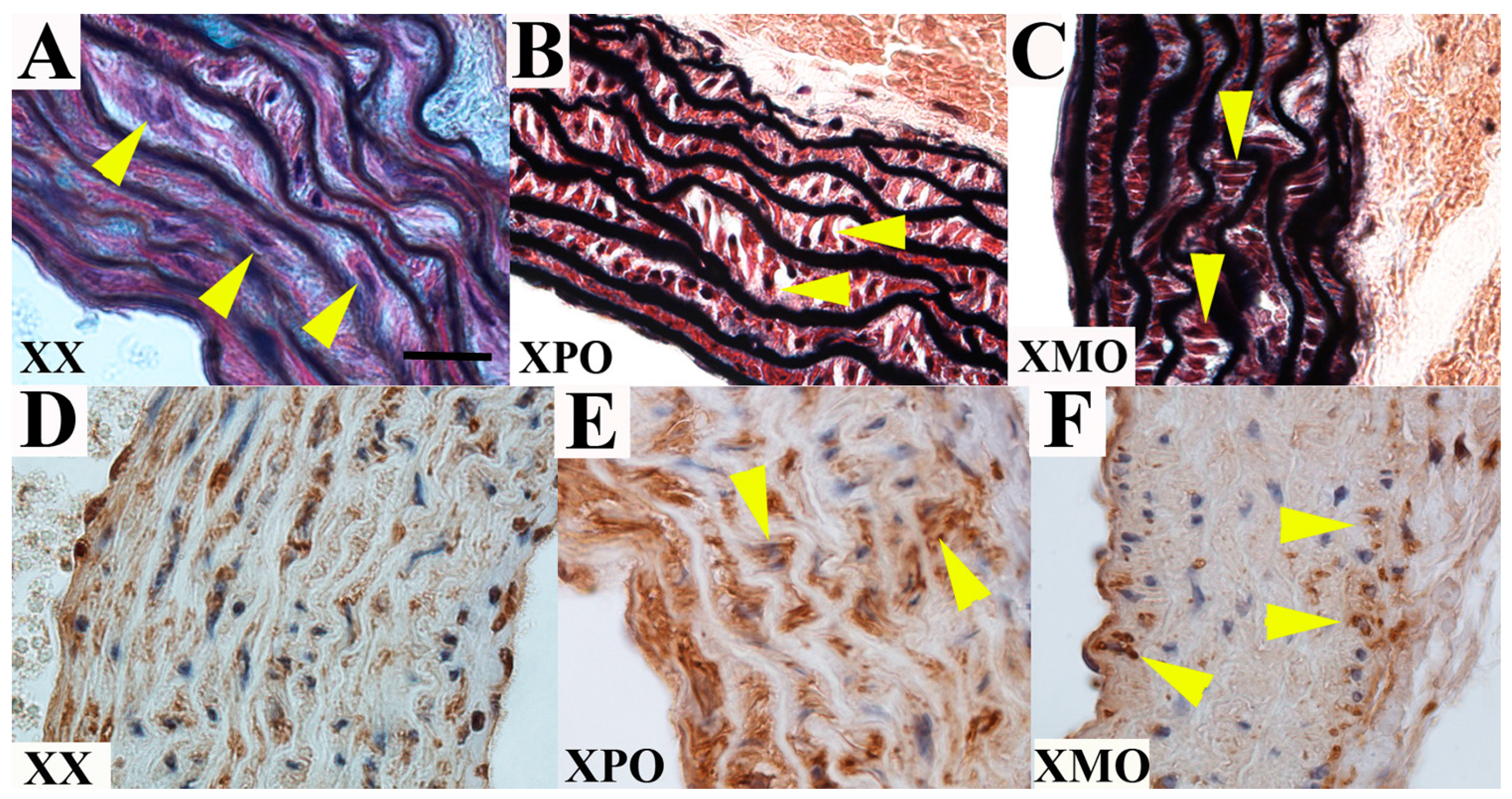
3.3. Discussion
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Sybert, V.P.; McCauley, E. Turner’s syndrome. N. Engl. J. Med. 2004, 351, 1227–1238. [Google Scholar] [CrossRef] [PubMed]
- Sachdev, V.; Matura, L.A.; Sidenko, S.; Ho, V.B.; Arai, A.E.; Rosing, D.R.; Bondy, C.A. Aortic valve disease in Turner syndrome. J. Am. Coll. Cardiol. 2008, 51, 1904–1909. [Google Scholar] [CrossRef] [PubMed]
- Sybert, V.P. Cardiovascular malformations and complications in Turner syndrome. Pediatrics 1998, 101, E11. [Google Scholar] [CrossRef] [PubMed]
- Lin, A.E.; Lippe, B.; Rosenfeld, R.G. Further delineation of aortic dilation, dissection, and rupture in patients with Turner syndrome. Pediatrics 1998, 102, e12. [Google Scholar] [CrossRef] [PubMed]
- Carrel, L.; Willard, H.F. X-inactivation profile reveals extensive variability in X-linked gene expression in females. Nature 2005, 434, 400–404. [Google Scholar] [CrossRef] [PubMed]
- Chu, C.E.; Donaldson, M.D.; Kelnar, C.J.; Smail, P.J.; Greene, S.A.; Paterson, W.F.; Connor, J.M. Possible role of imprinting in the Turner phenotype. J. Med. Genet. 1994, 31, 840–842. [Google Scholar] [CrossRef] [PubMed]
- Lepage, J.F.; Hong, D.S.; Mazaika, P.K.; Raman, M.; Sheau, K.; Marzelli, M.J.; Hallmayer, J.; Reiss, A.L. Genomic imprinting effects of the X chromosome on brain morphology. J. Neurosci. 2013, 33, 8567–8574. [Google Scholar] [CrossRef] [PubMed]
- Sagi, L.; Zuckerman-Levin, N.; Gawlik, A.; Ghizzoni, L.; Buyukgebiz, A.; Rakover, Y.; Bistritzer, T.; Admoni, O.; Vottero, A.; Baruch, O.; et al. Clinical significance of the parental origin of the X chromosome in turner syndrome. J. Clin. Endocrinol. Metab. 2007, 92, 846–852. [Google Scholar] [CrossRef] [PubMed]
- Skuse, D.H.; James, R.S.; Bishop, D.V.; Coppin, B.; Dalton, P.; Aamodt-Leeper, G.; Bacarese-Hamilton, M.; Creswell, C.; McGurk, R.; Jacobs, P.A. Evidence from Turner’s syndrome of an imprinted X-linked locus affecting cognitive function. Nature 1997, 387, 705–708. [Google Scholar] [CrossRef] [PubMed]
- Abd-Elmoniem, K.Z.; Bakalov, V.K.; Matta, J.R.; Muldoon, N.; Hanover, J.A.; Bondy, C.A.; Gharib, A.M. X Chromosome Parental Origin and Aortic Stiffness in Turner Syndrome. Clin. Endocrinol. 2014, 81, 467–470. [Google Scholar] [CrossRef] [PubMed]
- Davies, M.J.; Treasure, T.; Richardson, P.D. The pathogenesis of spontaneous arterial dissection. Heart 1996, 75, 434–435. [Google Scholar] [CrossRef] [PubMed]
- Bondy, C.A.; Matura, L.A.; Wooten, N.; Troendle, J.; Zinn, A.R.; Bakalov, V.K. The physical phenotype of girls and women with Turner syndrome is not X-imprinted. Hum. Genet. 2007, 121, 469–474. [Google Scholar] [CrossRef] [PubMed]
