Modeling Novel Putative Drugs and Vaccine Candidates against Tick-Borne Pathogens: A Subtractive Proteomics Approach

 , , and
, , and 
Abstract
:1. Introduction
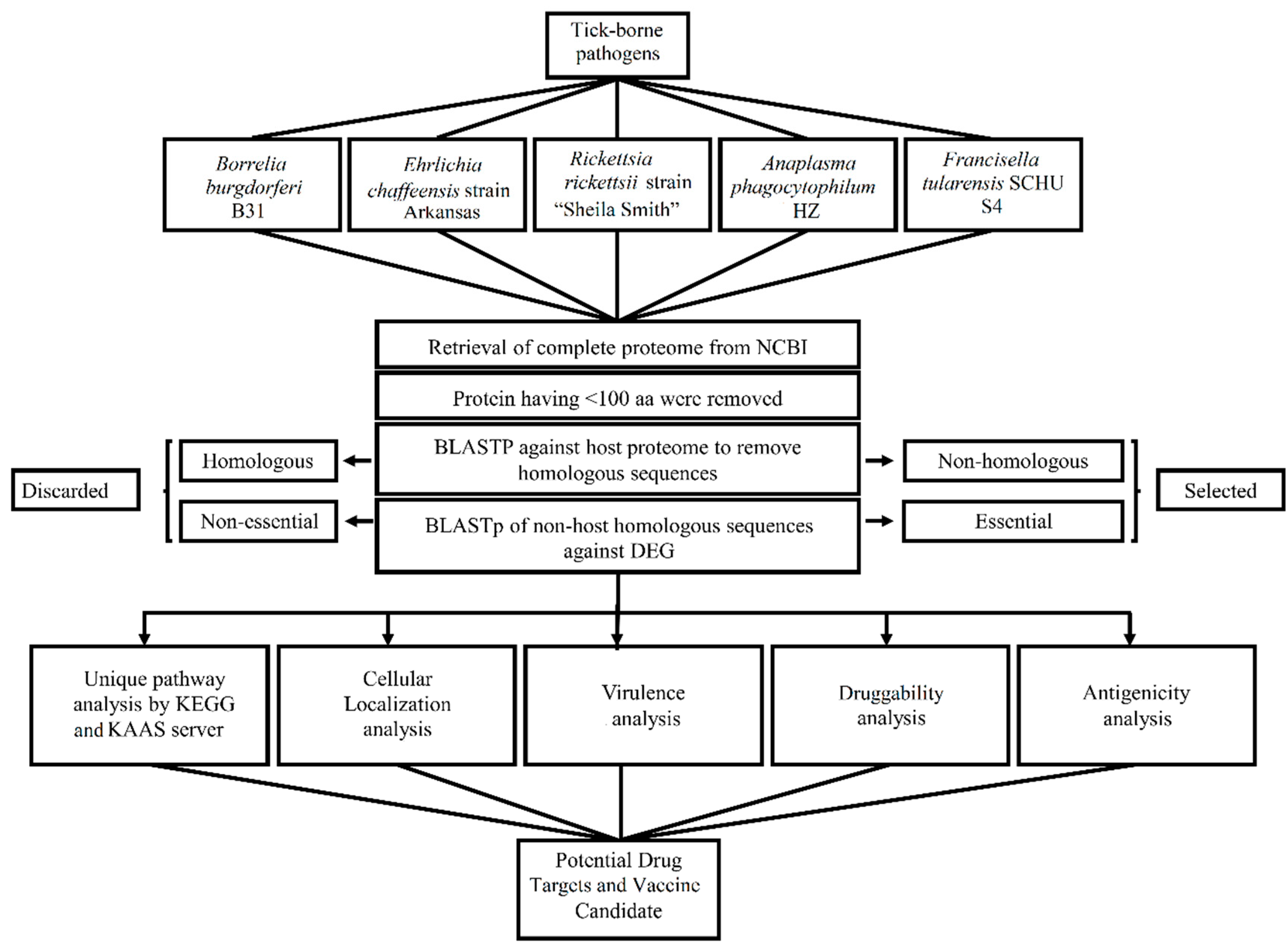
2. Methodology
2.1. Retrieval of Pathogens Proteome
2.2. Identification of Essential and Non-Host Homologous Proteins in Pathogens
2.3. Metabolic Pathways and Subcellular Localization Analysis
2.4. Druggability, Virulency Antigenicity, and Allergenicity Analysis
2.5. Human Gut-Metagenomes Screening and Secondary Structure Prediction
2.6. Phylogenetic Analysis
2.7. Homology Modeling and Molecular Dynamics Simulation
3. Results and Discussion
3.1. Identification of Essential and Non-Host Homologous Proteins in Pathogens
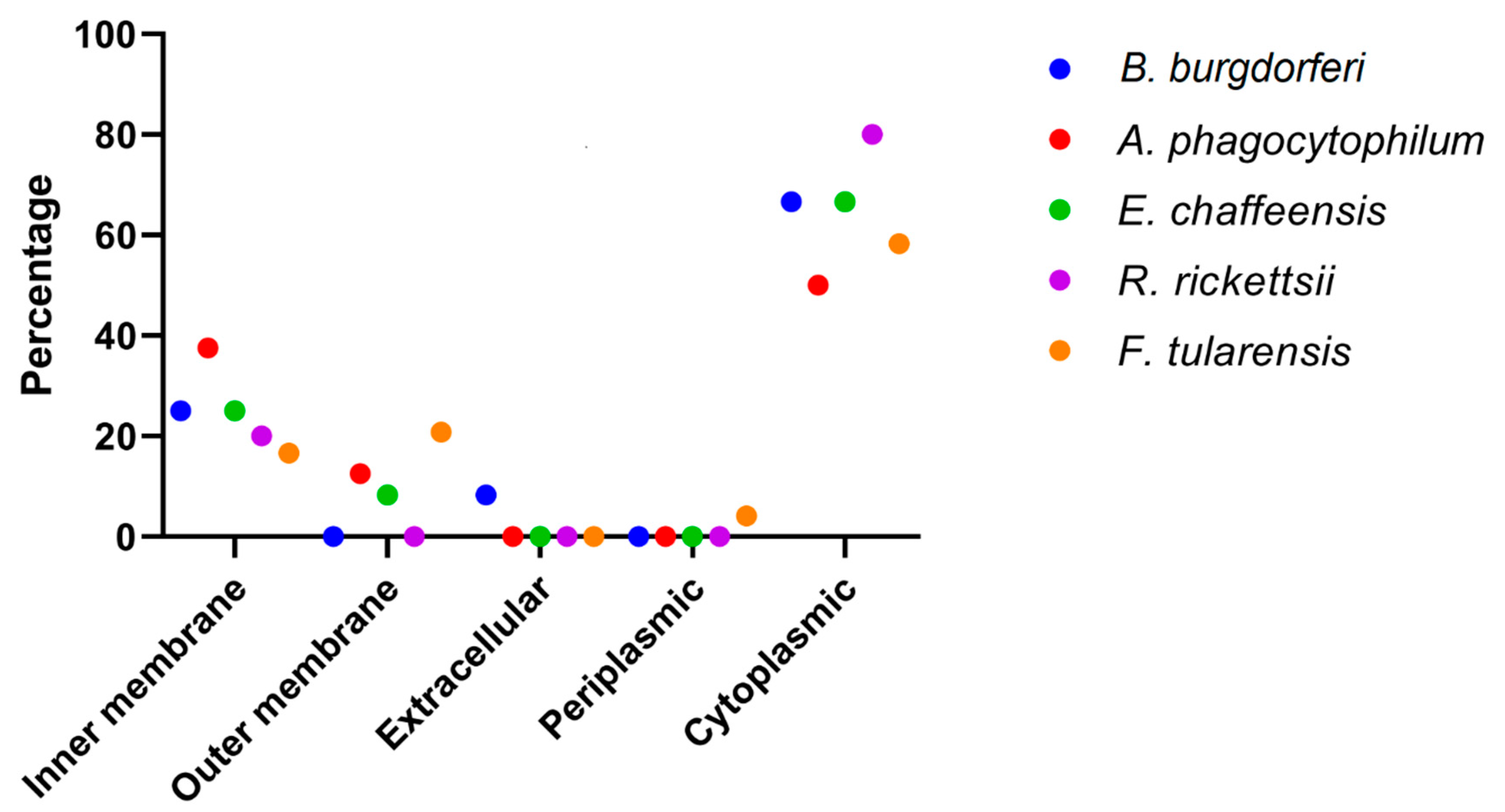
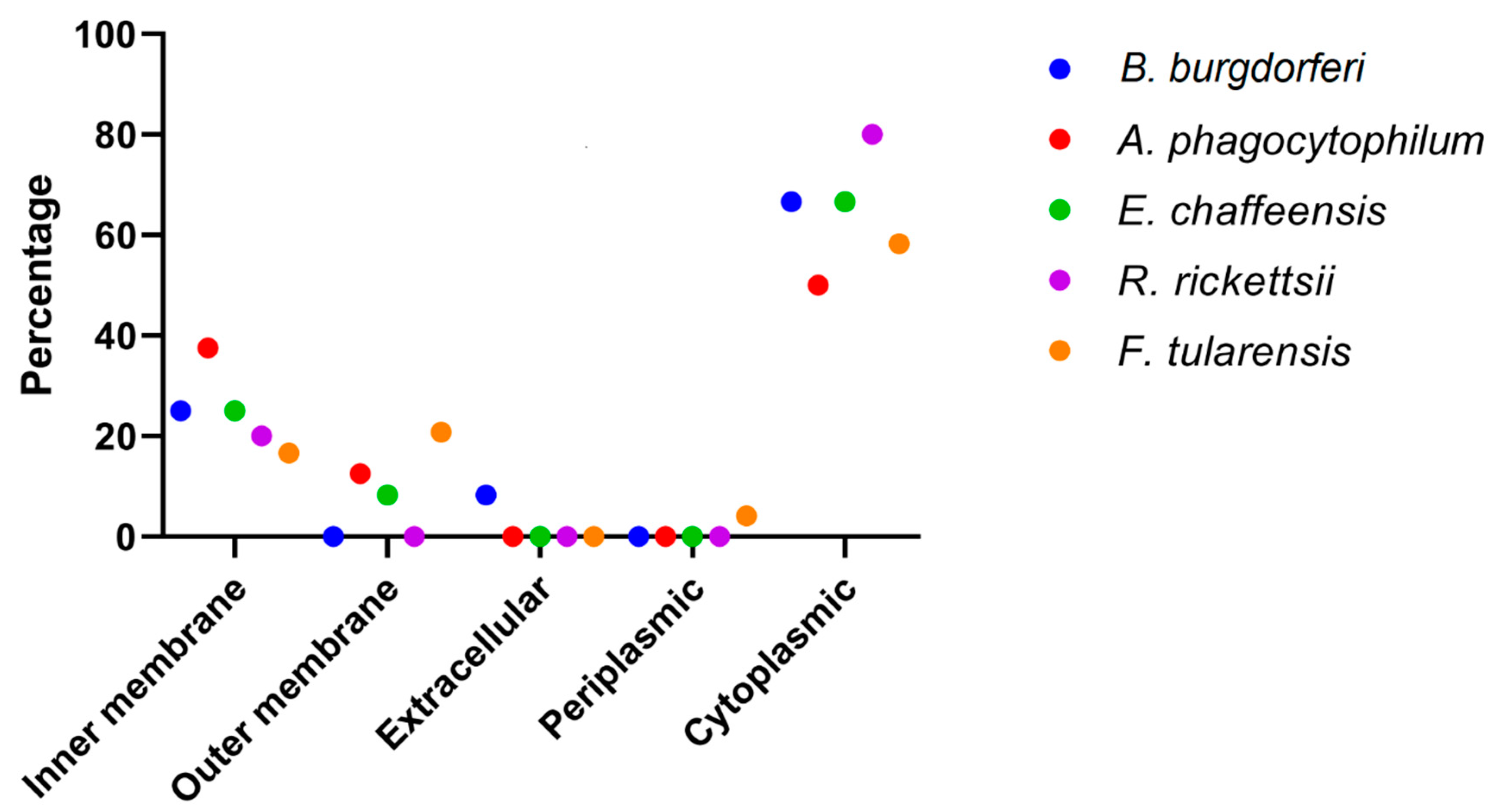
3.2. Pathogens Unique Metabolic Pathways and Subcellular Localization
3.3. Functional Analysis of Unique Pathways
3.4. Druggability and Virulence Analysis for the Identification of Potential Drug Targets and Vaccine Candidates
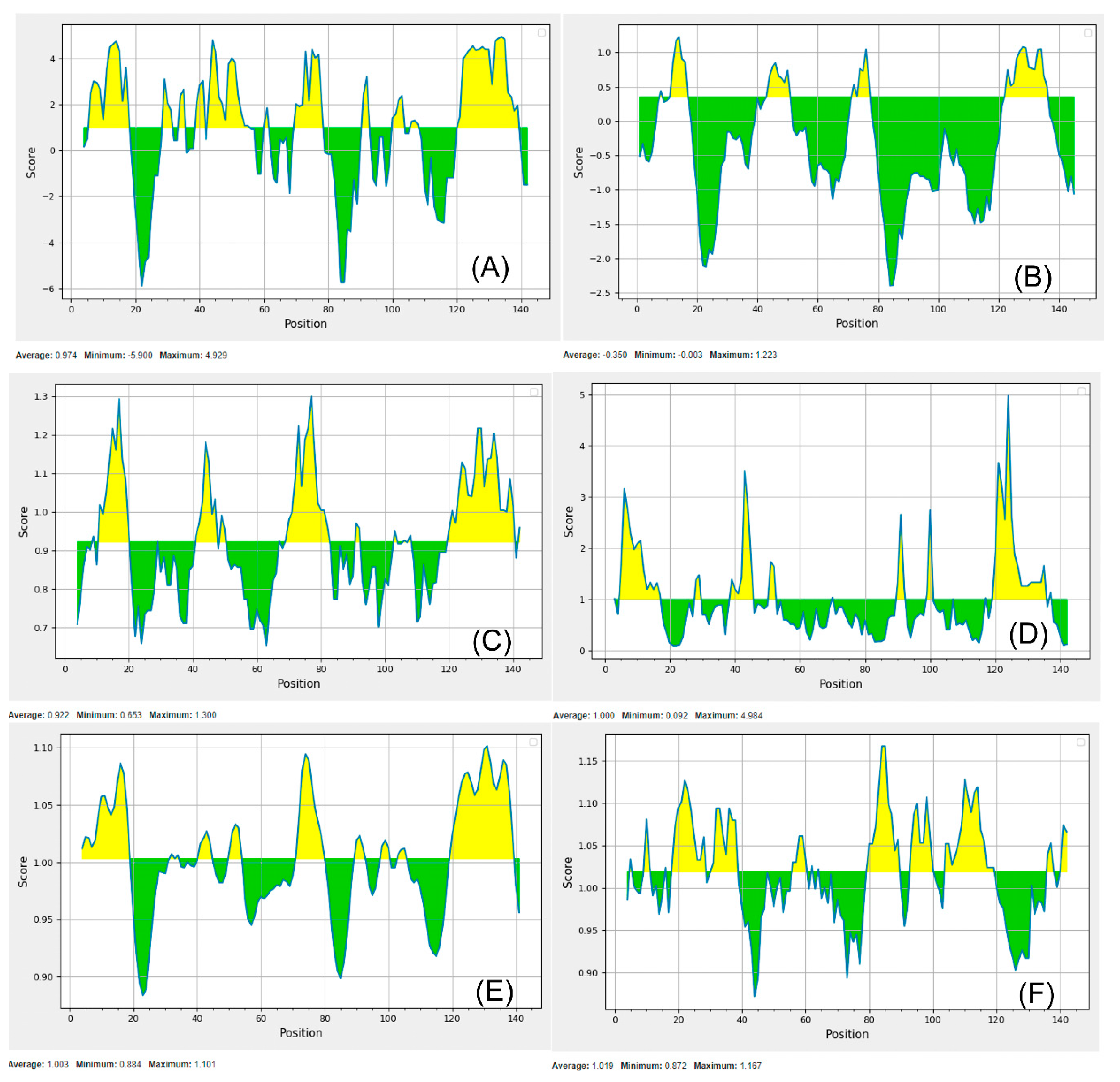
3.5. Screening of Essential, Non-Homologous Target Proteins Versus Gut Metagenome and Secondary Structure Analysis
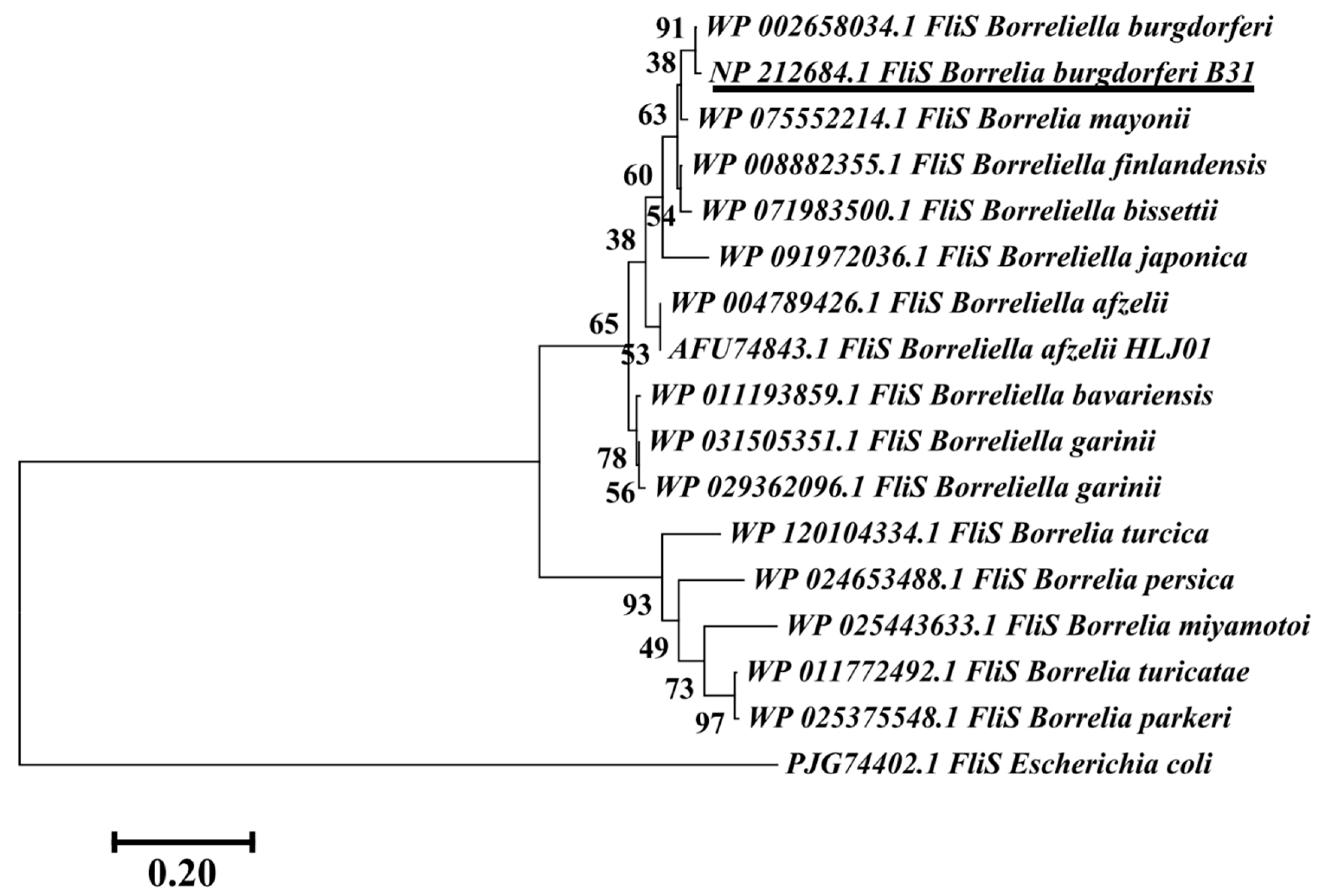
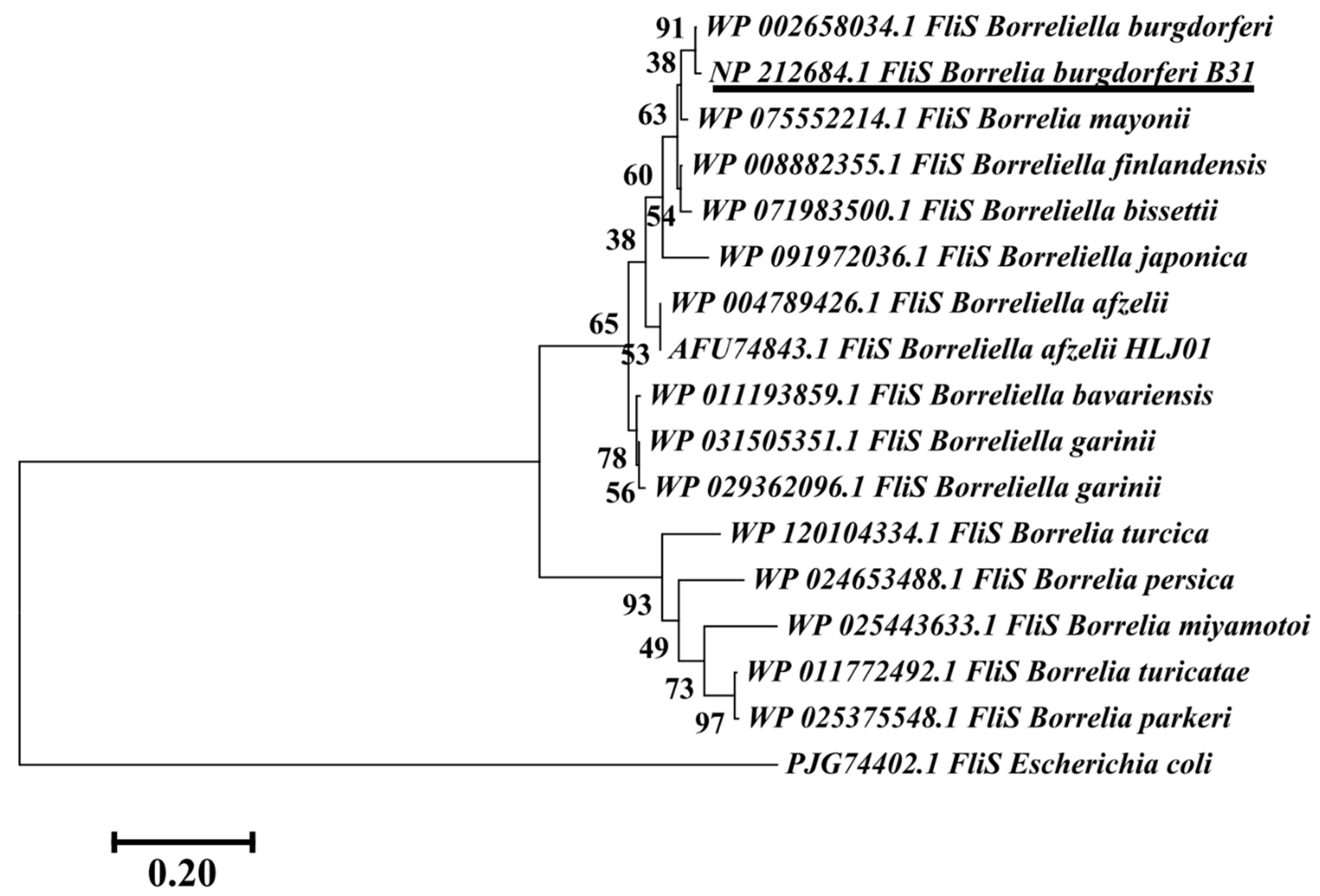
3.6. Phylogenetic Analysis
3.7. Characterization of Drug Targets and Vaccine Candidates
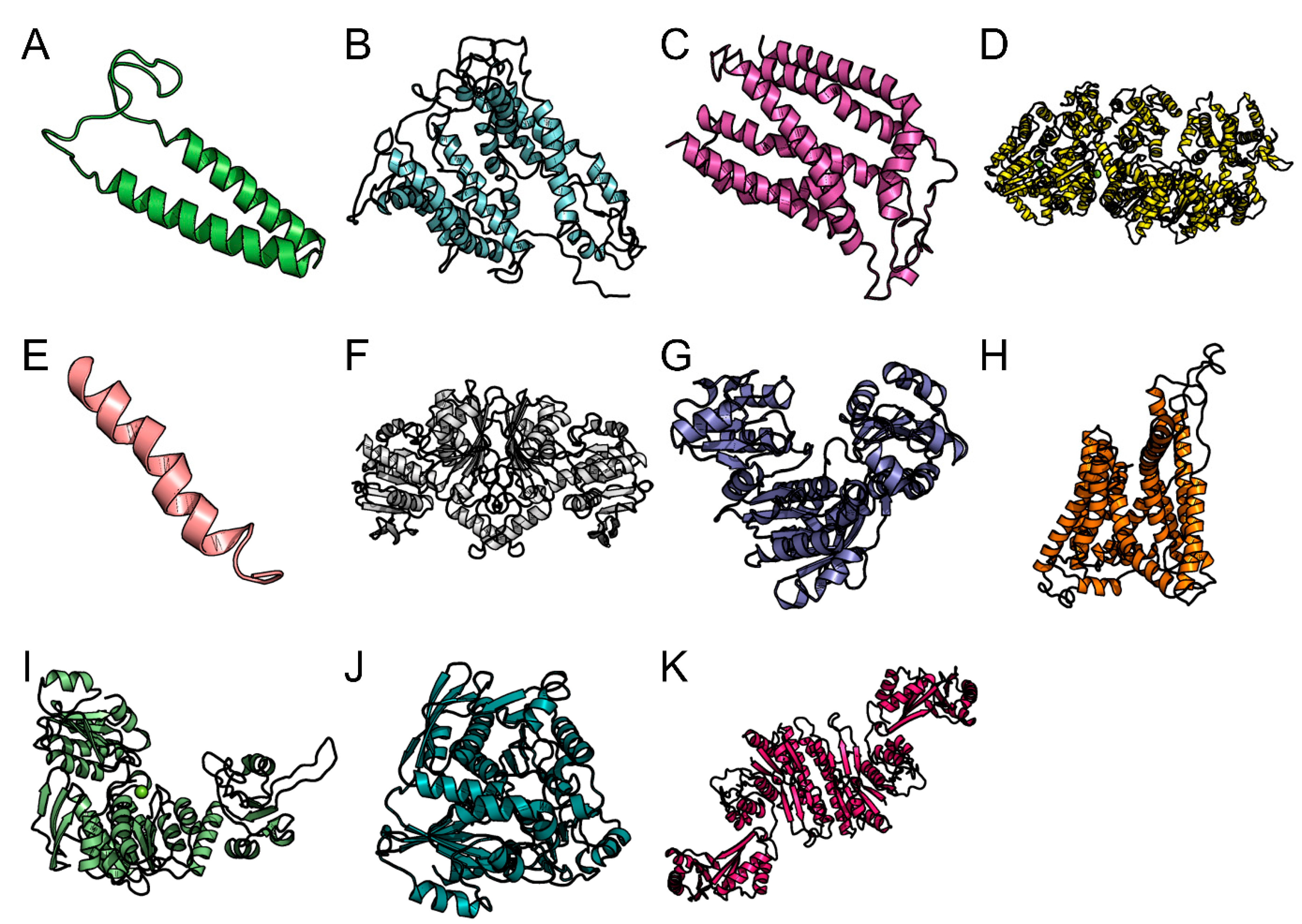
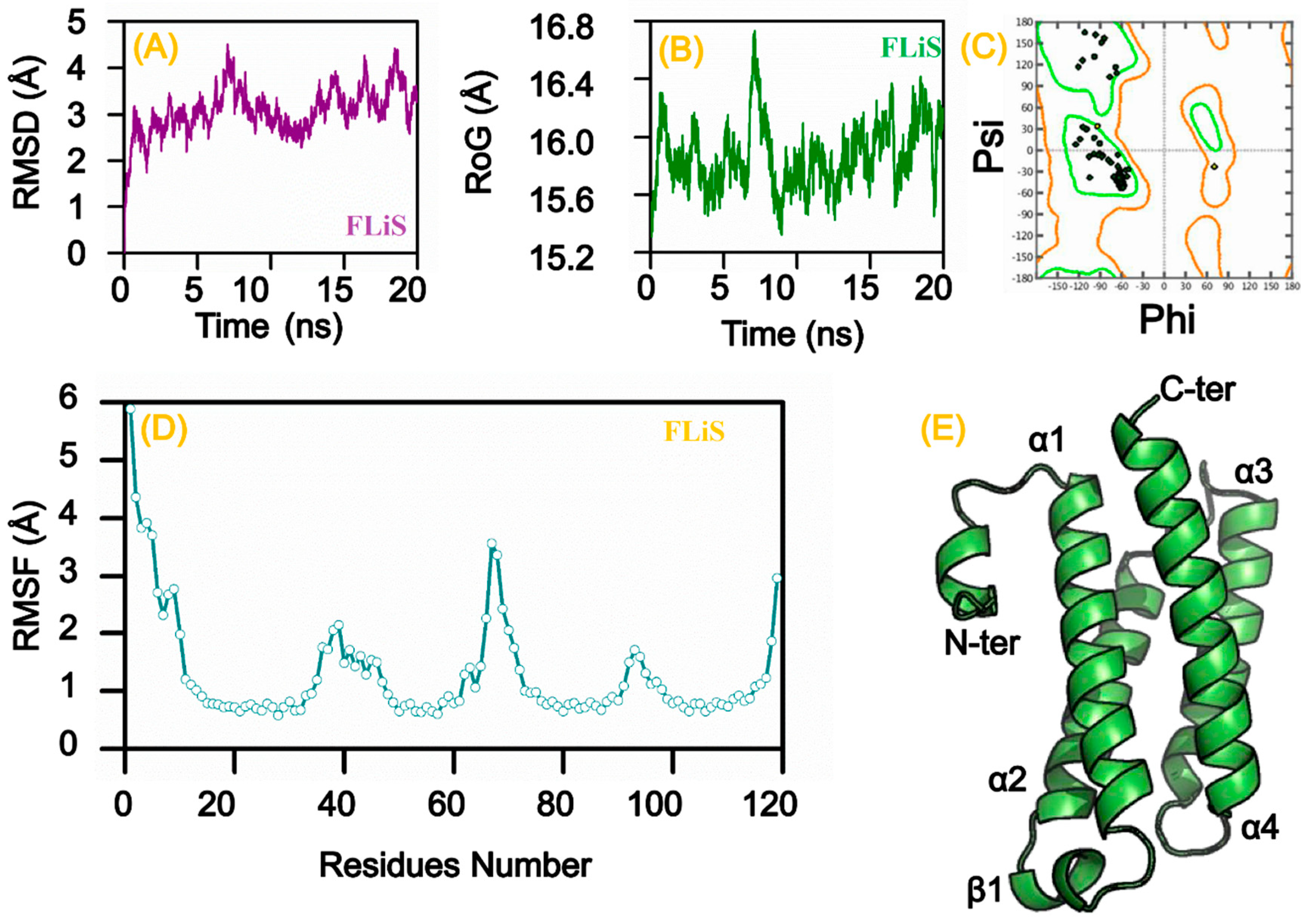
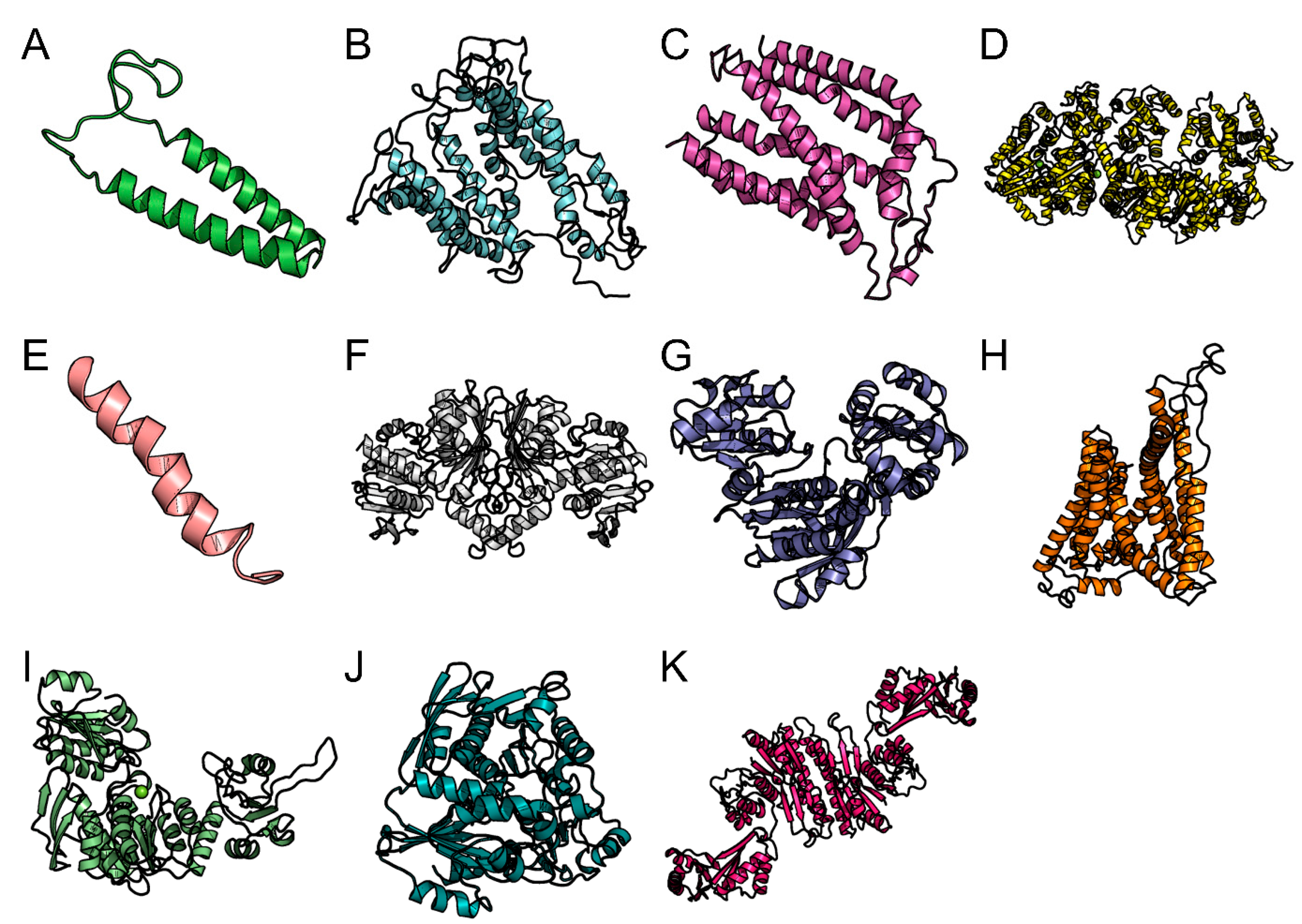
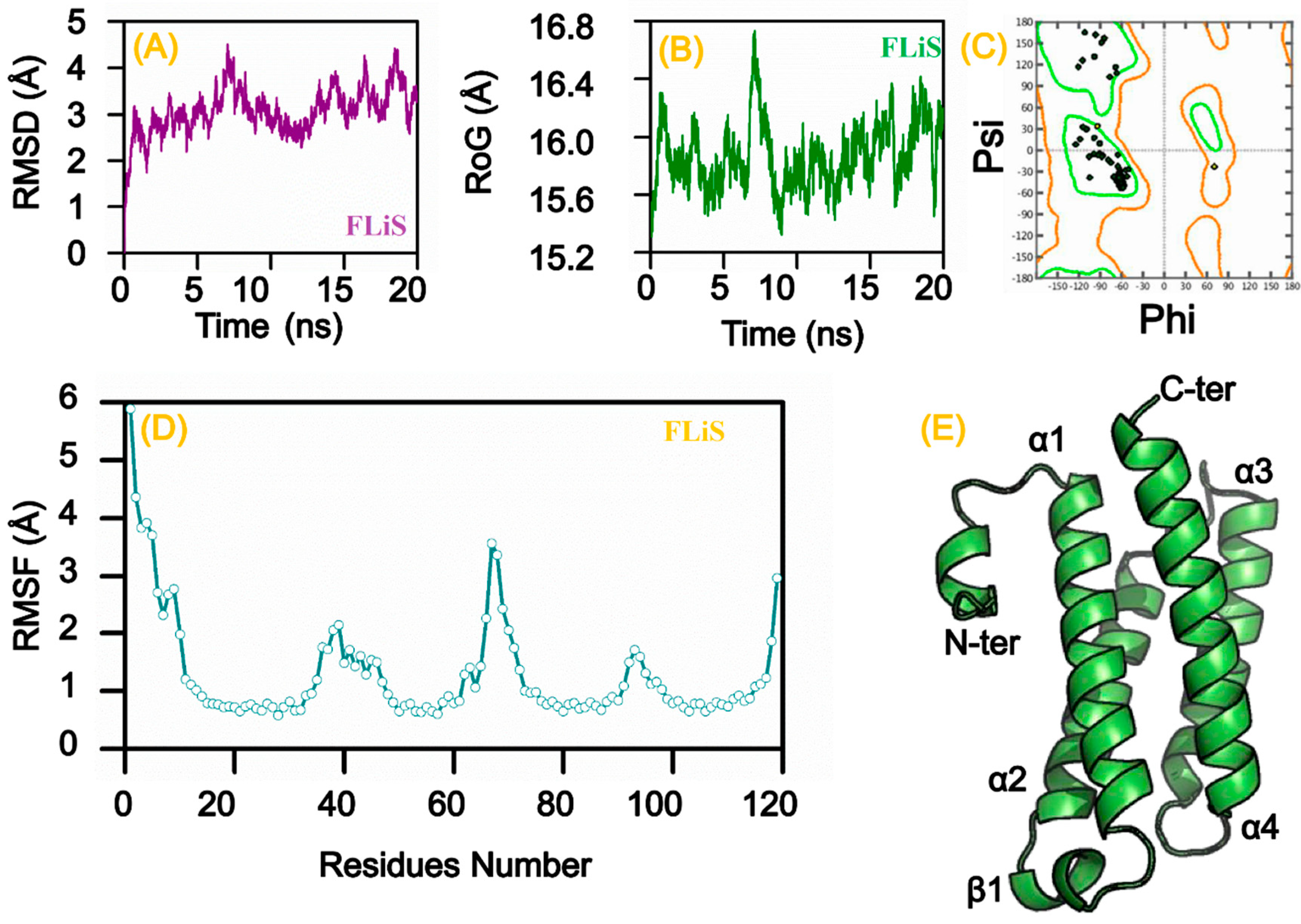
3.8. Homology Modeling and Molecular Dynamic Simulation
4. Conclusions and Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Merino, O.; Alberdi, P.; Pérez de la Lastra, J.M.; de la Fuente, J. Tick vaccines and the control of tick-borne pathogens. Front. Cell. Infect. Microbiol. 2013, 3, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogden, N. Changing geographic ranges of ticks and tick-borne pathogens: Drivers, mechanisms and consequences for pathogen diversity. Front. Cell. Infect. Microbiol. 2013, 3, 46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gondard, M.; Cabezas-Cruz, A.; Charles, R.A.; Vayssier-Taussat, M.; Albina, E.; Moutailler, S. Ticks and tick-borne pathogens of the Caribbean: Current understanding and future directions for more comprehensive surveillance. Front. Cell. Infect. Microbiol. 2017, 7, 490. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.; Mulenga, A.; Da Silva Vaz, I., Jr. Tick and Tick-Borne Pathogens: Molecular and Immune Targets for Control Strategies. Front. Physiol. 2020. [Google Scholar] [CrossRef]
- Labruna, M.B.; Jorge, R.S.; Sana, D.A.; Jácomo, A.T.A.; Kashivakura, C.K.; Furtado, M.M.; Ferro, C.; Perez, S.A.; Silveira, L.; Santos, T.S., Jr. Ticks (Acari: Ixodida) on wild carnivores in Brazil. Exp. Appl. Acarol. 2005, 36, 149–163. [Google Scholar] [CrossRef]
- de la Fuente, J.; Estrada-Pena, A.; Venzal, J.M.; Kocan, K.M.; Sonenshine, D.E. Overview: Ticks as vectors of pathogens that cause disease in humans and animals. Front. Biosci. 2008, 13, 6938–6946. [Google Scholar] [CrossRef] [Green Version]
- Socolovschi, C.; Mediannikov, O.; Raoult, D.; Parola, P. The relationship between spotted fever group Rickettsiae and Ixodid ticks. Vet. Res. 2009, 40, 34. [Google Scholar] [CrossRef] [Green Version]
- Estrada-Peña, A.; Ayllón, N.; De La Fuente, J. Impact of climate trends on tick-borne pathogen transmission. Front. Physiol. 2012, 3, 64. [Google Scholar] [CrossRef] [Green Version]
- Pujalte, G.G.; Chua, J.V. Tick-borne infections in the United States. Prim. Care 2013, 40, 619–635. [Google Scholar] [CrossRef]
- Bente, D.A.; Forrester, N.L.; Watts, D.M.; McAuley, A.J.; Whitehouse, C.A.; Bray, M. Crimean-Congo hemorrhagic fever: History, epidemiology, pathogenesis, clinical syndrome and genetic diversity. Antivir. Res. 2013, 100, 159–189. [Google Scholar] [CrossRef] [Green Version]
- Chmelař, J.; Kotál, J.; Kopeckỳ, J.; Pedra, J.H.; Kotsyfakis, M. All for one and one for all on the tick–host battlefield. Trends Parasitol. 2016, 32, 368–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Estrada-Peña, A. Climate, niche, ticks, and models: What they are and how we should interpret them. Parasitol. Res. 2008, 103, 87–95. [Google Scholar] [CrossRef]
