

Identification of Rare Genetic Variants in Familial Spontaneous Coronary Artery Dissection and Evidence for Shared Biological Pathways
 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
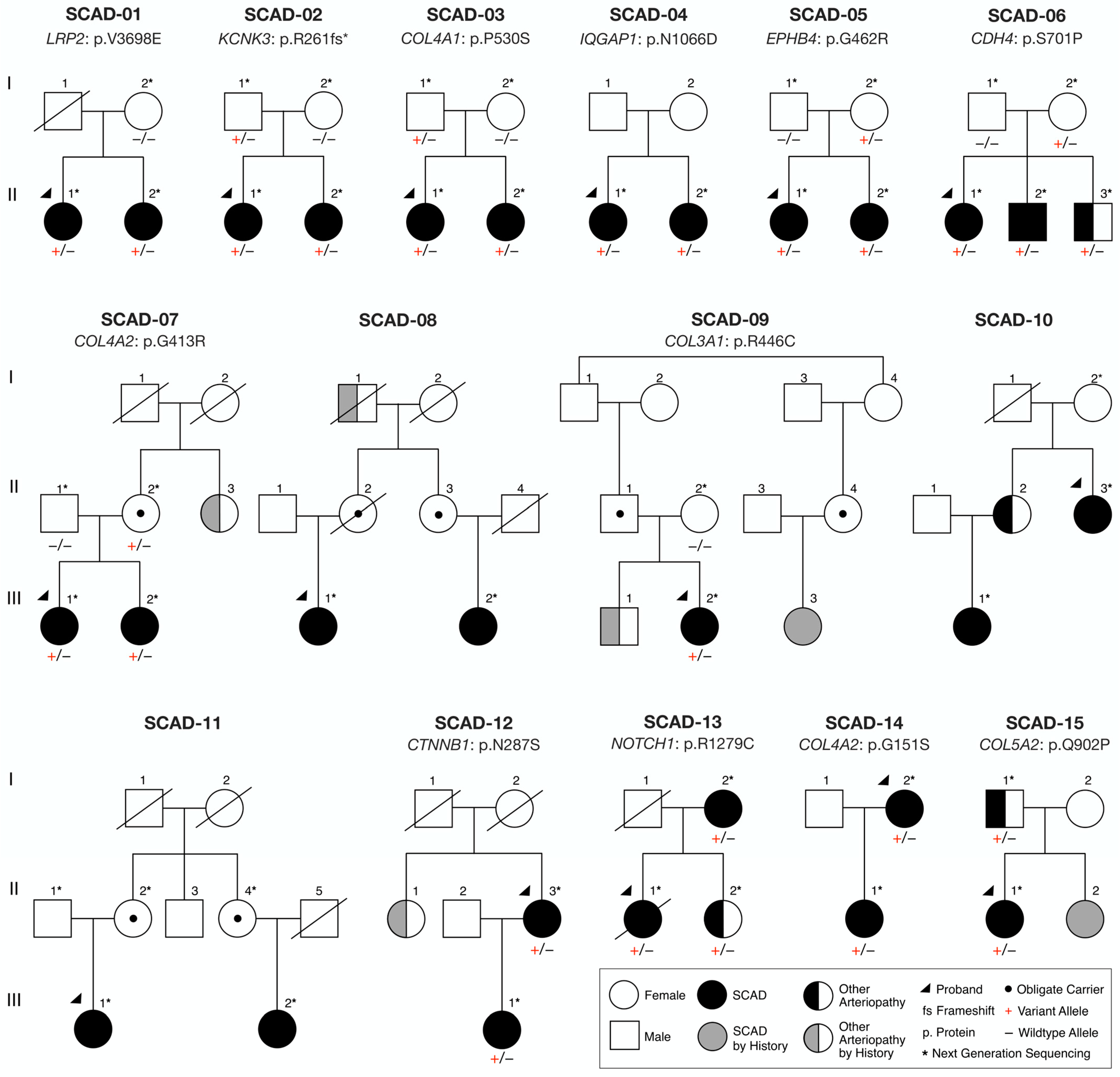
2.1. Study Subjects
2.2. Next-Generation Sequencing and Bioinformatics Analysis
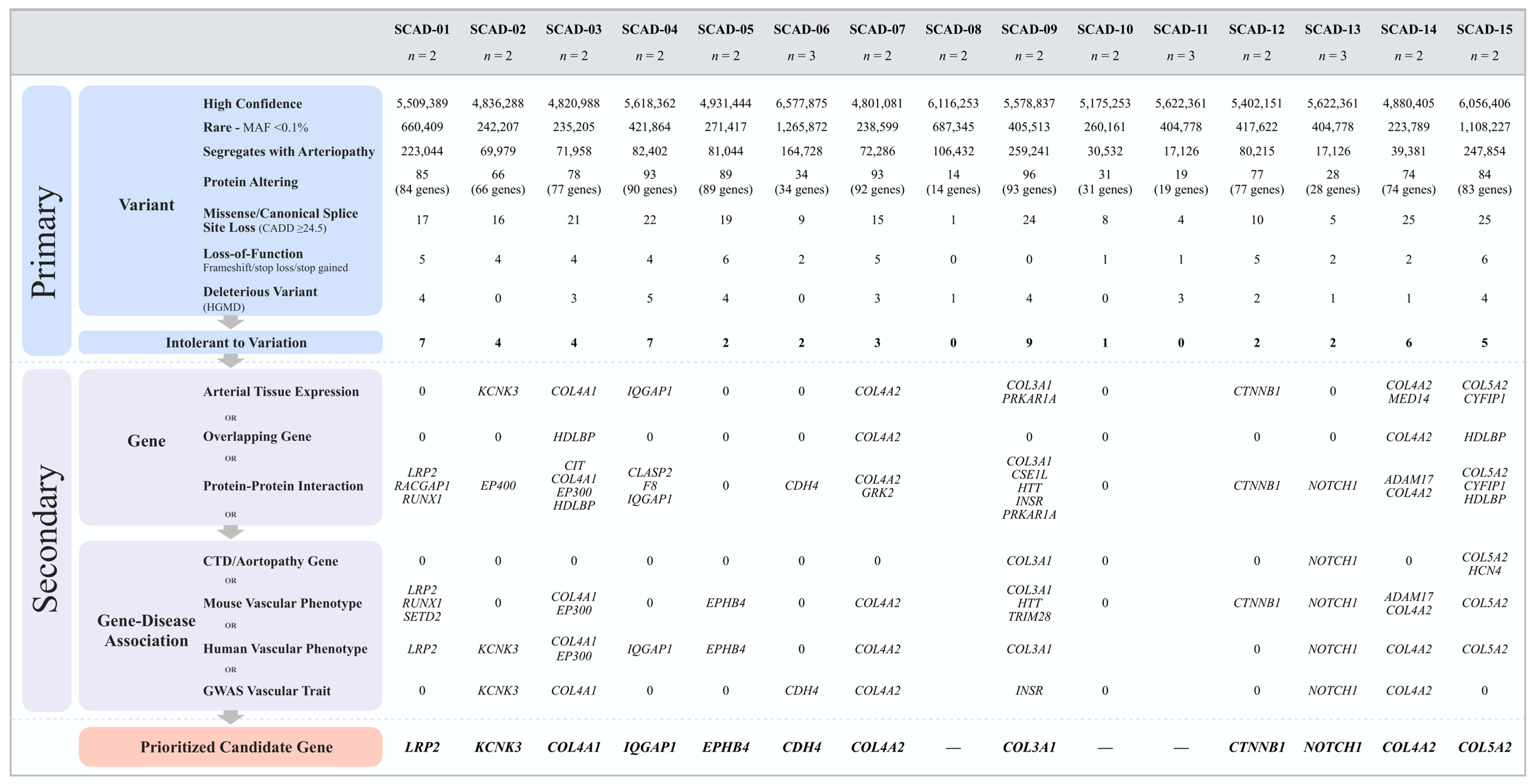
2.3. Genetic Variant Filtering and Prioritization
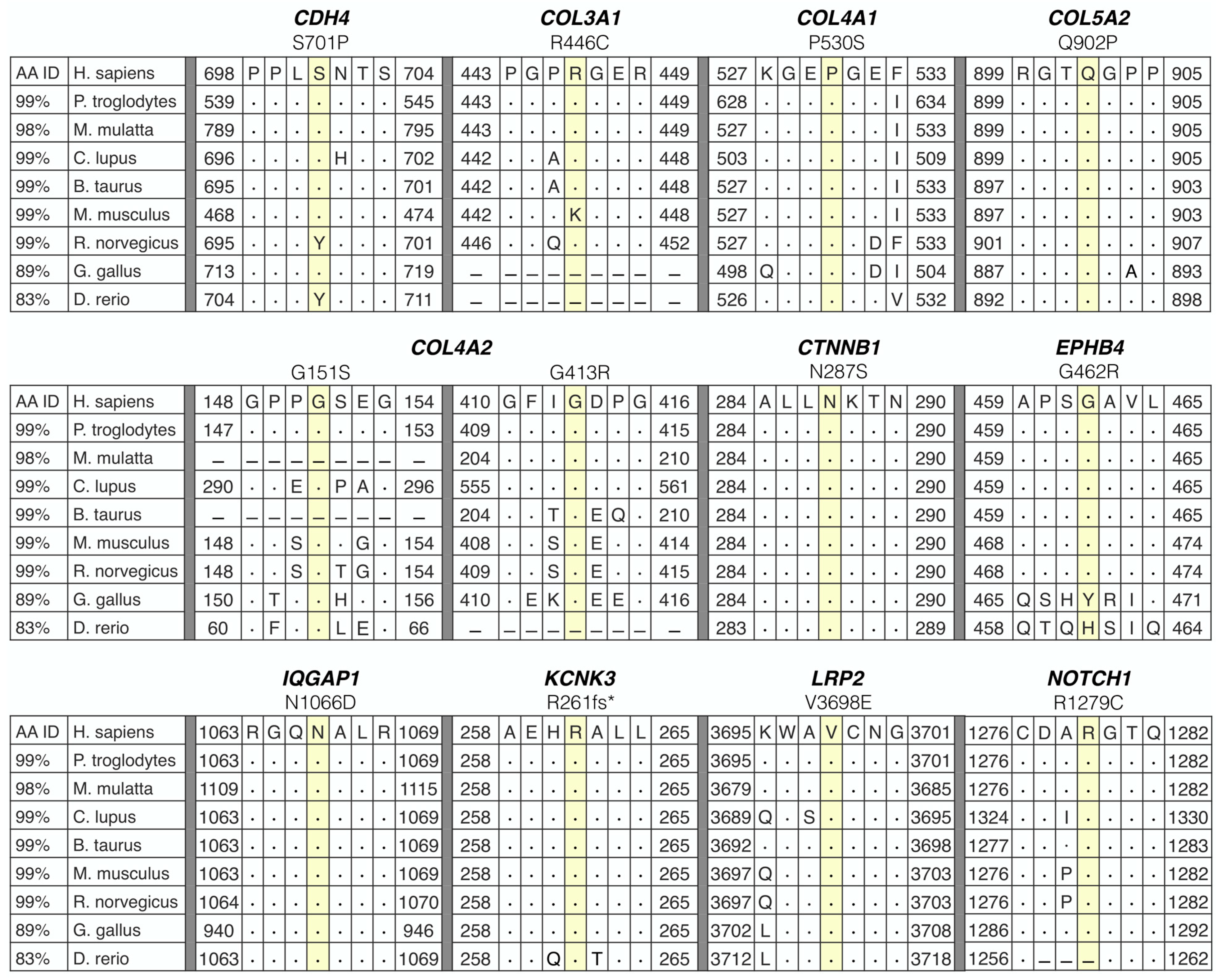
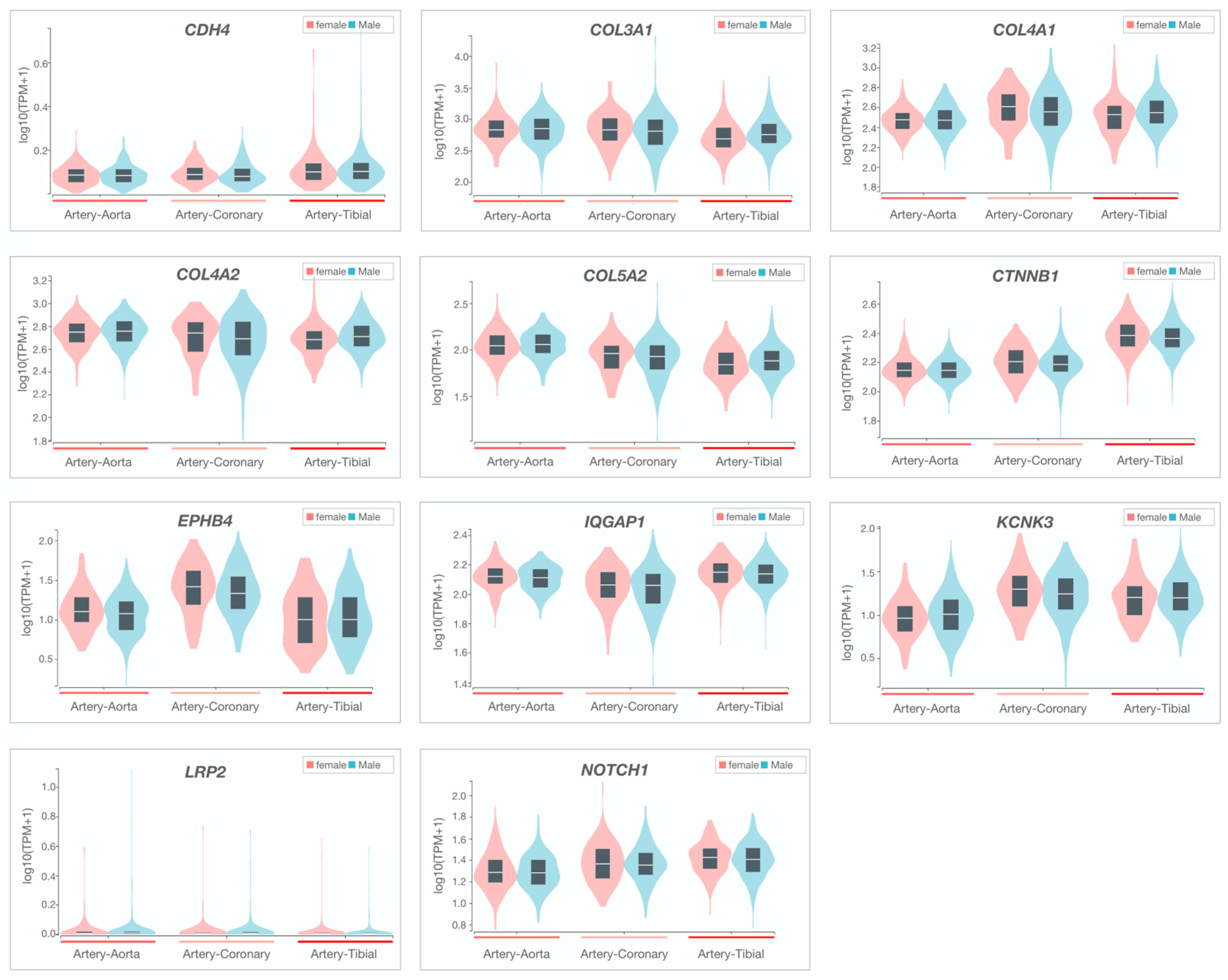
2.4. Candidate Gene Prioritization
2.5. In Silico Analysis of Shared Biological Networks and Processes
3. Results
3.1. Clinical Characteristics
3.2. Whole-Genome Sequencing Reveals Disease-Associated Candidate Genes in Familial SCAD
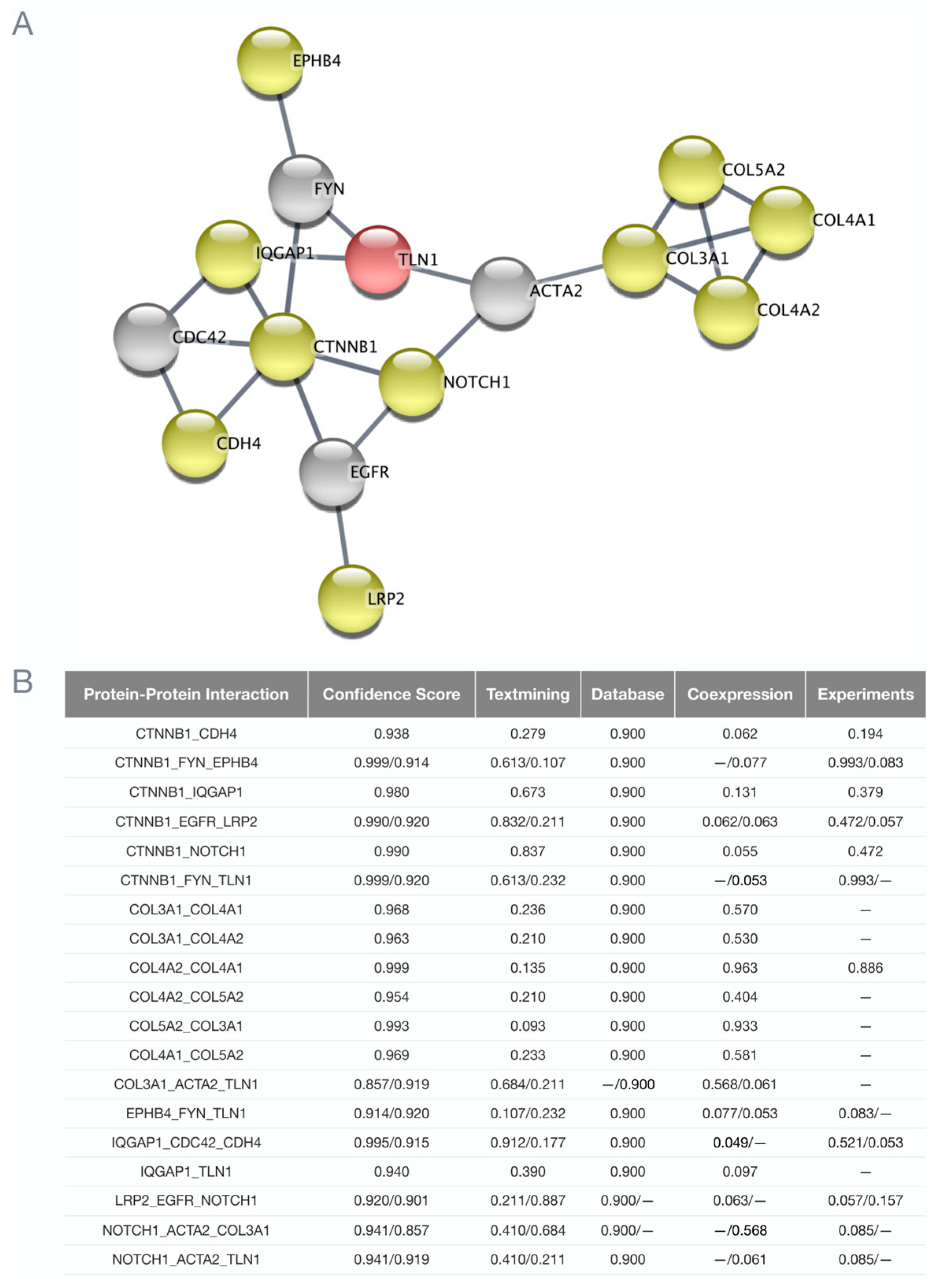
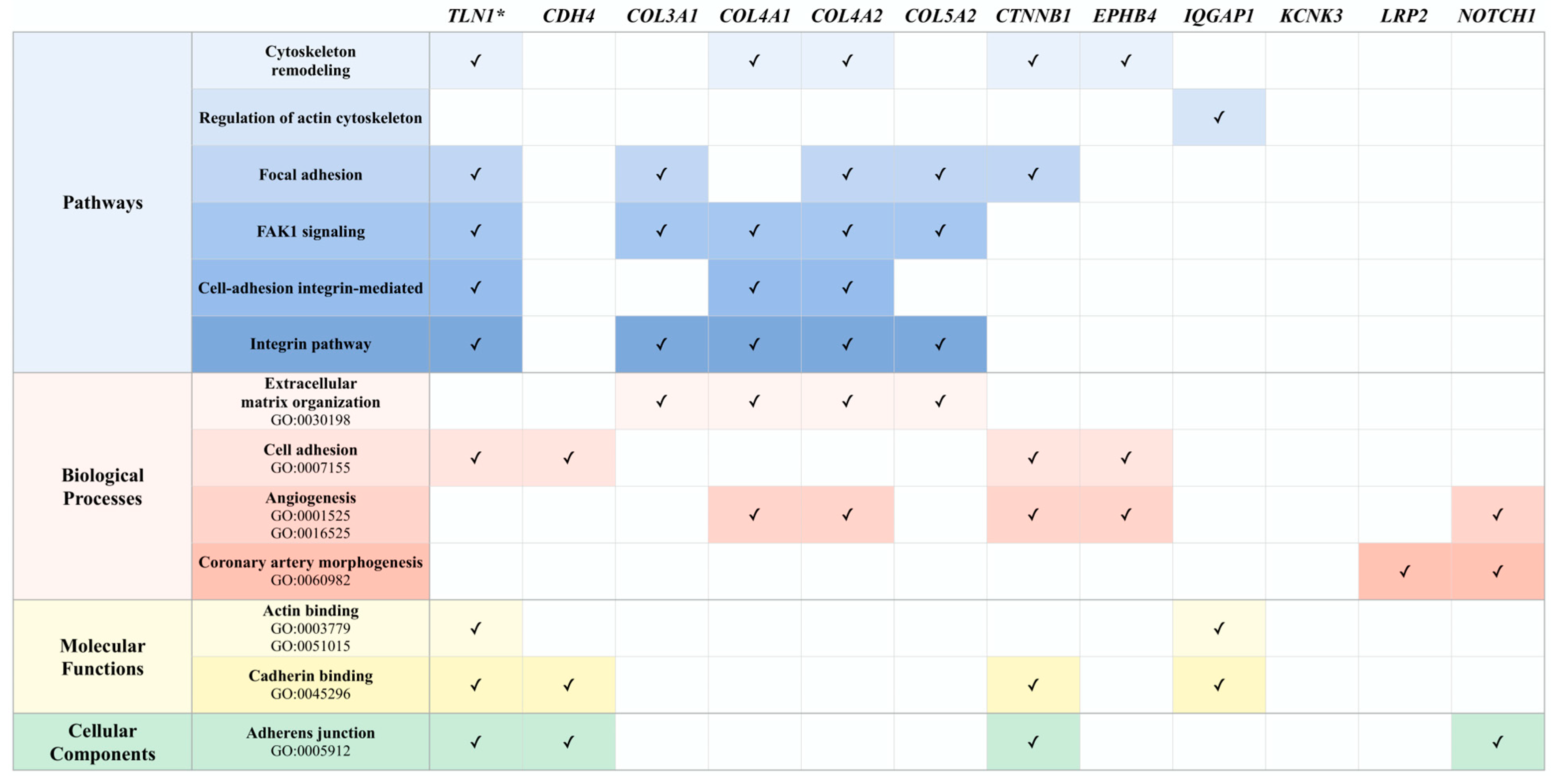
3.3. Potential Associations among Familial SCAD Candidate Genes
3.4. Ancillary Investigation of Non-Coding Variants
4. Discussion
4.1. Cell–Cell Adhesion
4.2. Cell–Extracellular Matrix Adhesion
4.3. Adhesion Signaling