- Ko, J.M.; Kim, J.M.; Kim, G.H.; Lee, B.H.; Yoo, H.W. Influence of parental origin of the X chromosome on physical phenotypes and GH responsiveness of patients with Turner syndrome. Clin. Endocrinol. 2010, 73, 66–71. [Google Scholar]
- Davies, W.; Isles, A.R.; Burgoyne, P.S.; Wilkinson, L.S. X-linked imprinting: Effects on brain and behaviour. Bioessays 2006, 28, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Henn, W.; Zang, K.D. Mosaicism in Turner’s syndrome. Nature 1997, 390, 569. [Google Scholar] [CrossRef] [PubMed]
- Davies, W.; Isles, A.; Smith, R.; Karunadasa, D.; Burrmann, D.; Humby, T.; Ojarikre, O.; Biggin, C.; Skuse, D.; Burgoyne, P.; et al. Xlr3b is a new imprinted candidate for X-linked parent-of-origin effects on cognitive function in mice. Nat. Genet. 2005, 37, 625–629. [Google Scholar] [CrossRef] [PubMed]
- Lynn, P.M.; Davies, W. The 39,XO mouse as a model for the neurobiology of Turner syndrome and sex-biased neuropsychiatric disorders. Behav. Brain Res. 2007, 179, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Hinton, R.B. Bicuspid aortic valve and thoracic aortic aneurysm: Three patient populations, two disease phenotypes, and one shared genotype. Cardiol. Res. Pract. 2012, 2012, 926975. [Google Scholar] [CrossRef] [PubMed]
- Hinton, R.B.; Alfieri, C.M.; Witt, S.A.; Glascock, B.J.; Khoury, P.R.; Benson, D.W.; Yutzey, K.E. Mouse heart valve structure and function: Echocardiographic and morphometric analyses from the fetus through the aged adult. Am. J. Physiol. Heart Circ. Physiol. 2008, 294, H2480–H2488. [Google Scholar] [CrossRef] [PubMed]
- Matura, L.A.; Ho, V.B.; Rosing, D.R.; Bondy, C.A. Aortic dilatation and dissection in Turner syndrome. Circulation 2007, 116, 1663–1670. [Google Scholar] [CrossRef] [PubMed]
- Carlson, M.; Airhart, N.; Lopez, L.; Silberbach, M. Moderate aortic enlargement and bicuspid aortic valve are associated with aortic dissection in Turner syndrome: Report of the international turner syndrome aortic dissection registry. Circulation 2012, 126, 2220–2226. [Google Scholar] [CrossRef] [PubMed]
- Hinton, R.B.; Lincoln, J.; Deutsch, G.H.; Osinska, H.; Manning, P.B.; Benson, D.W.; Yutzey, K.E. Extracellular matrix remodeling and organization in developing and diseased aortic valves. Circ. Res. 2006, 98, 1431–1438. [Google Scholar] [CrossRef] [PubMed]
- Bunton, T.E.; Biery, N.J.; Myers, L.; Gayraud, B.; Ramirez, F.; Dietz, H.C. Phenotypic alteration of vascular smooth muscle cells precedes elastolysis in a mouse model of Marfan syndrome. Circ. Res. 2001, 88, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Olivieri, L.J.; Baba, R.Y.; Arai, A.E.; Bandettini, W.P.; Rosing, D.R.; Bakalov, V.; Sachdev, V.; Bondy, C.A. Spectrum of aortic valve abnormalities associated with aortic dilation across age groups in Turner syndrome. Circ. Cardiovasc. Imaging 2013, 6, 1018–1023. [Google Scholar] [CrossRef] [PubMed]
