Measurement and Numerical Modeling of Cell-Free Protein Synthesis: Combinatorial Block-Variants of the PURE System


 ,
,  and
and 
Abstract
:
1. Summary
2. Data Description
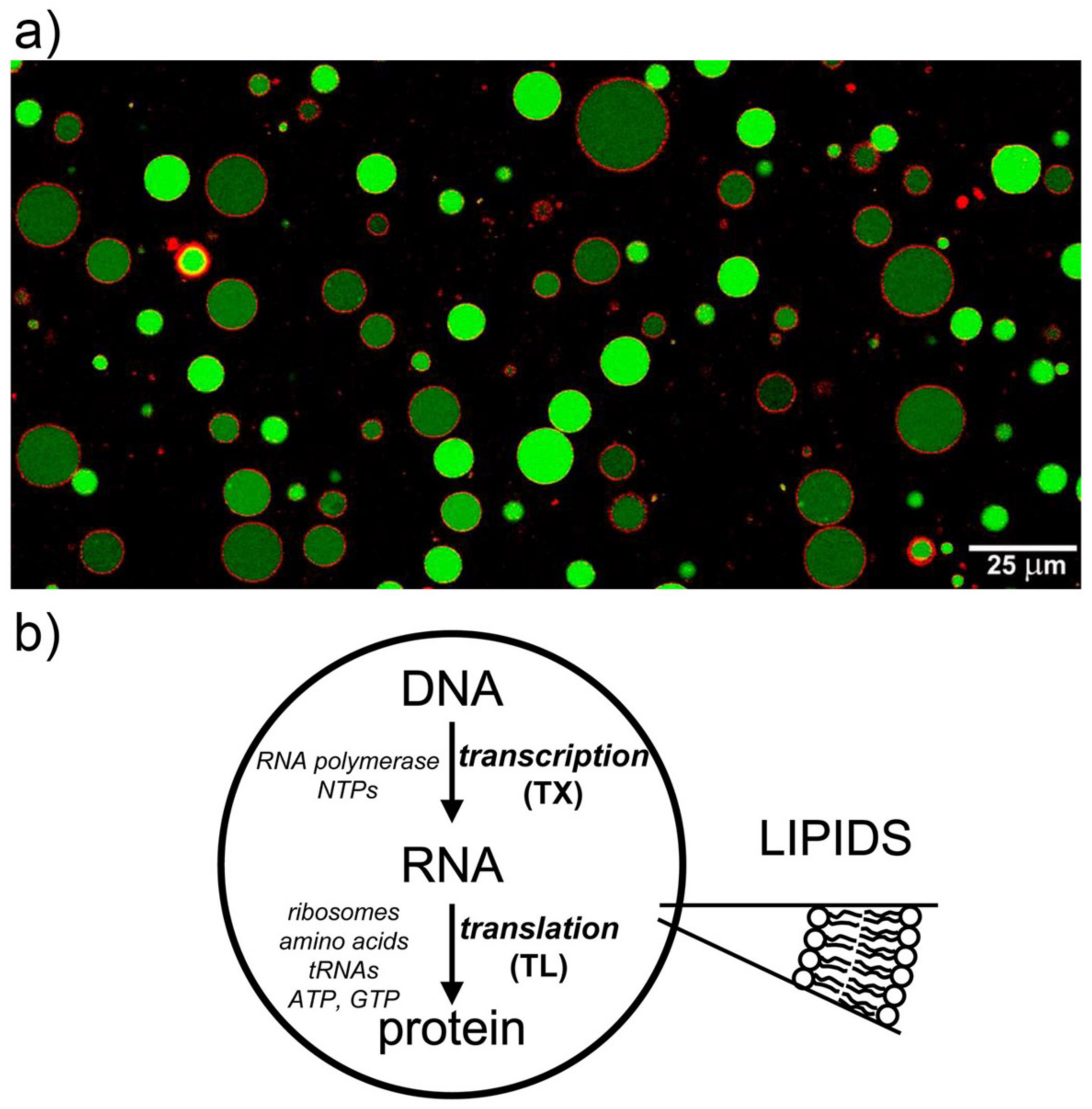