- Dantas-Torres, F. Climate change, biodiversity, ticks and tick-borne diseases: The butterfly effect. Int. J. Parasitol. Parasites Wildl. 2015, 4, 452–461. [Google Scholar] [CrossRef] [Green Version]
- Hildebrandt, A.; Fritzsch, J.; Franke, J.; Sachse, S.; Dorn, W.; Straube, E. Co-circulation of emerging tick-borne pathogens in Middle Germany. Vector Borne Zoonot. Dis. 2011, 11, 533–537. [Google Scholar] [CrossRef] [PubMed]
- Parola, P.; Paddock, C.D.; Socolovschi, C.; Labruna, M.B.; Mediannikov, O.; Kernif, T.; Abdad, M.Y.; Stenos, J.; Bitam, I.; Fournier, P.-E. Update on tick-borne rickettsioses around the world: A geographic approach. Clin. Microbiol. Rev. 2013, 26, 657–702. [Google Scholar] [CrossRef] [Green Version]
- Otranto, D.; Dantas-Torres, F.; Giannelli, A.; Latrofa, M.S.; Cascio, A.; Cazzin, S.; Ravagnan, S.; Montarsi, F.; Zanzani, S.A.; Manfredi, M.T. Ticks infesting humans in Italy and associated pathogens. Parasites Vectors 2014, 7, 328. [Google Scholar] [CrossRef] [Green Version]
- Brites-Neto, J.; Duarte, K.M.R.; Martins, T.F. Tick-borne infections in human and animal population worldwide. Vet. World 2015, 8, 301. [Google Scholar] [CrossRef] [PubMed]
- Barh, D.; Tiwari, S.; Jain, N.; Ali, A.; Santos, A.R.; Misra, A.N.; Azevedo, V.; Kumar, A. In silico subtractive genomics for target identification in human bacterial pathogens. Drug Dev. Res. 2011, 72, 162–177. [Google Scholar] [CrossRef]
- Ali, A.; Tirloni, L.; Isezaki, M.; Seixas, A.; Konnai, S.; Ohashi, K.; da Junior, I.S.V.; Termignoni, C. Reprolysin metalloproteases from Ixodes persulcatus, Rhipicephalus sanguineus and Rhipicephalus microplus ticks. Exp. Appl. Acarol. 2014, 63, 559–578. [Google Scholar] [CrossRef] [PubMed]
- Hosen, M.I.; Tanmoy, A.M.; Mahbuba, D.-A.; Salma, U.; Nazim, M.; Islam, M.T.; Akhteruzzaman, S. Application of a subtractive genomics approach for in silico identification and characterization of novel drug targets in Mycobacterium tuberculosis F11. Interdiscip. Sci. 2014, 6, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Solanki, V.; Tiwari, V. Subtractive proteomics to identify novel drug targets and reverse vaccinology for the development of chimeric vaccine against Acinetobacter baumannii. Sci. Rep. 2018, 8, 1–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, N.-H.; Lee, J.-A.; Park, S.-Y.; Song, C.-S.; Choi, I.-S.; Lee, J.-B. A review of vaccine development and research for industry animals in Korea. Clin. Exp. Vaccine Res. 2012, 1, 18–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakharkar, K.R.; Sakharkar, M.K.; Chow, V.T. A novel genomics approach for the identification of drug targets in pathogens, with special reference to Pseudomonas aeruginosa. In Silico Biol. 2004, 4, 355–360. [Google Scholar] [PubMed]
- Singh, S.; Malik, B.K.; Sharma, D.K. Metabolic pathway analysis of S. pneumoniae: An in silico approach towards drug-design. J. Bioinform. Comput. Biol. 2007, 5, 135–153. [Google Scholar] [CrossRef]
- Anishetty, S.; Pulimi, M.; Pennathur, G. Potential drug targets in Mycobacterium tuberculosis through metabolic pathway analysis. Comput. Biol. Chem. 2005, 29, 368–378. [Google Scholar] [CrossRef]
- Asif, S.M.; Asad, A.; Faizan, A.; Anjali, M.S.; Arvind, A.; Neelesh, K.; Hirdesh, K.; Sanjay, K. Dataset of potential targets for Mycobacterium tuberculosis H37Rv through comparative genome analysis. Bioinformation 2009, 4, 245. [Google Scholar] [CrossRef] [Green Version]
- de la Fuente, J.; Contreras, M. Tick vaccines: Current status and future directions. Expert. Rev. Vaccines 2015, 14, 1367–1376. [Google Scholar] [CrossRef] [Green Version]
- de la Fuente, J.; Kopáček, P.; Lew-Tabor, A.; Maritz-Olivier, C. Strategies for new and improved vaccines against ticks and tick-borne diseases. Parasite Immunol. 2016, 38, 754–769. [Google Scholar] [CrossRef]
- Allsop, A.E. Bacterial genome sequencing and drug discovery. Curr. Opin. Biotechnol. 1998, 9, 637–642. [Google Scholar] [CrossRef]
- Stumm, G.; Russ, A.; Nehls, M. Deductive Genomics: A Functional Approach to Identify Innovative Drug Targets in the Post-Genome Era. Am. J. Pharm. 2002, 2, 263–271. [Google Scholar] [CrossRef]