4.4. Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Hayes, S.N.; Tweet, M.S.; Adlam, D.; Kim, E.S.H.; Gulati, R.; Price, J.E.; Rose, C.H. Spontaneous coronary artery dissection: Jacc state-of-the-art review. J. Am. Coll. Cardiol. 2020, 76, 961–984. [Google Scholar] [CrossRef] [PubMed]
- Adlam, D.; Olson, T.M.; Combaret, N.; Kovacic, J.C.; Iismaa, S.E.; Al-Hussaini, A.; O’Byrne, M.M.; Bouajila, S.; Georges, A.; Mishra, K.; et al. Association of the PHACTR1/EDN1 genetic locus with spontaneous coronary artery dissection. J. Am. Coll. Cardiol. 2019, 73, 58–66. [Google Scholar] [CrossRef] [PubMed]
- Goel, K.; Tweet, M.; Olson, T.M.; Maleszewski, J.J.; Gulati, R.; Hayes, S.N. Familial spontaneous coronary artery dissection: Evidence for genetic susceptibility. JAMA Intern. Med. 2015, 175, 821–826. [Google Scholar] [CrossRef]
- Turley, T.N.; Theis, J.L.; Sundsbak, R.S.; Evans, J.M.; O’Byrne, M.M.; Gulati, R.; Tweet, M.S.; Hayes, S.N.; Olson, T.M. Rare missense variants in tln1 are associated with familial and sporadic spontaneous coronary artery dissection. Circ. Genom. Precis. Med. 2019, 12, e002437. [Google Scholar] [CrossRef] [PubMed]