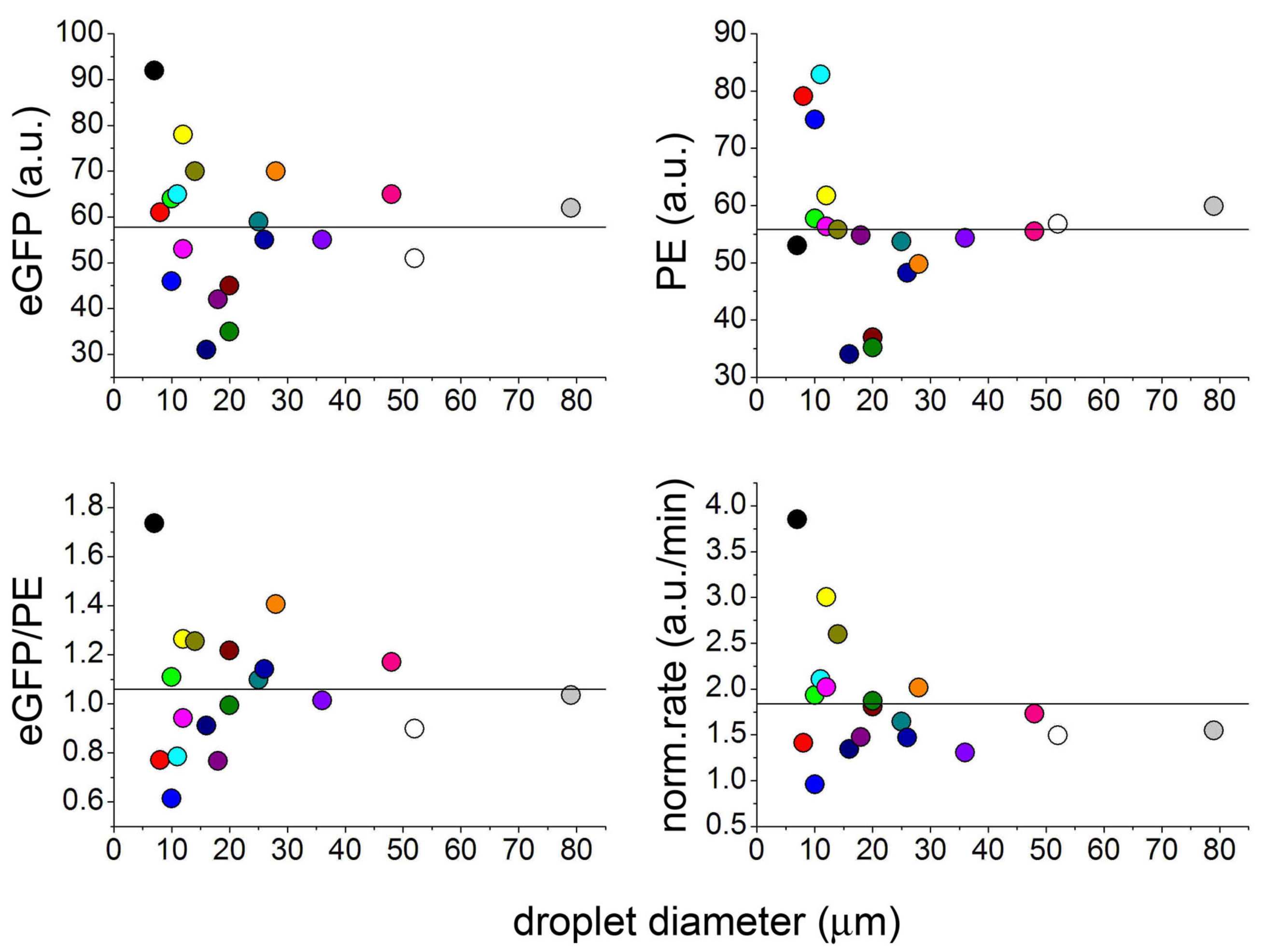
2.1. Protein Synthesis by TX–TL Reactions in Water-in-Oil Droplets Displays High Between-Droplet Diversity
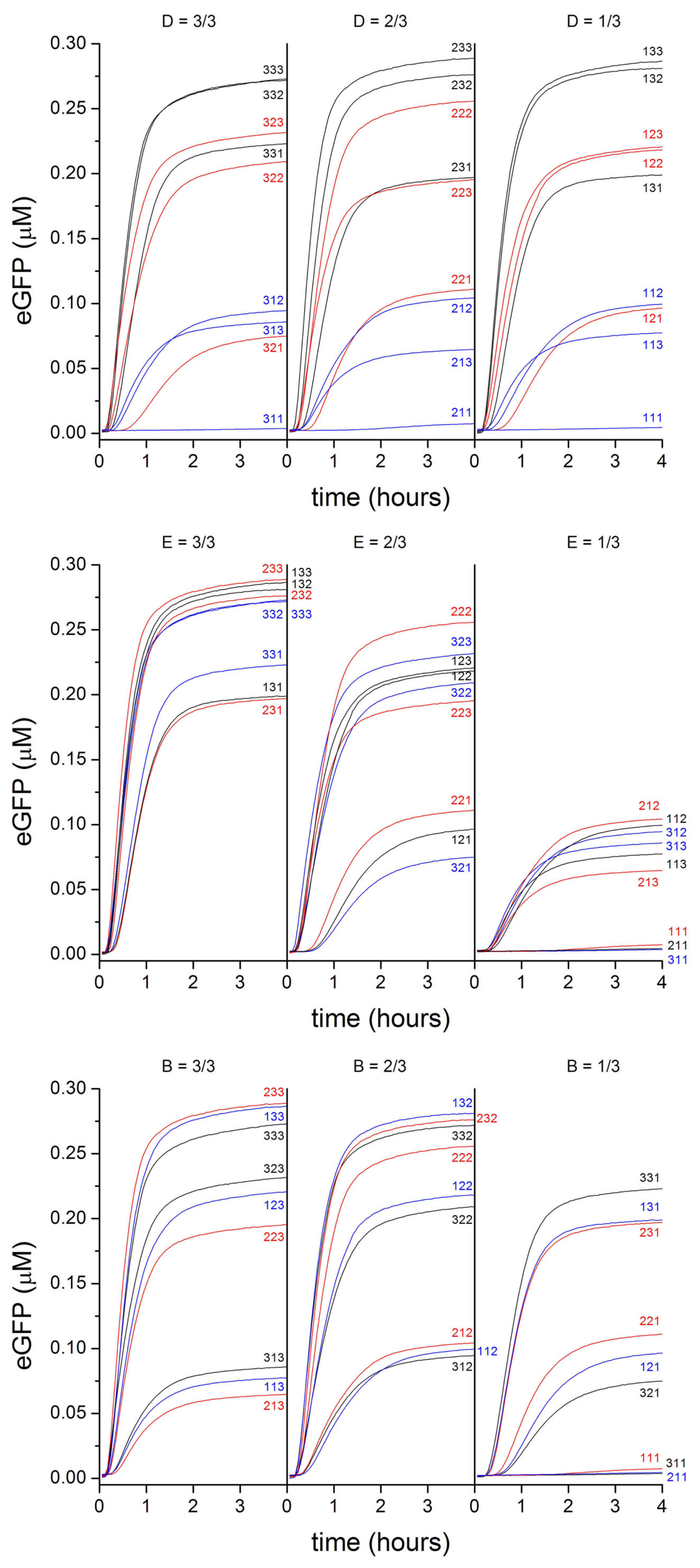
2.2. PURE Systems Block-Variants