- Barh, D.; Kumar, A. In silico identification of candidate drug and vaccine targets from various pathways in Neisseria gonorrhoeae. In Silico Biol. 2009, 9, 225–231. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.; Fernando Parizi, L.; Garcia Guizzo, M.; Tirloni, L.; Seixas, A.; da Silva Vaz, I., Jr.; Termignoni, C. Immunoprotective potential of a Rhipicephalus (Boophilus) microplus metalloprotease. Vet. Parasitol. 2015, 207, 107–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amineni, U.; Pradhan, D.; Marisetty, H. In silico identification of common putative drug targets in Leptospira interrogans. J. Chem. Biol. 2010, 3, 165–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar Jaiswal, A.; Tiwari, S.; Jamal, S.B.; Barh, D.; Azevedo, V.; Soares, S.C. An in silico identification of common putative vaccine candidates against Treponema pallidum: A reverse vaccinology and subtractive genomics based approach. Int. J. Mol. Sci. 2017, 18, 402. [Google Scholar] [CrossRef]
- Chan, J.N.; Nislow, C.; Emili, A. Recent advances and method development for drug target identification. Trends Pharmacol. Sci. 2010, 31, 82–88. [Google Scholar] [CrossRef]
- Madagi, S.; Patil, V.M.; Sadegh, S.; Singh, A.K.; Garwal, B.; Banerjee, A.; Talambedu, U.; Bhattacharjee, B. Identification of membrane associated drug targets in Borrelia burgdorferi ZS7-subtractive genomics approach. Bioinformation 2011, 6, 356. [Google Scholar] [CrossRef] [Green Version]
- Maurya, P.K.; Singh, S.; Mani, A. Comparative genomic analysis of Rickettsia rickettsii for identification of drug and vaccine targets: TolC as a proposed candidate for case study. Acta Trop. 2018, 182, 100–110. [Google Scholar] [CrossRef]
- Cornish-Bowden, A. The amino acid compositions of proteins are correlated with their molecular sizes. Biochem. J. 1983, 213, 271–274. [Google Scholar] [CrossRef] [Green Version]
- Sikic, K.; Carugo, O. Protein sequence redundancy reduction: Comparison of various method. Bioinformation 2010, 5, 234. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Niu, B.; Gao, Y.; Fu, L.; Li, W. CD-HIT Suite: A web server for clustering and comparing biological sequences. Bioinformatics 2010, 26, 680–682. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Zhang, R.; Lin, Y. DEG 5.0, a database of essential genes in both prokaryotes and eukaryotes. Nucleic Acids Res. 2009, 37, D455–D458. [Google Scholar] [CrossRef] [Green Version]
- Sharma, A.; Pan, A. Identification of potential drug targets in Yersinia pestis using metabolic pathway analysis: MurE ligase as a case study. Eur. J. Med. Chem. 2012, 57, 185–195. [Google Scholar] [CrossRef] [PubMed]
- Mondal, S.I.; Ferdous, S.; Jewel, N.A.; Akter, A.; Mahmud, Z.; Islam, M.M.; Afrin, T.; Karim, N. Identification of potential drug targets by subtractive genome analysis of Escherichia coli O157: H7: An in silico approach. Adv. Appl. Bioinform. Chem. AABC 2015, 8, 49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birhanu, B.T.; Lee, S.-J.; Park, N.-H.; Song, J.-B.; Park, S.-C. In silico analysis of putative drug and vaccine targets of the metabolic pathways of Actinobacillus pleuropneumoniae using a subtractive/comparative genomics approach. J. Vet. Sci. 2018, 19, 188–199. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Moriya, Y.; Itoh, M.; Okuda, S.; Yoshizawa, A.C.; Kanehisa, M. The metabolic pathways were examined through KAAS (KEGG automatic annotation server): An automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 2007, 35, W182–W185. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Goto, S.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. Data, information, knowledge and principle: Back to metabolism in KEGG. Nucleic Acids Res. 2014, 42, D199–D205. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016, 44, D457–D462. [Google Scholar] [CrossRef] [Green Version]
- Yu, C.-S.; Chen, Y.-C.; Lu, C.-H.; Hwang, J.-K. Prediction of protein subcellular localization. Proteins 2006, 64, 643–651. [Google Scholar] [CrossRef] [PubMed]
- Knox, C.; Law, V.; Jewison, T.; Liu, P.; Ly, S.; Frolkis, A.; Pon, A.; Banco, K.; Mak, C.; Neveu, V. DrugBank 3.0: A comprehensive resource for ‘omics’ research on drugs. Nucleic Acids Res. 2010, 39, D1035–D1041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Yang, J.; Yu, J.; Yao, Z.; Sun, L.; Shen, Y.; Jin, Q. VFDB: A reference database for bacterial virulence factors. Nucleic Acids Res. 2005, 33, D325–D328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larsen, J.E.P.; Lund, O.; Nielsen, M. Improved method for predicting linear B-cell epitopes. Immunome Res. 2006, 2, 2. [Google Scholar] [CrossRef] [Green Version]