- Turley, T.N.; O’Byrne, M.M.; Kosel, M.L.; de Andrade, M.; Gulati, R.; Hayes, S.N.; Tweet, M.S.; Olson, T.M. Identification of susceptibility loci for spontaneous coronary artery dissection. JAMA Cardiol. 2020, 5, 929–938. [Google Scholar] [CrossRef] [PubMed]
- Turley, T.N.; Kosel, M.L.; Bamlet, W.R.; Gulati, R.; Hayes, S.N.; Tweet, M.S.; Olson, T.M. Susceptibility locus for pregnancy-associated spontaneous coronary artery dissection. Circ. Genom. Precis. Med. 2021, 14, e003398. [Google Scholar] [CrossRef]
- Carss, K.J.; Baranowska, A.A.; Armisen, J.; Webb, T.R.; Hamby, S.E.; Premawardhana, D.; Al-Hussaini, A.; Wood, A.; Wang, Q.; Deevi, S.V.V.; et al. Spontaneous coronary artery dissection: Insights on rare genetic variation from genome sequencing. Circ. Genom. Precis. Med. 2020, 13, e003030. [Google Scholar] [CrossRef]
- Amrani-Midoun, A.; Adlam, D.; Bouatia-Naji, N. Recent advances on the genetics of spontaneous coronary artery dissection. Circ. Genomic Precis. Med. 2021, 14, e003393. [Google Scholar] [CrossRef]
- Hayes, S.N.; Kim, E.S.H.; Saw, J.; Adlam, D.; Arslanian-Engoren, C.; Economy, K.E.; Ganesh, S.K.; Gulati, R.; Lindsay, M.E.; Mieres, J.H.; et al. Spontaneous coronary artery dissection: Current state of the science: A scientific statement from the american heart association. Circulation 2018, 137, e523–e557. [Google Scholar] [CrossRef]
- Tweet, M.S.; Hayes, S.N.; Pitta, S.R.; Simari, R.D.; Lerman, A.; Lennon, R.J.; Gersh, B.J.; Khambatta, S.; Best, P.J.M.; Rihal, C.S.; et al. Clinical features, management, and prognosis of spontaneous coronary artery dissection. Circulation 2012, 126, 579–588. [Google Scholar] [CrossRef]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar] [CrossRef]
- Novocraft Technologies. NovoAlign. Available online: http://www.novocraft.com/ (accessed on 18 January 2021).
- Picard Tools—By Broad Institute. Available online: http://broadinstitute.github.io/picard/ (accessed on 6 October 2021).