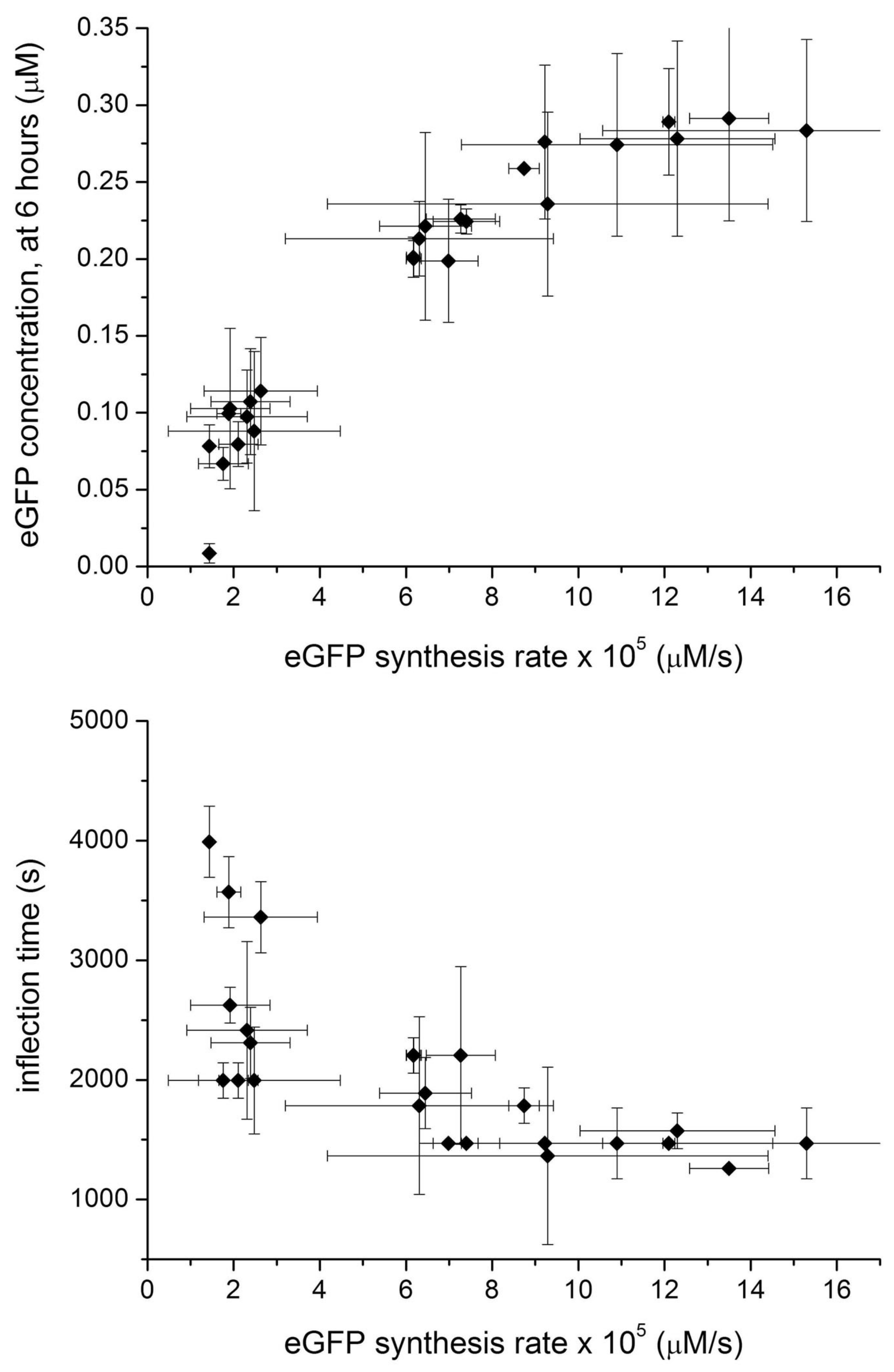
2.3. Commenting the Results
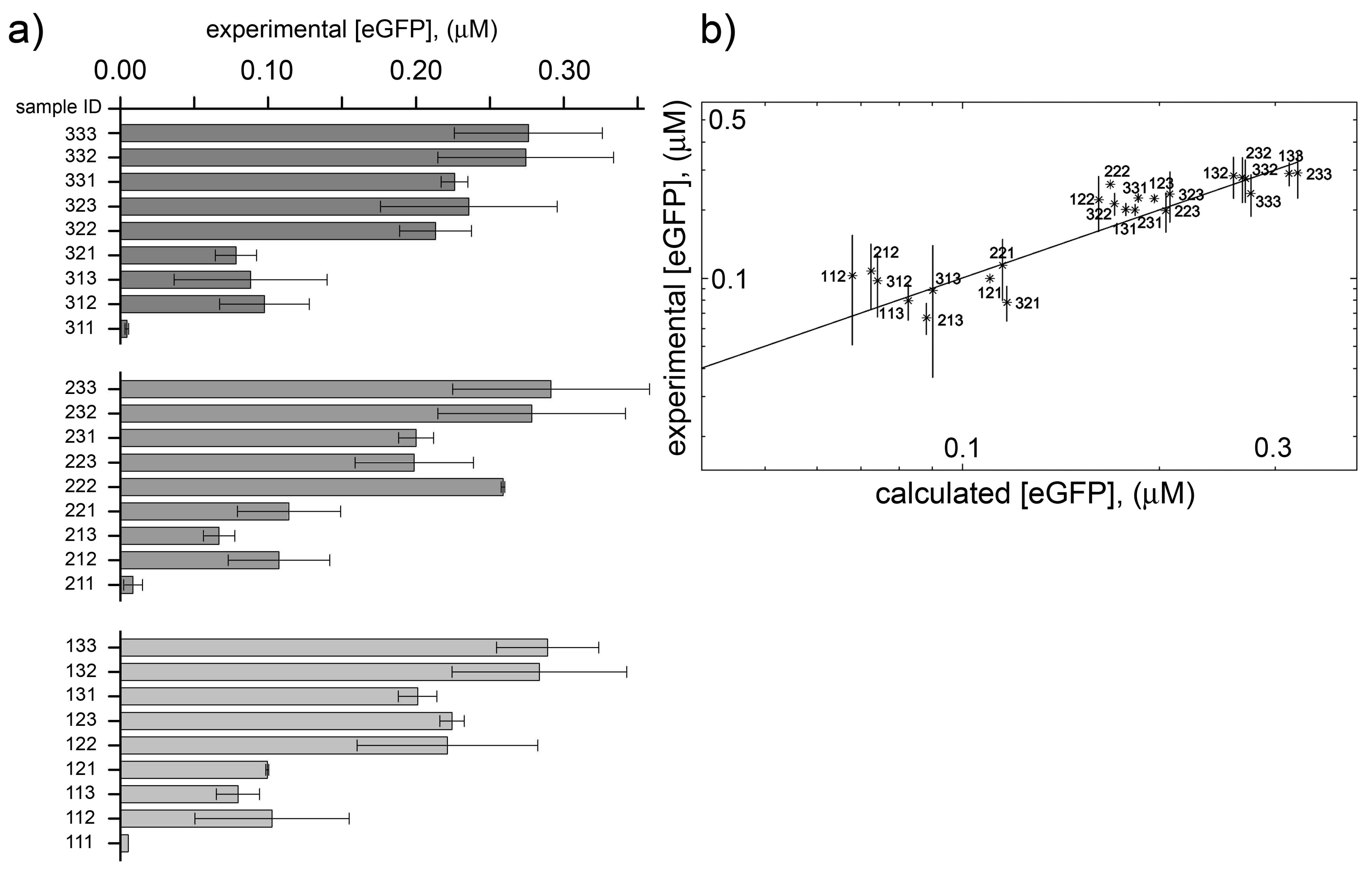
2.4. Analysis and Numerical Modeling
2.5. Comparison with Previously Published Dataset
- Block-variants with high production (>67% with respect to the maximal value): in the previous study [32], these are—typically —combination of high concentration of initiaton and elongation TL factors, as well as of ribosomes, generally low concentration of aminoacyl tRNA synthetase, and high concentration of tRNAs and energy recycling enzymes. In this study, the best combinations have medium-high concentration of all macromolecules (enzymes and tRNAs), and buffer/salts.