- Ward, C. Clinical significance of the bicuspid aortic valve. Heart 2000, 83, 81–85. [Google Scholar] [CrossRef] [PubMed]
- Lacro, R.V.; Jones, K.L.; Benirschke, K. Coarctation of the aorta in Turner syndrome: A pathologic study of fetuses with nuchal cystic hygromas, hydrops fetalis, and female genitalia. Pediatrics 1988, 81, 445–451. [Google Scholar] [PubMed]
- Iyer, N.P.; Tucker, D.F.; Roberts, S.H.; Moselhi, M.; Morgan, M.; Matthes, J.W. Outcome of fetuses with Turner syndrome: A 10-year congenital anomaly register based study. J. Matern. Fetal Neonatal Med. 2012, 25, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Davies, W. Genomic imprinting on the X chromosome: Implications for brain and behavioral phenotypes. Ann. N.Y. Acad. Sci. 2010, 1204, E14–E19. [Google Scholar] [CrossRef] [PubMed]
- Bondy, C.A.; Bakalov, V.K.; Cheng, C.; Olivieri, L.; Rosing, D.R.; Arai, A.E. Bicuspid aortic valve and aortic coarctation are linked to deletion of the X chromosome short arm in Turner syndrome. J. Med. Genet. 2013, 50, 662–665. [Google Scholar] [CrossRef] [PubMed][Green Version]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hinton, R.B.; Opoka, A.M.; Ojarikre, O.A.; Wilkinson, L.S.; Davies, W. Preliminary Evidence for Aortopathy and an X-Linked Parent-of-Origin Effect on Aortic Valve Malformation in a Mouse Model of Turner Syndrome. J. Cardiovasc. Dev. Dis. 2015, 2, 190-199. https://doi.org/10.3390/jcdd2030190
Hinton RB, Opoka AM, Ojarikre OA, Wilkinson LS, Davies W. Preliminary Evidence for Aortopathy and an X-Linked Parent-of-Origin Effect on Aortic Valve Malformation in a Mouse Model of Turner Syndrome. Journal of Cardiovascular Development and Disease. 2015; 2(3):190-199. https://doi.org/10.3390/jcdd2030190
Chicago/Turabian StyleHinton, Robert B., Amy M. Opoka, Obah A. Ojarikre, Lawrence S. Wilkinson, and William Davies. 2015. "Preliminary Evidence for Aortopathy and an X-Linked Parent-of-Origin Effect on Aortic Valve Malformation in a Mouse Model of Turner Syndrome" Journal of Cardiovascular Development and Disease 2, no. 3: 190-199. https://doi.org/10.3390/jcdd2030190
APA StyleHinton, R. B., Opoka, A. M., Ojarikre, O. A., Wilkinson, L. S., & Davies, W. (2015). Preliminary Evidence for Aortopathy and an X-Linked Parent-of-Origin Effect on Aortic Valve Malformation in a Mouse Model of Turner Syndrome. Journal of Cardiovascular Development and Disease, 2(3), 190-199. https://doi.org/10.3390/jcdd2030190