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The genome analysis toolkit: A mapreduce framework for analyzing next-generation dna sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- DePristo, M.A.; Banks, E.; Poplin, R.; Garimella, K.V.; Maguire, J.R.; Hartl, C.; Philippakis, A.A.; del Angel, G.; Rivas, M.A.; Hanna, M.; et al. A framework for variation discovery and genotyping using next-generation dna sequencing data. Nat. Genet. 2011, 43, 491–498. [Google Scholar] [CrossRef] [PubMed]
- Poplin, R.; Ruano-Rubio, V.; DePristo, M.; Fennell, T.; Carneiro, M.; Van der Auwera, G.; Kling, D.; Gauthier, L.; Levy-Moonshine, A.; Roazen, D.; et al. Scaling accurate genetic variant discovery to tens of thousands of samples. bioRxiv 2018. [Google Scholar] [CrossRef]
- Benson, G. Tandem repeats finder: A program to analyze dna sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.S.; Thormann, A.; Flicek, P.; Cunningham, F. The ensembl variant effect predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef]
- Rentzsch, P.; Witten, D.; Cooper, G.M.; Shendure, J.; Kircher, M. CADD: Predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019, 47, D886–D894. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Stenson, P.D.; Mort, M.; Ball, E.V.; Shaw, K.; Phillips, A.; Cooper, D.N. The human gene mutation database: Building a comprehensive mutation repository for clinical and molecular genetics, diagnostic testing and personalized genomic medicine. Hum. Genet. 2014, 133, 1–9. [Google Scholar] [CrossRef]
- Wang, J.; Zhuang, J.; Iyer, S.; Lin, X.-Y.; Greven, M.C.; Kim, B.-H.; Moore, J.; Pierce, B.G.; Dong, X.; Virgil, D.; et al. Factorbook.Org: A wiki-based database for transcription factor-binding data generated by the encode consortium. Nucleic Acids Res. 2013, 41, D171–D176. [Google Scholar] [CrossRef] [PubMed]
- Boyle, A.P.; Hong, E.L.; Hariharan, M.; Cheng, Y.; Schaub, M.A.; Kasowski, M.; Karczewski, K.J.; Park, J.; Hitz, B.C.; Weng, S.; et al. Annotation of functional variation in personal genomes using regulomeDB. Genome Res. 2012, 22, 1790–1797. [Google Scholar] [CrossRef] [PubMed]
- Thorvaldsdóttir, H.; Robinson, J.T.; Mesirov, J.P. Integrative genomics viewer (IGV): High-performance genomics data visualization and exploration. Brief. Bioinform. 2013, 14, 178–192. [Google Scholar] [CrossRef]
- Shihab, H.A.; Gough, J.; Cooper, D.N.; Stenson, P.D.; Barker, G.L.A.; Edwards, K.J.; Day, I.N.M.; Gaunt, T.R. Predicting the functional, molecular, and phenotypic consequences of amino acid substitutions using hidden markov models. Hum. Mutat. 2013, 34, 57–65. [Google Scholar] [CrossRef] [PubMed]
- GTEx Consortium human genomics. the genotype-tissue expression (gtex) pilot analysis: Multitissue gene regulation in humans. Science 2015, 348, 648–660. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING V11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Smith, C.L.; Blake, J.A.; Kadin, J.A.; Richardson, J.E.; Bult, C.J. Mouse genome database group mouse genome database (mgd)-2018: Knowledgebase for the laboratory mouse. Nucleic Acids Res. 2018, 46, D836–D842. [Google Scholar] [CrossRef]
- Buniello, A.; MacArthur, J.A.L.; Cerezo, M.; Harris, L.W.; Hayhurst, J.; Malangone, C.; McMahon, A.; Morales, J.; Mountjoy, E.; Sollis, E.; et al. The NHGRI-EBI GWAS catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 2019, 47, D1005–D1012. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Jassal, B.; Matthews, L.; Viteri, G.; Gong, C.; Lorente, P.; Fabregat, A.; Sidiropoulos, K.; Cook, J.; Gillespie, M.; Haw, R.; et al. The reactome pathway knowledgebase. Nucleic Acids Res. 2020, 48, D498–D503. [Google Scholar] [CrossRef] [PubMed]
- Geer, L.Y.; Marchler-Bauer, A.; Geer, R.C.; Han, L.; He, J.; He, S.; Liu, C.; Shi, W.; Bryant, S.H. The NCBI BioSystems database. Nucleic Acids Res. 2010, 38, D492–D496. [Google Scholar] [CrossRef] [PubMed]
- Harel, A.; Inger, A.; Stelzer, G.; Strichman-Almashanu, L.; Dalah, I.; Safran, M.; Lancet, D. GIFtS: Annotation landscape analysis with genecards. BMC Bioinform. 2009, 10, 348. [Google Scholar] [CrossRef] [PubMed]
- Zekavat, S.M.; Chou, E.L.; Zekavat, M.; Pampana, A.; Paruchuri, K.; Lino Cardenas, C.L.; Koyama, S.; Ghazzawi, Y.; Kii, E.; Uddin, M.M.; et al. Fibrillar collagen variants in spontaneous coronary artery dissection. JAMA Cardiol. 2022, 7, 396–406. [Google Scholar] [CrossRef] [PubMed]
- Tarr, I.; Hesselson, S.; Iismaa, S.E.; Rath, E.; Monger, S.; Troup, M.; Mishra, K.; Wong, C.M.Y.; Hsu, P.-C.; Junday, K.; et al. Exploring the genetic architecture of spontaneous coronary artery dissection using whole-genome sequencing. Circ. Genom. Precis. Med. 2022, 15, e003527. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Starovoytov, A.; Murad, A.M.; Hunker, K.L.; Brunham, L.R.; Li, J.Z.; Saw, J.; Ganesh, S.K. Burden of rare genetic variants in spontaneous coronary artery dissection with high-risk features. JAMA Cardiol. 2022, 7, 1045–1055. [Google Scholar] [CrossRef]
- Henkin, S.; Negrotto, S.M.; Tweet, M.S.; Kirmani, S.; Deyle, D.R.; Gulati, R.; Olson, T.M.; Hayes, S.N. Spontaneous coronary artery dissection and its association with heritable connective tissue disorders. Heart 2016, 102, 876–881. [Google Scholar] [CrossRef]