- Block-variants with small production (<33% with respect to the maximal value): in the previous study [32], these are—typically—combinations of high concentration of buffer, low concentration of tRNAs, medium concentration of ribosomes, generally low concentration of aminoacyl tRNA synthetase, and high concentration of energy recycling enzymes. In this study, the worst combinations have low concentration of all enzymes (and in minor way, tRNAs, buffer/salts).
3. Methods
3.1. Materials
3.2. TX–TL Reaction Inside Soybean Lecithin w/o Droplet
3.3. PURE System Block-Variants
3.4. PURE System Numerical Model
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Luisi, P.L. Toward the engineering of minimal living cells. Anat. Rec. 2002, 268, 208–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pohorille, A.; Deamer, D. Artificial cells: Prospects for biotechnology. Trends Biotechnol. 2002, 20, 123–128. [Google Scholar] [CrossRef]
- Luisi, P.L.; Ferri, F.; Stano, P. Approaches to semi-synthetic minimal cells: A review. Naturwissenschaften 2006, 93, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Mansy, S.S.; Szostak, J.W. Reconstructing the emergence of cellular life through the synthesis of model protocells. Cold Spring Harbor Symp. Quant. Biol. 2009, 74, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Ichihashi, N.; Matsuura, T.; Kita, H.; Sunami, T.; Suzuki, H.; Yomo, T. Constructing partial models of cells. Cold Spring Harbor Perspect. Biol. 2010, 2, a004945. [Google Scholar] [CrossRef] [PubMed]
- Kurihara, K.; Tamura, M.; Shohda, K.-I.; Toyota, T.; Suzuki, K.; Sugawara, T. Self-reproduction of supramolecular giant vesicles combined with the amplification of encapsulated DNA. Nat. Chem. 2011, 3, 775–781. [Google Scholar] [CrossRef] [PubMed]
- Nourian, Z.; Roelofsen, W.; Danelon, C. Triggered gene expression in fed-vesicle microreactors with a multifunctional membrane. Angew. Chem. Int. Ed. 2012, 51, 3114–3118. [Google Scholar] [CrossRef] [PubMed]
- Blain, J.C.; Szostak, J.W. Progress Toward Synthetic Cells. Annu. Rev. Biochem. 2014, 83, 615–640. [Google Scholar] [CrossRef] [PubMed]
- Luisi, P.L.; Varela, F.J. Self-replicating micelles—A chemical version of a minimal autopoietic system. Origins Life Evol. Biosphere 1989, 19, 633–643. [Google Scholar] [CrossRef]
- Bachmann, P.; Luisi, P.; Lang, J. Autocatalytic Self-Replicating Micelles as Models for Prebiotic Structures. Nature 1992, 357, 57–59. [Google Scholar] [CrossRef]
- Oberholzer, T.; Wick, R.; Luisi, P.L.; Biebricher, C.K. Enzymatic RNA replication in self-reproducing vesicles: An approach to a minimal cell. Biochem. Biophys. Res. Commun. 1995, 207, 250–257. [Google Scholar] [CrossRef] [PubMed]
- Stano, P.; Mavelli, F. Protocells Models in Origin of Life and Synthetic Biology. Life 2015, 5, 1700–1702. [Google Scholar] [CrossRef]
- Oberholzer, T.; Nierhaus, K.H.; Luisi, P.L. Protein expression in liposomes. Biochem. Biophys. Res. Commun. 1999, 261, 238–241. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Sato, K.; Wakabayashi, M.; Nakaishi, T.; Ko-Mitamura, E.P.; Shima, Y.; Urabe, I.; Yomo, T. Synthesis of functional protein in liposome. J. Biosci. Bioeng. 2001, 92, 590–593. [Google Scholar] [CrossRef]