- Kolaskar, A.S.; Tongaonkar, P.C. A semi-empirical method for prediction of antigenic determinants on protein antigens. FEBS Lett. 1990, 276, 172–174. [Google Scholar] [CrossRef] [Green Version]
- Emini, E.A.; Hughes, J.V.; Perlow, D.; Boger, J. Induction of hepatitis A virus-neutralizing antibody by a virus-specific synthetic peptide. J. Virol. 1985, 55, 836–839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karplus, P.A.; Schulz, G.E. Prediction of chain flexibility in proteins. Naturwissenschaften 1985, 72, 212–213. [Google Scholar] [CrossRef]
- Chou, P.Y.; Fasman, G.D. Empirical Predictions of Protein Conformation. Annu. Rev. Biochem. 1978, 47, 251–276. [Google Scholar] [CrossRef]
- Rini, J.M.; Schulze-Gahmen, U.; Wilson, I.A. Structural evidence for induced fit as a mechanism for antibody-antigen recognition. Science 1992, 255, 959–965. [Google Scholar] [CrossRef]
- Gasteiger, E.; Gattiker, A.; Hoogland, C.; Ivanyi, I.; Appel, R.D.; Bairoch, A. ExPASy: The proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res. 2003, 31, 3784–3788. [Google Scholar] [CrossRef] [Green Version]
- Wu, G.D.; Bushmanc, F.D.; Lewis, J.D. Diet, the human gut microbiota, and IBD. Anaerobe 2013, 24, 117–120. [Google Scholar] [CrossRef] [PubMed]
- Geourjon, C.; Deleage, G. SOPMA: Significant improvements in protein secondary structure prediction by consensus prediction from multiple alignments. Bioinformatics 1995, 11, 681–684. [Google Scholar] [CrossRef] [PubMed]
- Hall, T.A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. In Proceedings of the Nucleic Acids Symposium Series; Information Retrieval Ltd.: London, UK, 1999; Volume 41, pp. 95–98. [Google Scholar]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis Version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Roy, A.; Yang, J.; Zhang, Y. COFACTOR: An accurate comparative algorithm for structure-based protein function annotation. Nucleic Acids Res. 2012, 40, W471–W477. [Google Scholar] [CrossRef] [Green Version]
- Salomon-Ferrer, R.; Götz, A.W.; Poole, D.; Le Grand, S.; Walker, R.C. Routine microsecond molecular dynamics simulations with AMBER on GPUs. 2. Explicit solvent particle mesh Ewald. J. Chem. 2013, 9, 3878–3888. [Google Scholar] [CrossRef]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J.C. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef] [Green Version]
- Sharma, V.; Gupta, P.; Dixit, A. In silico identification of putative drug targets from different metabolic pathways of Aeromonas hydrophila. In Silico Biol. 2008, 8, 331–338. [Google Scholar]
- Dutta, A.; Singh, S.K.; Ghosh, P.; Mukherjee, R.; Mitter, S.; Bandyopadhyay, D. In silico identification of potential therapeutic targets in the human pathogen Helicobacter pylori. In Silico Biol. 2006, 6, 43–47. [Google Scholar]
- Georrge, J.J.; Umrania, V. In silico identification of putative drug targets in Klebsiella pneumonia MGH78578. Indian J. Biotechnol. 2011, 10, 432–439. [Google Scholar]
- H Reddy, P.; P Reddy, T. Mitochondria as a therapeutic target for aging and neurodegenerative diseases. Curr. Alzheimer Res. 2011, 8, 393–409. [Google Scholar] [CrossRef]
- Shoukat, K.; Rasheed, N.; Sajid, M. Subtractive genome analysis for in silico identification and characterization of novel drug targets in C. trachomatis STRAIN D/UW-3/Cx. Int. J. Curr. Res. 2012, 4, 6. [Google Scholar]
- Sarkar, M.; Maganti, L.; Ghoshal, N.; Dutta, C. In silico quest for putative drug targets in Helicobacter pylori HPAG1: Molecular modeling of candidate enzymes from lipopolysaccharide biosynthesis pathway. J. Mol. Model. 2012, 18, 1855–1866. [Google Scholar] [CrossRef] [PubMed]
- Rahman, H.; King, R.M.; Shewell, L.K.; Semchenko, E.A.; Hartley-Tassell, L.E.; Wilson, J.C.; Day, C.J.; Korolik, V. Characterisation of a multi-ligand binding chemoreceptor CcmL (Tlp3) of Campylobacter jejuni. PLoS Pathog. 2014, 10, e1003822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasan, M.A.; Khan, M.A.; Sharmin, T.; Mazumder, M.H.H.; Chowdhury, A.S. Identification of putative drug targets in Vancomycin-resistant Staphylococcus aureus (VRSA) using computer aided protein data analysis. Gene 2016, 575, 132–143. [Google Scholar] [CrossRef] [PubMed]