- Kaadan, M.I.; MacDonald, C.; Ponzini, F.; Duran, J.; Newell, K.; Pitler, L.; Lin, A.; Weinberg, I.; Wood, M.J.; Lindsay, M.E. Prospective cardiovascular genetics evaluation in spontaneous coronary artery dissection. Circ. Genom. Precis. Med. 2018, 11, e001933. [Google Scholar] [CrossRef]
- Gough, R.E.; Goult, B.T. The tale of two talins—Two isoforms to fine-tune integrin signalling. FEBS Lett. 2018, 592, 2108–2125. [Google Scholar] [CrossRef]
- Bax, D.V.; Bernard, S.E.; Lomas, A.; Morgan, A.; Humphries, J.; Shuttleworth, C.A.; Humphries, M.J.; Kielty, C.M. Cell adhesion to fibrillin-1 molecules and microfibrils is mediated by alpha 5 beta 1 and alpha v beta 3 integrins. J. Biol. Chem. 2003, 278, 34605–34616. [Google Scholar] [CrossRef]
- Wujak, L.; Böttcher, R.T.; Pak, O.; Frey, H.; El Agha, E.; Chen, Y.; Schmitt, S.; Bellusci, S.; Schaefer, L.; Weissmann, N.; et al. Low density lipoprotein receptor-related protein 1 couples β1 integrin activation to degradation. Cell. Mol. Life Sci. CMLS 2018, 75, 1671–1685. [Google Scholar] [CrossRef] [PubMed]
- Koyama, Y.; Baba, A. Endothelin-induced cytoskeletal actin re-organization in cultured astrocytes: Inhibition by c3 adp-ribosyltransferase. Glia 1996, 16, 342–350. [Google Scholar] [CrossRef]
- Cunnick, J.M.; Kim, S.; Hadsell, J.; Collins, S.; Cerra, C.; Reiser, P.; Flynn, D.C.; Cho, Y. Actin filament-associated protein 1 is required for csrc activity and secretory activation in the lactating mammary gland. Oncogene 2015, 34, 2640–2649. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Na, C.-Y.; Choi, S.Y.; Kim, H.W.; Du Kim, Y.; Kwon, J.B.; Chung, M.Y.; Hong, J.M.; Park, C.B. Integration of gene-expression profiles and pathway analysis in ascending thoracic aortic aneurysms. Ann. Vasc. Surg. 2010, 24, 538–549. [Google Scholar] [CrossRef]
- Bi, S.; Liu, R.; He, L.; Li, J.; Gu, J. Bioinformatics analysis of common key genes and pathways of intracranial, abdominal, and thoracic aneurysms. BMC Cardiovasc. Disord. 2021, 21, 14. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Du, Y.; Lok, J.; Lo, E.H.; Xing, C. Lipocalin-2 Enhances angiogenesis in rat brain endothelial cells via reactive oxygen species and iron-dependent mechanisms. J. Neurochem. 2015, 132, 622–628. [Google Scholar] [CrossRef]
- Wei, X.; Sun, Y.; Wu, Y.; Zhu, J.; Gao, B.; Yan, H.; Zhao, Z.; Zhou, J.; Jing, Z. Downregulation of talin-1 expression associates with increased proliferation and migration of vascular smooth muscle cells in aortic dissection. BMC Cardiovasc. Disord. 2017, 17, 162. [Google Scholar] [CrossRef]
- Guo, D.-C.; Pannu, H.; Tran-Fadulu, V.; Papke, C.L.; Yu, R.K.; Avidan, N.; Bourgeois, S.; Estrera, A.L.; Safi, H.J.; Sparks, E.; et al. Mutations in smooth muscle alpha-actin (ACTA2) lead to thoracic aortic aneurysms and dissections. Nat. Genet. 2007, 39, 1488–1493. [Google Scholar] [CrossRef]
- Weber, G.F.; Bjerke, M.A.; DeSimone, D.W. Integrins and cadherins join forces to form adhesive networks. J. Cell Sci. 2011, 124, 1183–1193. [Google Scholar] [CrossRef]
- Dorrell, M.I.; Aguilar, E.; Friedlander, M. Retinal Vascular development is mediated by endothelial filopodia, a preexisting astrocytic template and specific r-cadherin adhesion. Investig. Ophthalmol. Vis. Sci. 2002, 43, 3500–3510. [Google Scholar]
- Oda, H.; Takeichi, M. Evolution: Structural and functional diversity of cadherin at the adherens junction. J. Cell Biol. 2011, 193, 1137–1146. [Google Scholar] [CrossRef] [PubMed]
- Kemler, R. From cadherins to catenins: Cytoplasmic protein interactions and regulation of cell adhesion. Trends Genet. TIG 1993, 9, 317–321. [Google Scholar] [CrossRef] [PubMed]
- Lampugnani, M.G.; Dejana, E. Adherens junctions in endothelial cells regulate vessel maintenance and angiogenesis. Thromb. Res. 2007, 120 (Suppl. 2), S1–S6. [Google Scholar] [CrossRef] [PubMed]
- Slater, S.C.; Koutsouki, E.; Jackson, C.L.; Bush, R.C.; Angelini, G.D.; Newby, A.C.; George, S.J. R-Cadherin:Beta-Catenin complex and its association with vascular smooth muscle cell proliferation. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1204–1210. [Google Scholar] [CrossRef] [PubMed]
- Cattelino, A.; Liebner, S.; Gallini, R.; Zanetti, A.; Balconi, G.; Corsi, A.; Bianco, P.; Wolburg, H.; Moore, R.; Oreda, B.; et al. The conditional inactivation of the beta-catenin gene in endothelial cells causes a defective vascular pattern and increased vascular fragility. J. Cell Biol. 2003, 162, 1111–1122. [Google Scholar] [CrossRef] [PubMed]
- Fukata, M.; Watanabe, T.; Noritake, J.; Nakagawa, M.; Yamaga, M.; Kuroda, S.; Matsuura, Y.; Iwamatsu, A.; Perez, F.; Kaibuchi, K. Rac1 and Cdc42 capture microtubules through IQGAP1 and CLIP-170. Cell 2002, 109, 873–885. [Google Scholar] [CrossRef]
- Fukata, M.; Kuroda, S.; Nakagawa, M.; Kawajiri, A.; Itoh, N.; Shoji, I.; Matsuura, Y.; Yonehara, S.; Fujisawa, H.; Kikuchi, A.; et al. Cdc42 and Rac1 regulate the interaction of IQGAP1 with Beta-Catenin. J. Biol. Chem. 1999, 274, 26044–26050. [Google Scholar] [CrossRef]
- Yamaoka-Tojo, M.; Ushio-Fukai, M.; Hilenski, L.; Dikalov, S.I.; Chen, Y.E.; Tojo, T.; Fukai, T.; Fujimoto, M.; Patrushev, N.A.; Wang, N.; et al. IQGAP1, a novel vascular endothelial growth factor receptor binding protein, is involved in reactive oxygen species--dependent endothelial migration and proliferation. Circ. Res. 2004, 95, 276–283. [Google Scholar] [CrossRef]