- Stano, P.; Carrara, P.; Kuruma, Y.; de Souza, T.P.; Luisi, P.L. Compartmentalized reactions as a case of soft-matter biotechnology: Synthesis of proteins and nucleic acids inside lipid vesicles. J. Mater. Chem. 2011, 21, 18887–18902. [Google Scholar] [CrossRef]
- Altamura, E.; Carrara, P.; D’Angelo, F.; Mavelli, F.; Stano, P. Extrinsic stochastic factors (solute partition) in gene expression inside lipid vesicles and lipid-stabilized water-in-oil droplets: A review. Synth. Biol. 2018, 3, ysy011. [Google Scholar] [CrossRef]
- Mavelli, F.; Stano, P. Experiments on and Numerical Modeling of the Capture and Concentration of Transcription–translation Machinery inside Vesicles. Artif. Life 2015, 21, 445–463. [Google Scholar] [CrossRef] [PubMed]
- Paradisi, P.; Allegrini, P.; Chiarugi, D. A renewal model for the emergence of anomalous solute crowding in liposomes. BMC Syst. Biol. 2015, 9, S7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimizu, Y.; Inoue, A.; Tomari, Y.; Suzuki, T.; Yokogawa, T.; Nishikawa, K.; Ueda, T. Cell-free translation reconstituted with purified components. Nat. Biotechnol. 2001, 19, 751–755. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, Y.; Kanamori, T.; Ueda, T. Protein synthesis by pure translation systems. Methods 2005, 36, 299–304. [Google Scholar] [CrossRef] [PubMed]
- Sunami, T.; Sato, K.; Matsuura, T.; Tsukada, K.; Urabe, I.; Yomo, T. Femtoliter compartment in liposomes for in vitro selection of proteins. Anal. Biochem. 2006, 357, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Murtas, G.; Kuruma, Y.; Bianchini, P.; Diaspro, A.; Luisi, P.L. Protein synthesis in liposomes with a minimal set of enzymes. Biochem. Biophys. Res. Commun. 2007, 363, 12–17. [Google Scholar] [CrossRef] [PubMed]
- Weitz, M.; Kim, J.; Kapsner, K.; Winfree, E.; Franco, E.; Simmel, F.C. Diversity in the dynamical behaviour of a compartmentalized programmable biochemical oscillator. Nat. Chem. 2014, 6, 295–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, H.; Nilsson, L.; Kurland, C.G. Co-variation of tRNA abundance and codon usage in Escherichia coli at different growth rates. J. Mol. Biol. 1996, 260, 649–663. [Google Scholar] [CrossRef] [PubMed]
- Carrara, P.; Stano, P.; Luisi, P.L. Giant Vesicles and w/o Emulsions as Biochemical Reactors. Orig. Life Evol. Biosph. 2008, 39, 179–392. [Google Scholar]
- Siegal-Gaskins, D.; Tuza, Z.A.; Kim, J.; Noireaux, V.; Murray, R.M. Gene circuit performance characterization and resource usage in a cell-free “breadboard”. ACS Synth. Biol. 2014, 3, 416–425. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, M.K.; Hayes, C.A.; Chappell, J.; Sun, Z.Z.; Murray, R.M.; Noireaux, V.; Lucks, J.B. Characterizing and prototyping genetic networks with cell-free transcription–translation reactions. Methods 2015, 86, 60–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garamella, J.; Marshall, R.; Rustad, M.; Noireaux, V. The All E. coli TX–TL Toolbox 2.0: A Platform for Cell-Free Synthetic Biology. ACS Synth. Biol. 2016, 5, 344–355. [Google Scholar] [CrossRef] [PubMed]
- Stano, P.; D’Aguanno, E.; Bolz, J.; Fahr, A.; Luisi, P.L. A remarkable self-organization process as the origin of primitive functional cells. Angew. Chem. Int. Ed. Engl. 2013, 52, 13397–13400. [Google Scholar] [CrossRef] [PubMed]
- Mavelli, F.; Marangoni, R.; Stano, P. A Simple Protein Synthesis Model for the PURE System Operation. Bull. Math. Biol. 2015, 77, 1185–1212. [Google Scholar] [CrossRef] [PubMed]