- Hobohm, U.; Scharf, M.; Schneider, R.; Sander, C. Selection of representative protein data sets. Protein Sci. 1992, 1, 409–417. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Xiao, J.; Pan, L.; Yang, M.; Zhang, G.; Jin, S.; Yu, J. A systematic survey of mini-proteins in bacteria and archaea. PLoS ONE 2008, 3, e4027. [Google Scholar] [CrossRef] [Green Version]
- Gupta, S.K.; Sarita, S.; Gupta, M.K.; Pant, K.K.; Seth, P.K. Definition of potential targets in Mycoplasma pneumoniae through subtractive genome analysis. J. Antivir. Antiretrovir. 2010, 2, 38–41. [Google Scholar]
- Haag, N.L.; Velk, K.K.; Wu, C. In silico identification of drug targets in methicillin/multidrug-resistant Staphylococcus aureus. Int. J. Adv. Life Sci. 2012, 4, 21–32. [Google Scholar]
- Uddin, R.; Saeed, K. Identification and characterization of potential drug targets by subtractive genome analyses of methicillin resistant Staphylococcus aureus. Comput. Biol. Chem. 2014, 48, 55–63. [Google Scholar] [CrossRef]
- Zhang, C.; Xia, Y. Identification of genes differentially expressed in vivo by Metarhizium anisopliae in the hemolymph of Locusta migratoria using suppression-subtractive hybridization. Curr. Genet. 2009, 55, 399–407. [Google Scholar] [CrossRef] [PubMed]
- Restrepo-Montoya, D.; Vizcaíno, C.; Niño, L.F.; Ocampo, M.; Patarroyo, M.E.; Patarroyo, M.A. Validating subcellular localization prediction tools with mycobacterial proteins. BMC Bioinform. 2009, 10, 134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duffield, M.; Cooper, I.; McAlister, E.; Bayliss, M.; Ford, D.; Oyston, P. Predicting conserved essential genes in bacteria: In silico identification of putative drug targets. Mol. Biosyst. 2010, 6, 2482–2489. [Google Scholar] [CrossRef] [PubMed]
- Novick, R.P.; Geisinger, E. Quorum sensing in staphylococci. Annu. Rev. Genet. 2008, 42, 541–564. [Google Scholar] [CrossRef]
- Ng, W.-L.; Bassler, B.L. Bacterial quorum-sensing network architectures. Annu. Rev. Genet. 2009, 43, 197–222. [Google Scholar] [CrossRef] [Green Version]
- Williams, P.; Cámara, M. Quorum sensing and environmental adaptation in Pseudomonas aeruginosa: A tale of regulatory networks and multifunctional signal molecules. Cur. Opin. Microbiol. 2009, 12, 182–191. [Google Scholar] [CrossRef]
- Barrett, J.F.; Hoch, J.A. Two-component signal transduction as a target for microbial anti-infective therapy. Antimicrob. Agents Chemother. 1998, 42, 1529–1536. [Google Scholar] [CrossRef] [Green Version]
- Cai, X.-H.; Zhang, Q.; Shi, S.-Y.; Ding, D.-F. Searching for potential drug targets in two-component and phosphorelay signal-transduction systems using three-dimensional cluster analysis. Acta Biochim. Biophys. Sin. (Shanghai) 2005, 37, 293–302. [Google Scholar] [CrossRef] [Green Version]
- Vollmer, W.; Bertsche, U. Murein (peptidoglycan) structure, architecture and biosynthesis in Escherichia coli. Biochim. Biophys. Acta 2008, 1778, 1714–1734. [Google Scholar] [CrossRef] [Green Version]
- de Pedro, M.A.; Cava, F. Structural constraints and dynamics of bacterial cell wall architecture. Front. Microbiol. 2015, 6, 449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basavannacharya, C.; Robertson, G.; Munshi, T.; Keep, N.H.; Bhakta, S. ATP-dependent MurE ligase in Mycobacterium tuberculosis: Biochemical and structural characterisation. Tuberculosis 2010, 90, 16–24. [Google Scholar] [CrossRef]
- Natale, P.; Brüser, T.; Driessen, A.J. Sec-and Tat-mediated protein secretion across the bacterial cytoplasmic membrane—Distinct translocases and mechanisms. Biochim. Biophys. Acta 2008, 1778, 1735–1756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ochsner, U.A.; Snyder, A.; Vasil, A.I.; Vasil, M.L. Effects of the twin-arginine translocase on secretion of virulence factors, stress response, and pathogenesis. Proc. Natl. Acad. Sci. USA 2002, 99, 8312–8317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pradel, N.; Ye, C.; Livrelli, V.; Xu, J.; Joly, B.; Wu, L.-F. Contribution of the twin arginine translocation system to the virulence of enterohemorrhagic Escherichia coli O157: H7. Infect. Immun. 2003, 71, 4908–4916. [Google Scholar] [CrossRef] [Green Version]
- Lavander, M.; Ericsson, S.K.; Bröms, J.E.; Forsberg, A. The twin arginine translocation system is essential for virulence of Yersinia pseudotuberculosis. Infect. Immun. 2006, 74, 1768–1776. [Google Scholar] [CrossRef] [Green Version]