- Kadler, K.E.; Baldock, C.; Bella, J.; Boot-Handford, R.P. Collagens at a glance. J. Cell Sci. 2007, 120, 1955–1958. [Google Scholar] [CrossRef]
- Myllyharju, J.; Kivirikko, K.I. Collagens and collagen-related diseases. Ann. Med. 2001, 33, 7–21. [Google Scholar] [CrossRef]
- Frank, M.; Albuisson, J.; Ranque, B.; Golmard, L.; Mazzella, J.-M.; Bal-Theoleyre, L.; Fauret, A.-L.; Mirault, T.; Denarié, N.; Mousseaux, E.; et al. The Type of Variants at the COL3A1 Gene Associates with the Phenotype and Severity of Vascular Ehlers-Danlos Syndrome. Eur. J. Hum. Genet. EJHG 2015, 23, 1657–1664. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, M.; Nakagawa, R.; Naruto, T.; Kohmoto, T.; Suga, K.; Goji, A.; Kagami, S.; Masuda, K.; Imoto, I. A Novel Missense Mutation of COL5A2 in a patient with ehlers–danlos syndrome. Hum. Genome Var. 2016, 3, 16030. [Google Scholar] [CrossRef]
- Yoneda, Y.; Haginoya, K.; Arai, H.; Yamaoka, S.; Tsurusaki, Y.; Doi, H.; Miyake, N.; Yokochi, K.; Osaka, H.; Kato, M.; et al. De Novo and inherited mutations in COL4A2, encoding the Type IV collagen A2 chain cause porencephaly. Am. J. Hum. Genet. 2012, 90, 86–90. [Google Scholar] [CrossRef] [PubMed]
- van de Luijtgaarden, K.M.; Heijsman, D.; Maugeri, A.; Weiss, M.M.; Verhagen, H.J.M.; IJpma, A.; Brüggenwirth, H.T.; Majoor-Krakauer, D. First genetic analysis of aneurysm genes in familial and sporadic abdominal aortic aneurysm. Hum. Genet. 2015, 134, 881–893. [Google Scholar] [CrossRef] [PubMed]
- Plaisier, E.; Gribouval, O.; Alamowitch, S.; Mougenot, B.; Prost, C.; Verpont, M.C.; Marro, B.; Desmettre, T.; Cohen, S.Y.; Roullet, E.; et al. COL4A1 mutations and hereditary angiopathy, nephropathy, aneurysms, and muscle cramps. N. Engl. J. Med. 2007, 357, 2687–2695. [Google Scholar] [CrossRef]
- Huveneers, S.; Danen, E.H.J. Adhesion signaling—Crosstalk between integrins, Src and Rho. J. Cell Sci. 2009, 122, 1059–1069. [Google Scholar] [CrossRef]
- Sakamoto, H.; Zhang, X.-Q.; Suenobu, S.; Ohbo, K.; Ogawa, M.; Suda, T. Cell Adhesion to Ephrinb2 Is induced by EphB4 independently of its kinase activity. Biochem. Biophys. Res. Commun. 2004, 321, 681–687. [Google Scholar] [CrossRef]
- Luxán, G.; Stewen, J.; Díaz, N.; Kato, K.; Maney, S.K.; Aravamudhan, A.; Berkenfeld, F.; Nagelmann, N.; Drexler, H.C.; Zeuschner, D.; et al. Endothelial EphB4 maintains vascular integrity and transport function in adult heart. eLife 2019, 8, e45863. [Google Scholar] [CrossRef]
- Polacheck, W.J.; Kutys, M.L.; Yang, J.; Eyckmans, J.; Wu, Y.; Vasavada, H.; Hirschi, K.K.; Chen, C.S. A Non-canonical notch complex regulates adherens junctions and vascular barrier function. Nature 2017, 552, 258–262. [Google Scholar] [CrossRef]
- McKellar, S.H.; Tester, D.J.; Yagubyan, M.; Majumdar, R.; Ackerman, M.J.; Sundt, T.M. Novel NOTCH1 mutations in patients with bicuspid aortic valve disease and thoracic aortic aneurysms. J. Thorac. Cardiovasc. Surg. 2007, 134, 290–296. [Google Scholar] [CrossRef]
- Dabral, S.; Tian, X.; Kojonazarov, B.; Savai, R.; Ghofrani, H.A.; Weissmann, N.; Florio, M.; Sun, J.; Jonigk, D.; Maegel, L.; et al. Notch1 signalling regulates endothelial proliferation and apoptosis in pulmonary arterial hypertension. Eur. Respir. J. 2016, 48, 1137–1149. [Google Scholar] [CrossRef] [PubMed]
- Bai, B.; Zhang, M.; Zhuang, Y.; Zhu, J.; Li, W.; Ma, W.; Chen, H. The pregnancy-associated spontaneous coronary artery dissection in a young woman with a novel missense mutation in NOTCH1: A case report. BMC Med. Genet. 2020, 21, 119. [Google Scholar] [CrossRef] [PubMed]
- Olschewski, A.; Li, Y.; Tang, B.; Hanze, J.; Eul, B.; Bohle, R.M.; Wilhelm, J.; Morty, R.E.; Brau, M.E.; Weir, E.K.; et al. Impact of TASK-1 in human pulmonary artery smooth muscle cells. Circ. Res. 2006, 98, 1072–1080. [Google Scholar] [CrossRef] [PubMed]
- Navas Tejedor, P.; Tenorio Castaño, J.; Palomino Doza, J.; Arias Lajara, P.; Gordo Trujillo, G.; López Meseguer, M.; Román Broto, A.; Lapunzina Abadía, P.; Escribano Subía, P. An homozygous mutation in kcnk3 is associated with an aggressive form of hereditary pulmonary arterial hypertension. Clin. Genet. 2017, 91, 453–457. [Google Scholar] [CrossRef] [PubMed]
- Tweet, M.S.; Miller, V.M.; Hayes, S.N. The evidence on estrogen, progesterone, and spontaneous coronary artery dissection. JAMA Cardiol. 2019, 4, 403–404. [Google Scholar] [CrossRef] [PubMed]
- Zuo, C.; Shin, S.; Keles, S. atSNP: Transcription factor binding affinity testing for regulatory SNP detection. Bioinformatics 2015, 31, 3353–3355. [Google Scholar] [CrossRef]
- McGeary, S.; Lin, K.; Shi, C.; Pham, T.; Bisaria, N.; Kelley, G.; Bartel, D. The biochemical basis of microRNA targeting efficacy. Science 2019, 366, eaav1741. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Family | ID | Sex | Race | Age at Event/ Diagnosis, Yrs | Clinical Features | Affected Coronary Artery |
|---|---|---|---|---|---|---|
| SCAD-01 | II.1 | F | W | 42 | SCAD, MH | MV: LM, LCx, OM2, D1 |
| II.2 | F | 47 | SCAD, FMD, P, ES | OM | ||
| SCAD-02 | II.1 | F | W | 35 | SCAD, CT, ES | LAD |
| II.2 | F | 45 | SCAD | LAD | ||
| SCAD-03 | II.1 | F | W | 43 | SCAD, FMD, CT | LAD |
| II.2 | F | 45 | SCAD, MH | LAD | ||
| SCAD-04 | II.1 | F | W | 43 | SCAD, ES | MV: LCx, OM |
| II.2 | F | 59 | SCAD | LAD | ||