- Stögbauer, T.; Windhager, L.; Zimmer, R.; Rädler, J.O. Experiment and mathematical modeling of gene expression dynamics in a cell-free system. Integr. Biol. 2012, 4, 494–501. [Google Scholar] [CrossRef] [PubMed]
- Matsuura, T.; Kazuta, Y.; Aita, T.; Adachi, J.; Yomo, T. Quantifying epistatic interactions among the components constituting the protein translation system. Mol. Syst. Biol. 2009, 5, 297. [Google Scholar] [CrossRef] [PubMed]
- Fischer, A.; Franco, A.; Oberholzer, T. Giant vesicles as microreactors for enzymatic mRNA synthesis. ChemBioChem 2002, 3, 409–417. [Google Scholar] [CrossRef]
- Gallo, V.; Stano, P.; Luisi, P.L. Protein Synthesis in Sub-Micrometer Water-in-Oil Droplets. ChemBioChem 2015, 16, 2073–2079. [Google Scholar] [CrossRef] [PubMed]
- Fiordemondo, D.; Stano, P. Lecithin-based water-in-oil compartments as dividing bioreactors. ChemBioChem 2007, 8, 1965–1973. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pereira de Souza, T.; Stano, P.; Luisi, P.L. The minimal size of liposome-based model cells brings about a remarkably enhanced entrapment and protein synthesis. ChemBioChem 2009, 10, 1056–1063. [Google Scholar] [CrossRef] [PubMed]
- Iizuka, R.; Yamagishi-Shirasaki, M.; Funatsu, T. Kinetic study of de novo chromophore maturation of fluorescent proteins. Anal. Biochem. 2011, 414, 173–178. [Google Scholar] [CrossRef] [PubMed]
- Macdonald, P.J.; Chen, Y.; Mueller, J.D. Chromophore maturation and fluorescence fluctuation spectroscopy of fluorescent proteins in a cell-free expression system. Anal. Biochem. 2012, 421, 291–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group D | Group E | Group B | |
|---|---|---|---|
| 1 [19,20] | DNA | Transcription T7 RNA polymerase Translation Ribosomes, MTF,IF1, IF2, IF3, EF-G, EF-Tu, EF-Ts, RF1, RF2, RF3, RRF tRNA charging 20 aa-tRNA synthetase Energy recycling creatine kinase, myokinase, nucleoside-diphosphate kinase, pyrophosphatase | 20 amino acids tRNA mix (46) 2 ATP, GTP, CTP, UTP creatine phosphate Hepes–KOH, pH 7.6 potassium glutamate magnesium acetate spermidine DTT 10-formyl-5,6,7,8-tetrahydrofolic acid |
| Numerical model [Mavelli et al., 2015] | DNA | Txcat Tlcat Rscat ENcat | A T NTP CP |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carrara, P.; Altamura, E.; D’Angelo, F.; Mavelli, F.; Stano, P. Measurement and Numerical Modeling of Cell-Free Protein Synthesis: Combinatorial Block-Variants of the PURE System. Data 2018, 3, 41. https://doi.org/10.3390/data3040041
Carrara P, Altamura E, D’Angelo F, Mavelli F, Stano P. Measurement and Numerical Modeling of Cell-Free Protein Synthesis: Combinatorial Block-Variants of the PURE System. Data. 2018; 3(4):41. https://doi.org/10.3390/data3040041
Chicago/Turabian StyleCarrara, Paolo, Emiliano Altamura, Francesca D’Angelo, Fabio Mavelli, and Pasquale Stano. 2018. "Measurement and Numerical Modeling of Cell-Free Protein Synthesis: Combinatorial Block-Variants of the PURE System" Data 3, no. 4: 41. https://doi.org/10.3390/data3040041
APA StyleCarrara, P., Altamura, E., D’Angelo, F., Mavelli, F., & Stano, P. (2018). Measurement and Numerical Modeling of Cell-Free Protein Synthesis: Combinatorial Block-Variants of the PURE System. Data, 3(4), 41. https://doi.org/10.3390/data3040041