- Young, G.M.; Schmiel, D.H.; Miller, V.L. A new pathway for the secretion of virulence factors by bacteria: The flagellar export apparatus functions as a protein-secretion system. Proc. Natl. Acad. Sci. USA 1999, 96, 6456–6461. [Google Scholar] [CrossRef] [Green Version]
- Mühlen, S.; Dersch, P. Anti-virulence strategies to target bacterial infections. In How to Overcome the Antibiotic Crisis; Springer: Berlin/Heidelberg, Germany, 2015; pp. 147–183. [Google Scholar]
- Xing, X.; Bi, S.; Fan, X.; Jin, M.; Liu, W.; Wang, B. Intranasal Vaccination with Multiple Virulence Factors Promotes Mucosal Clearance of Streptococcus suis Across Serotypes and Protects Against Meningitis in Mice. J. Infect. Dis. 2019, 220, 1679–1687. [Google Scholar] [CrossRef]
- Guarner, F.; Malagelada, J.-R. Gut flora in health and disease. Lancet 2003, 361, 512–519. [Google Scholar] [CrossRef]
- Sears, C.L. A dynamic partnership: Celebrating our gut flora. Anaerobe 2005, 11, 247–251. [Google Scholar] [CrossRef]
- Senes, A.; Ubarretxena-Belandia, I.; Engelman, D.M. The Cα—H⋯ O hydrogen bond: A determinant of stability and specificity in transmembrane helix interactions. Proc. Natl. Acad. Sci. USA 2001, 98, 9056–9061. [Google Scholar] [CrossRef] [Green Version]
- Adamian, L.; Liang, J. Interhelical hydrogen bonds and spatial motifs in membrane proteins: Polar clamps and serine zippers. Proteins 2002, 47, 209–218. [Google Scholar] [CrossRef]
- Curran, A.R.; Engelman, D.M. Sequence motifs, polar interactions and conformational changes in helical membrane proteins. Curr. Opin. Struct. Biol. 2003, 13, 412–417. [Google Scholar] [CrossRef]
- Xiong, J. Essential Bioinformatics; Cambridge University Press: Cambridge, UK, 2006. [Google Scholar]
- Chu, K.H.; Wong, S.H.; Leung, P.S. Tropomyosin is the major mollusk allergen: Reverse transcriptase polymerase chain reaction, expression and IgE reactivity. Mar. Biotechnol. 2000, 2, 499–509. [Google Scholar] [CrossRef] [PubMed]
- Nisbet, A.J.; Huntley, J.F. Progress and opportunities in the development of vaccines against mites, fleas and myiasis-causing flies of veterinary importance. Parasite Immunol. 2006, 28, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Rappuoli, R. Reverse vaccinology. Curr. Opin. Struct. Biol. 2000, 3, 445–450. [Google Scholar] [CrossRef]
- Bhavsar, A.P.; Guttman, J.A.; Finlay, B.B. Manipulation of host-cell pathways by bacterial pathogens. Nature 2007, 449, 827–834. [Google Scholar] [CrossRef]
- Stavrinides, J.; McCann, H.C.; Guttman, D.S. Host–pathogen interplay and the evolution of bacterial effectors. Cell. Microbiol. 2007, 10, 285–292. [Google Scholar] [CrossRef]
- Simeone, R.; Bottai, D.; Brosch, R. ESX/type VII secretion systems and their role in host–pathogen interaction. Curr. Opin. Struct. Biol. 2009, 12, 4–10. [Google Scholar] [CrossRef]
- Parizi, L.F.; Ali, A.; Tirloni, L.; Oldiges, D.P.; Sabadin, G.A.; Coutinho, M.L.; Seixas, A.; Logullo, C.; Termignoni, C.; Da Silva Vaz, I., Jr. Peptidase inhibitors in tick physiology. Med. Vet. Entomol. 2018, 32, 129–144. [Google Scholar] [CrossRef] [Green Version]
- Ali, A.; Khan, S.; Ali, I.; Karim, S.; da Silva Vaz, I., Jr.; Termignoni, C. Probing the functional role of tick metalloproteases. Physiol. Entomol. 2015, 40, 177–188. [Google Scholar] [CrossRef] [Green Version]
- Durrant, J.D.; McCammon, J.A. Molecular dynamics simulations and drug discovery. BMC Biol. 2011, 9, 71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.; Kim, M.I.; Park, J.; Jeon, B.-Y.; Yoon, S.; Hong, M. Crystal structure of the flagellar chaperone FliS from Bacillus cereus and an invariant proline critical for FliS dimerization and flagellin recognition. Biochem. Biophys. Res. Commun. 2017, 487, 381–387. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Step Number | Steps | Borrelia burgdorferi B31 | Ehrlichia chaffeensis Arkansas | Rickettsia rickettsii Strain “Sheila Smith” | Anaplasma phagocytophilum HZ | Francisella tularensis SCHU S4 |
|---|---|---|---|---|---|---|
| Host | ||||||
| Homo sapiens | Homo sapiens | Bos taurus | Homo sapiens | Homo sapiens | ||
| 1. | Total proteome | 1391 | 889 | 1246 | 1048 | 1556 |
| 2. | Duplicates (>60% identical) in CD-HIT a | 1181 | 846 | 830 | 712 | 1295 |
| 3. | Non-homologs | 765 | 793 | 409 | 453 | 788 |