| SCAD-05 | II.1 | F | W | 34 | SCAD, FMD, P, PE | OM2 |
| II.2 | F | 36 | SCAD, FMD | LAD | ||
| SCAD-06 | II.1 | F | W | 50 | SCAD, FMD, ICAA | MV: LAD, LCx |
| II.2 | M | 47 | SCAD | DA | ||
| II.3 | M | 40 | SMAD | — | ||
| SCAD-07 | III.1 | F | W | 41 | SCAD, FMD, ES, CA | LAD |
| III.2 | F | 37 | SCAD, FMD, CT | LAD | ||
| II.3 | F | 65 | CA, ICAA | — | ||
| SCAD-08 | III.1 | F | W | 50, 59 | SCAD, R, ES | 1. LAD, 2. OM1 |
| III.2 | F | 50 | SCAD, CT | OM1 | ||
| I.1 | M | 71 | CA | — | ||
| SCAD-09 | III.2 | F | W | 42 | SCAD, ES | MV: RCA, PDA |
| III.3 | F | Unk | SCAD, FMD | |||
| III.1 | M | 25 | CVA | — | ||
| SCAD-10 | II.3 | F | W | 42, 45, 56 | SCAD, R, ES | 1. LCx, 2. MV: LAD, LM, 3. RCA |
| III.1 | F | 39 | SCAD, MH | LAD | ||
| II.2 | F | Unk | CT | — | ||
| SCAD-11 | III.1 | F | W | 36 | SCAD, FMD | OM1 |
| III.2 | F | 44 | SCAD, CT, MH, PE | RCA | ||
| SCAD-12 | II.3 | F | W | 47 | SCAD, FMD | LCx |
| III.1 | F | 38 | SCAD, P | OM | ||
| II.1 | F | Unk | CA | — | ||
| SCAD-13 | II.1 | F | W | 44 | SCAD | LAD |
| I.2 | F | 54 | SCAD | LAD | ||
| II.2 | F | 45 | CD | — | ||
| SCAD-14 | I.2 | F | W | 59, 61 | SCAD, FMD, R | 1. MV: LCx, OM2, 2. LAD |
| II.1 | F | 32 | SCAD, PE | LAD | ||
| SCAD-15 | II.1 | F | W | 33 | SCAD, FMD, MH | LAD |
| II.2 | F | Unk | SCAD | |||
| I.1 | F | 65 | AAA, IAA, PAA | — |
| Gene Symbol (Name) | rsID | Variant | Translation Impact | gnomAD MAF (%) | CADD Score (Percentile) | Deleterious Classification (HGMD) | Gene Constraint (Z-Score/pLI †) | Arterial Tissue Expression—Rank | PPI Talin 1 = 1, GWAS = 2 | CTD/ Aor Gene | Abnormal Vascular Phenotype (MGI) | Vascular Phenotype (HGMD) | GWAS Trait (GWAS Cat) | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Coronary | Aorta | Tibial | |||||||||||||
| CDH4 (cadherin 4) | rs1305825960 | c.2101T > C p.S701P | Missense | 0.0004 | 25.1 (75) | — | 2.02 | 47 | 46 | 43 | 2 | — | — | — | CAC, ICH |
| COL3A1 (collagen type III alpha 1 chain) | rs1238066761 | c.1336C > T p.R446C | Missense | 0.0004 | 32 (95) | — | 4.09 | 6 | 5 | 7 | 1, 2 | Yes | AD, aorta smooth muscle morphology | TAD, AA, CSVD, AAA, SCAD | — |
| COL4A1 (collagen type IV alpha 1 chain) | rs145172612 | c.1588C > T p.P530S | Missense | 0.03 | 29.4 (90) | CM1818194 | 3.02 | 1 | 3 | 2 | 1, 2 | — | ICH, retinal vascular morphology | ceAD, CSVD, ICH, CAD, TGA | Arterial stiffness, CVD, CAD |
| COL4A2 (collagen type IV alpha 2 chain) | rs746743018 | c.451G > A p.G151S | Missense | 0.002 | 24.8 (75) | — | 2.19 | 2 | 1 | 3 | 1, 2 | — | Aorta stenosis, cranial blood vascular morphology | CSVD, ICH | Arterial stiffness, carotid artery thickness, CAC, CAD |
| rs1464563247 | c.1237G > A p.G413R | Missense | 0.001 | 26.6 (85) | — | ||||||||||
| COL5A2 (collagen type V alpha 2 chain) | — | c.2705A > C p.Q902P | Missense | NR | 27.5 (90) | — | 2.44 | 6 | 4 | 8 | 1, 2 | Yes | Vascular congestion, cardiovascular system physiology | ceAD, CTHD, AD, IA, SCAD | — |
| CTNNB1 (catenin beta 1) | rs35288908 | c.860A > G p.N287S | Missense | 0.09 | 21.2 (30) | CM043757 | 3.85 | 13 | 15 | 3 | 1, 2 | — | Pulmonary artery, vascular endothelial cell and vitelline morphology | — | — |
| EPHB4 (ephrin type-B receptor 4) | rs146674844 | c.1384G > A p.G462R | Missense | 0.03 | 24.8 (75) | — | 2.30 | 30 | 37 | 38 | — | — | Angiogenesis, vascular branching, pulmonary artery and vitelline morphology | PAH, LVOTO | — |
| IQGAP1 (IQ-motif-containing GTPase-activating protein1) | — | c.3196A > G p.N1066D | Missense | NR | 27.7 (90) | — | 2.44 | 5 | 4 | 3 | 1, 2 | — | — | TOF and other cardiac abnormalities | — |
| KCNK3 (potassium channel subfamily K member 3) | — | c.781_794delCGCGCGCTGCTCAC p.R261fsTer | Frameshift * | NR | — | — | 0.90 † | 5 | 13 | 7 | — | — | — | PAH | CVD, ICH |
| LRP2 (LDL-receptor-related protein 2) | — | c.11093T > A p.V3698E | Missense | NR | 24.6 (75) | — | 2.07 | 35 | 31 | 34 | 2 | — | Aortic arch morphology | PAH | — |
| NOTCH1 (neurogenic locus notch homolog protein 1) | rs182330532 | c.3835C > T p.R1279C | Missense * | 0.06 | 34 (99) | — | 3.45 | 26 | 28 | 22 | 1, 2 | Yes | Angiogenesis, vasculogenesis, vitelline morphology | TAD, PAH, VAD, CoA, TOF, ceAD | CAC |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Turley, T.N.; Theis, J.L.; Evans, J.M.; Fogarty, Z.C.; Gulati, R.; Hayes, S.N.; Tweet, M.S.; Olson, T.M. Identification of Rare Genetic Variants in Familial Spontaneous Coronary Artery Dissection and Evidence for Shared Biological Pathways. J. Cardiovasc. Dev. Dis. 2023, 10, 393. https://doi.org/10.3390/jcdd10090393
Turley TN, Theis JL, Evans JM, Fogarty ZC, Gulati R, Hayes SN, Tweet MS, Olson TM. Identification of Rare Genetic Variants in Familial Spontaneous Coronary Artery Dissection and Evidence for Shared Biological Pathways. Journal of Cardiovascular Development and Disease. 2023; 10(9):393. https://doi.org/10.3390/jcdd10090393
Chicago/Turabian StyleTurley, Tamiel N., Jeanne L. Theis, Jared M. Evans, Zachary C. Fogarty, Rajiv Gulati, Sharonne N. Hayes, Marysia S. Tweet, and Timothy M. Olson. 2023. "Identification of Rare Genetic Variants in Familial Spontaneous Coronary Artery Dissection and Evidence for Shared Biological Pathways" Journal of Cardiovascular Development and Disease 10, no. 9: 393. https://doi.org/10.3390/jcdd10090393