| 4. | Essential proteins in DEG b | 34 | 113 | 76 | 105 | 185 |
| 5. | Unique metabolic pathway KEGG c | 13 | 8 | 14 | 6 | 25 |
| 6. | Essential proteins involved KEGG and KAAS d | 12 | 12 | 5 | 8 | 24 |
| 7. | Druggability with cut-off E-value 10−5 | 4 | 3 | 1 | 3 | 4 |
| 8. * | Gut metagenomics with cut-off E-value 10−5 | 2 | 2 | - | 3 | 4 |
| Name of Protein | NCBI Protein ID | Pathways | KO Number | KEGG ID | Biological Process | Molecular Function | Drug Bank ID | Drug Name | Chemical Formulae | Drug Group | Virulent | Localization | Antigenicity Score | Antigenicity | Allergenicity |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Preprotein translocase subunit SecY A. phagocytophilum | WP_011450434.1 | Quorum-sensing, A. phagocytophilum HZ | K03076 | aph02024 | Protein transport, translocation | None predicted evidence | DB06292 DB08907 DB09038 | Dapagliflozin Canagliflozin Empagliflozin | C21H25ClO6 C24H25FO5S C23H27ClO7 | Approved Approved Approved | Yes | Inner membrane | 0.4083 | Antigen | Non-allergen |
| Chromosomal replication initiator protein DnaA. A. phagocytophilum | WP_011450597.1 | Two-component system, A. phagocytophilum HZ | K02313 | aph02020 | DNA replication | DNA-binding | DB00173 | Adenine | C5H5N5 | Approved, nutraceutical | Yes | Cytoplasmic | 0.3982 | Non-Antigen | Non-allergen |
| Aspartate-semialdehyde dehydrogenase A. phagocytophilum | WP_011451356.1 | Lysine biosynthesis, A. phagocytophilum HZ | K00133 | aph00300 | Cellular amino acid biosynthetic process | Oxidoreductase activity | DB00181 DB00996 DB02530 | Baclofen Gabapentin gamma-Aminobutyric acid | C10H12ClNO2 C9H17NO2 C4H9NO2 | Approved Approved, investigational Approved, investigational | Yes | Outer membrane | 0.3987 | Non-Antigen | Non-allergen |
| 1UDP-n-acetylmuramoyl-tripeptide--D-alanyl-D-alanine ligas B. burgdorferi B31 | NP_212438.1 | Flagellar assembly, B. burgdorferi B31 | K01929 | bbu02040 | Peptidoglycan biosynthetic process | ATP binding | DB04272 | Citric acid | C6H8O7 | Approved, nutraceutical, Vet approved | Yes | Cytoplasmic | 0.2503 | Non-Antigen | Non-allergen |
| FliS B. burgdorferi B31 | NP_212684.1 | Flagellar assembly, B. burgdorferi B31 | K02422 | bbu02040 | Bacterial-type flagellum assembly | None predicted evidence | DB00120 | l-phenylalanine | C9H11NO2 | Approved, Investigational, nutraceutical | Yes | Extracellular | 0.4200 | Antigen | Non-allergen |
| Twin-arginine translocase subunit TatC Ehrlichia chaffeensis | WP_011452677.1 | Bacterial secretion system, Ehrlichia chaffeensis Arkansas | K03118 | ech03070 | Protein transport by the Tat complex | Protein transmembrane transporter activity | DB01277 DB13173 | Mecasermin cerliponase alfa | C331H518N94O101S7 Not Available | Approved, investigational Approved, investigational | No | Inner membrane | 0.8915 | Antigen | Non-allergen |
| Aspartate kinase E. chaffeensis | WP_011452940.1 | Monobactam biosynthesis, E. chaffeensis Arkansas | K00928 | ech00261 | Lysine biosynthetic process via diaminopimelate | Aspartate kinase activity | DB11638 | Artenimol | C15H24O5 | Experimental, investigational | Yes | Cytoplasmic | 0.5049 | Antigen | Non-allergen |
| Preprotein translocase subunit SecG F. tularensis SCHU S4 | YP_169156.1 | Quorum sensing, F. tularensis. S4 | K03075 | ftu02024 | Protein secretion | Protein translocase activity | DB00887 DB01016 DB01050 DB04941 DB08820 DB09213 DB09280 | Bumetanide Glyburide Ibuprofen Crofelemer Ivacaftor Dexibuprofen Lumacaftor | C17H20N2O5S C23H28ClN3O5S C13H18O2 Not Available C24H28N2O3 C13H18O2 C24H18F2N2O5 | Approved Approved Approved Approved Approved, investigational Approved, investigational Approved | Yes | Inner membrane | 0.7645 | Antigen | Non-allergen |
| UDP-N-acetylmuramate-l-alanine ligase F. tularensis SCHU S4 | YP_169292.1 | Peptidoglycan biosynthesis, F. tularensis SCHU S4 | K01924 | ftu00550 | Murein biosynthesis | Ligase activity | DB00157 DB09092 | NADH Xanthinol | C21H29N7O14P2 C13H21N5O4 | Approved, nutraceutical Approved, withdrawn | Yes | Cytoplasmic | 0.4349 | Antigen | Non-allergen |
| Preprotein translocase subunit SecY F. tularensis SCHU S4 | YP_169394.1 | Quorum sensing, F. tularensis SCHU S4 | K03076 | ftu02024 | Protein transport | None predicted evidence | DB00313; DB05541 | Valproic acid brivaracetam | C8H16O2 C11H20N2O2 | Approved, investigational Approved, investigational | Yes | Cytoplasmic | 0.6388 | Antigen | Non-allergen |
| UDP-N-acetylmuramoylalanyl-d-glutamate-2,6-diaminopimelate ligase F. tularensis SCHU S4 | YP_169464.1 | Peptidoglycan biosynthesis, F. tularensis SCHU S4 | K01928 | ftu00550 | Murein biosynthesis | Ligase activity | DB11638 | Artenimol | C15H24O5 | Experimental, investigational | Yes | Inner membrane | 0.3851 | Non-antigen | Non-allergen |
| Cytochrome d ubiquinol oxidase subunit II- R. rickettsii Sheila Smith | WP_012150506.1 | Two-component system, R. rickettsii “Sheila Smith” | K00426 | rri02020 | Oxidation-reduction process | None predicted evidence | DB01221 | Ketamine | C13H16ClNO | Approved, vet approved | Yes | Inner membrane | 0.6850 | Antigen | Non-allergen |
| SOPMA a | Flagellar Protein (FLiS, B. burgdorferi) | UDP-N-acetylmuramoyl-tripeptide-d-alanyl-d-alanine ligase B. burgdorferi | Preprotein Translocase Subunit SecY (A. phagocytophilum) | Chromosomal Replication Initiator Protein DnaA (A. phagocytophilum) | Aspartate-Semialdehyde Dehydrogenase (A. phagocytophilum) | Twin-Arginine Translocase Subunit TatC (E. chaffeensis) | Aspartate Kinase (E. chaffeensis) | Cytochrome d Ubiquinol Oxidase Subunit II (R. rickettsii) | Preprotein Translocase Subunit SecG (F. tularensis) | Preprotein Translocase Subunit SecY (F. tularensis) | UDP-N-acetylmuramate-l-alanine ligase (F. tularensis) | UDP-N-acetylmuramoylalanyl-d-glutamate-2,6-diaminopimelate ligase (F. tularensis) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| α-helix | 59.31% | 38.36% | 47.24% | 54.03% | 36.80% | 55.42% | 37.65% | 50.74% | 33.33% | 47.62% | 37.69% | 42.17% |
| 310 helices | 0.00% | 0.00% | 0.00% | 0.00% | 0.00% | 0.00% | 0.00% | 0.00% | 0.00% | 0.00% | 0.00% | 0.00% |
| Pi helix | 0.00% | 0.00% | 0.00% | 0.00% | 0.00% | 0.00% | 0.00% | 0.00% | 0.00% | 0.00% | 0.00% | 0.00% |
| Beta bridge | 0.00% | 0.00% | 0.00% | 0.00% | 0.00% | 0.00% | 0.00% | 0.00% | 0.00% | 0.00% | 0.00% | 0.00% |
| Extended strand | 13.10% | 22.84% | 18.66% | 12.64% | 22.26% | 15.66% | 22.54% | 15.34% | 16.24% | 17.91% | 21.95% | 18.79% |
| β-turn | 3.45% | 7.11% | 8.06% | 2.83% | 6.53% | 4.42% | 7.19% | 4.72% | 2.56% | 8.62% | 7.98% | 5.22% |
| Bend region | 0.00% | 0.00% | 0.00% | 0.00% | 0.00% | 0.00% | 0.00% | 0.00% | 0.00% | 0.00% | 0.00% | 0.00% |
| Random coil | 24.14% | 31.68% | 26.04% | 30.50% | 34.42% | 24.50% | 32.61% | 29.20% | 47.86% | 25.85% | 32.37% | 33.82% |
| Ambiguous states | 0.00% | 0.00% | 0.00% | 0.00% | 0.00% | 0.00% | 0.00% | 0.00% | 0.00% | 0.00% | 0.00% | 0.00% |
| Other states | 0.00% | 0.00% | 0.00% | 0.00% | 0.00% | 0.00% | 0.00% | 0.00% | 0.00% | 0.00% | 0.00% | 0.00% |
| Protein Name | Cscore LB | PDB Hit | TM-Score | RMSD a | IDEN a | Cov. | BS-Score | Lig. Name | Predicted Binding Site Residues |
|---|---|---|---|---|---|---|---|---|---|
| NP_2126841 | 0.30 | 3asoC | 0.697 | 2.53 | 0.034 | 0.891 | 0.94 | CDL | 24, 35, 112, 116 |
| 0.10 | 3ag4C | 0.696 | 2.54 | 0.034 | 0.891 | 1.01 | CDL | 20, 24, 28, 31, 119 | |
| 0.08 | 1ory0 | 0.952 | 0.95 | 0.336 | 0.975 | 1.47 | III | 8, 10, 13, 14, 19, 22, 24, 25, 28, 57, 60, 61, 64, 65, 68, 69, 70, 73, 74, 76, 77, 80, 83, 84, 85, 87, 106, 107, 109, 110, 113, 114, 117, 118, 120, 121 | |
| 0.02 | 2eikC | 0.697 | 2.53 | 0.034 | 0.891 | 0.84 | CD | 26, 30, 108, 112 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ali, A.; Ahmad, S.; Wadood, A.; Rehman, A.U.; Zahid, H.; Qayash Khan, M.; Nawab, J.; Rahman, Z.U.; Alouffi, A.S. Modeling Novel Putative Drugs and Vaccine Candidates against Tick-Borne Pathogens: A Subtractive Proteomics Approach. Vet. Sci. 2020, 7, 129. https://doi.org/10.3390/vetsci7030129
Ali A, Ahmad S, Wadood A, Rehman AU, Zahid H, Qayash Khan M, Nawab J, Rahman ZU, Alouffi AS. Modeling Novel Putative Drugs and Vaccine Candidates against Tick-Borne Pathogens: A Subtractive Proteomics Approach. Veterinary Sciences. 2020; 7(3):129. https://doi.org/10.3390/vetsci7030129
Chicago/Turabian StyleAli, Abid, Shabir Ahmad, Abdul Wadood, Ashfaq U. Rehman, Hafsa Zahid, Muhammad Qayash Khan, Javed Nawab, Zia Ur Rahman, and Abdulaziz S. Alouffi. 2020. "Modeling Novel Putative Drugs and Vaccine Candidates against Tick-Borne Pathogens: A Subtractive Proteomics Approach" Veterinary Sciences 7, no. 3: 129. https://doi.org/10.3390/vetsci7030129